

Abstract
Cardiomyocyte proteostasis is mediated by the ubiquitin/proteasome system (UPS) and autophagy/lysosome system and is fundamental for cardiac adaptation to both physiologic (e.g., exercise) and pathologic (e.g., pressure overload) stresses. Both the UPS and autophagy/lysosome system exhibit reduced efficiency as a consequence of aging, and dysfunction in these systems is associated with cardiomyopathies. The muscle-specific ubiquitin ligase atrogin-1 targets signaling proteins involved in cardiac hypertrophy for degradation. Here, using atrogin-1 KO mice in combination with in vivo pulsed stable isotope labeling of amino acids in cell culture proteomics and biochemical and cellular analyses, we identified charged multivesicular body protein 2B (CHMP2B), which is part of an endosomal sorting complex (ESCRT) required for autophagy, as a target of atrogin-1–mediated degradation. Mice lacking atrogin-1 failed to degrade CHMP2B, resulting in autophagy impairment, intracellular protein aggregate accumulation, unfolded protein response activation, and subsequent cardiomyocyte apoptosis, all of which increased progressively with age. Cellular proteostasis alterations resulted in cardiomyopathy characterized by myocardial remodeling with interstitial fibrosis, with reduced diastolic function and arrhythmias. CHMP2B downregulation in atrogin-1 KO mice restored autophagy and decreased proteotoxicity, thereby preventing cell death. These data indicate that atrogin-1 promotes cardiomyocyte health through mediating the interplay between UPS and autophagy/lysosome system and its alteration promotes development of cardiomyopathies.
Introduction
Cardiac muscle mass adapts as a consequence of functional requirements. Increased workload, such as during exercise or chronic disease stresses causing pressure overload, induces cardiomyocyte hypertrophy, while hemodynamic unloading leads to heart atrophy (1–3). Such changes in cell size depend on the equilibrium between the rates of protein synthesis and degradation, both of which are finely regulated by a number of signaling pathways and require a tight control over protein turnover and protein quality control to avoid accumulation of unfolded/misfolded proteins and subsequent ER stress (4–6). Unfolded/misfolded proteins are degraded by the ubiquitin/proteasome system (UPS) and autophagy/lysosome system (7–9). Proteins degraded via UPS are linked covalently by a polyubiquitin chain through a multistep reaction involving different enzymes, named ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin ligase (E3) enzymes, the last of which determines substrate specificity and the rate of ubiquitination (5, 10, 11). Atrogin-1 (also known as MAFbx) is a muscle-specific ubiquitin-ligase (12, 13), originally identified as a mediator of muscle atrophy under the control of FoxO transcription factors (14, 15). Similar to skeletal muscle (12), atrogin-1 upregulation in the heart leads to atrophy (16) and regression from calcineurin-dependent cardiac hypertrophy (17–19). However, it has been proposed recently that atrogin-1 inhibition prevents pressure overload induced cardiac hypertrophy (20).
Parallel to the UPS, autophagy is a conserved mechanism, which is upregulated in response to several stressors (21). Autophagy begins with the engulfment of a portion of cytoplasm by a double membrane to form the autophagosome, which, by fusing to lysosome, culminates in the degradation of the sequestered material. Among the key regulators of autophagy, the endosomal sorting complex required for transport (ESCRT) protein complexes have been reported to affect lysosome-autophagosome fusion (22–24). The functionality of both UPS and autophagy/lysosome system independently and of their interplay is essential for cell and tissue homeostasis. Indeed, both primary (e.g., mutations in autophagy-related genes) and secondary (e.g., unfolded protein accumulation) dysfunction in either the UPS or autophagy/lysosome system have been associated with cardiomyopathies and neurodegenerative diseases (4, 25, 26), mainly because the failure of one of the two systems leads to substrate overload and consequent impairment of the other. A common finding in aging is the reduced exercise tolerance due to cardiac dysfunction, which has been attributed to a number of concurrent mechanisms, including increased oxidative damage, activation of unfolded protein response (UPR), and increased rates of apoptotic cardiomyocyte death (27). All these observations suggest that alterations in the UPS and autophagy might be centrally involved in cardiac age-related dysfunction. Here, we demonstrate that atrogin-1 mediates the cross-talk between the UPS and autophagy/lysosome system by targeting a key effector of autophagy, the ESCRT-III protein charged multivesicular body protein 2B (CHMP2B), for proteasome degradation. CHMP2B accumulation, in the absence of atrogin-1, causes a block in the autophagy/lysosome system that leads to apoptosis and, with time, to cardiomyopathy and premature death.
Results
Genetic ablation of atrogin-1 causes myocardial interstitial fibrosis and cardiomyopathy.
Atrogin-1 is a muscle-specific ubiquitin ligase that has been associated with heart diseases, which represents an attractive novel target of cardiovascular therapy. However, the role of atrogin-1 in cardiomyocyte biology as well as its involvement in the molecular mechanism of cardiac dysfunction is largely unexplored. To address the role of atrogin-1 in the regulation of cardiomyocyte proteostasis and function, we monitored the cardiac phenotype of adult atrogin-1 KO mice (6 months old) onward until death and compared it with that of age- and sex-matched atrogin-1 haploinsufficient (atrogin-1+/–) mice and C57BL/6J WT controls. At 6 and 9 months old, both cardiac morphology and size were unchanged in atrogin-1 KO mice compared with that in controls (heart weight/body weight: 6 month, atrogin-1 KO vs. controls: 0.0048 ± 0.0004 vs. 0.0047 ± 0.0005; 9 month, atrogin-1 KO vs. controls: 0.0048 ± 0.0004 vs. 0.0047 ± 0.0002) (Figure 1, A and B), and consistently cardiomyocyte cross-sectional areas were identical to those of controls (Figure 1, C and D). At 16 months old, however, hearts of atrogin-1 KO mice appeared abnormal in shape with rounded ventricles, grossly dilated left atrium (LA), and increased heart weight, resulting in higher heart weight/body weight ratios (16 month, atrogin-1 KO vs. controls: 0.0090 ± 0.0003 vs. 0.0046 ± 0.0002) (Figure 1, A and B). In these hearts, cardiomyocyte cross-sectional areas were significantly increased (RV, atrogin-1 KO vs. controls: 279.85 ± 1.52 μm2 vs. 230.82 ± 1.58 μm2; LV, atrogin-1 KO vs. controls: 289.34 ± 2.23 μm2 vs. 236.77 ± 1.64 μm2) (Figure 1, C and D), and, interestingly, coexistence of hypertrophic and normally sized cardiomyocytes was observed in the LV wall (Figure 1, E and F). Such hypertrophic remodeling in aged atrogin-1 KO hearts was accompanied by an increase in Akt and S6 kinase phosphorylation, both of which regulate protein synthesis (Figure 1G). Reactivation of the fetal isoforms β-myosin heavy chain and atrial natriuretic factor, which is a hallmark of pathologic myocardial hypertrophy (28, 29), was evident in the aged atrogin-1 KO hearts (Figure 1, H and I). Echocardiography demonstrated that 16-month-old atrogin-1 KO hearts had thickened LV wall, without significant systolic dysfunction (ejection fraction, atrogin-1 KO vs. controls: 60.47% ± 10.98% vs. 55.20% ± 12.36%) (Figure 2, A and B, and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI66339DS1). Remarkably, echo-Doppler measurements showed that atrogin-1 KO mice, which had normal diastolic function at 6 and 9 months of age, developed significant impairment in diastolic relaxation at 16 months of age, as demonstrated by a reduction of the E wave deceleration time in the mitral valve inflow pattern measurements (E wave deceleration time, atrogin-1 KO vs. controls: 20.7 ± 2.7 ms vs. 27.8 ± 5.7 ms) (Figure 2B). We next used telemetry monitoring of the ECG to analyze heart rhythm in freely moving mice. Aged atrogin-1 KO mice had unchanged heart rhythm compared with that of controls but showed frequent ventricular ectopies and runs of ventricular tachycardia, both of which indicate increased arrhythmia vulnerability (Figure 2, C–E). These morphological and functional data were well supported by the observation that the myocardial interstitium, which was normal in 6-month-old atrogin-1 KO hearts, showed rare foci of fibrosis at 9 months of age and progressed into diffuse LV and LA fibrosis in the aged KO hearts (Figure 3, A–F, and Supplemental Figure 1, A and B). Such fibrotic remodeling was due to abundant interstitial collagen I and collagen VI deposition, which was predominant in the LV and interventricular septum (IVS) (atrogin-1 KO: collagen I, RV: 5.0% ± 1.4% collagen area per total area; IVS: 12.0% ± 1.6% collagen area per total area; LV: 16.0% ± 2.3% collagen area per total area; controls: collagen I, RV: 1.2% ± 0.3% collagen area per total area; IVS: 1.5% ± 0.5% collagen area per total area; LV: 1.5% ± 0.5% collagen area per total area) (Figure 3, G and H, and Supplemental Figure 1, A–D). Our findings indicate that atrogin-1 KO mice progressively develop cardiomyopathy with interstitial fibrosis, diastolic dysfunction accompanied by intraventricular conduction defects, and secondary LA and ventricular remodeling, all of which are common features of cardiomyopathies with a restrictive pattern (30). In further support of this, aged atrogin-1 KO mice had reduced tolerance to treadmill exercise compared with that of age-matched controls and shortened life span (Figure 3I and Supplemental Figure 1E).
Figure 1. Atrogin-1 KO mice develop an age-related cardiomyopathy.

(A) Hearts from 6-, 9-, and 16-month-old control and atrogin-1 KO mice. (B) Evaluation of the heart weight/body weight (HW/BW) ratio in 6-, 9-, and 16-month-old control (C) and atrogin-1 KO mice. Error bars indicate SEM (**P < 0.01; n = 6 hearts for each group). (C) Confocal immunofluorescence on ventricular cryosections from 6-, 9-, and 16-month-old control and atrogin-1 KO mice stained with an antibody against dystrophin. Images show details from the RV. Scale bar: 50 μm. The morphometric evaluation of RV cardiomyocyte cross-sectional area is also shown. Error bars indicate SEM (**P < 0.01; n = 6 hearts for each group). (D) Evaluation of LV cardiomyocyte area in 6-, 9-, and 16-month-old control and atrogin-1 KO mice. Error bars indicate SEM (**P < 0.01; n = 6 hearts for each group). (E) Confocal immunofluorescence on LV cryosections from aged control and atrogin-1 KO hearts stained with an antibody against dystrophin, showing the mixture of normal sized and hypertrophic cardiomyocytes. Scale bar: 50 μm. (F) Evaluation of cardiomyocyte size distribution in the LV of 16-month-old control and atrogin-1 KO hearts. (G) Western blotting on ventricular extracts from aged control and atrogin-1 KO hearts. (H and I) Confocal immunofluorescence analysis of ventricular cryosections from aged control and atrogin-1 KO mice stained with antibodies to (H) β-myosin heavy chain and (I) atrial natriuretic factor. Scale bar: 50 μm.
Figure 2. Aged atrogin-1 KO mice show impaired heart function.

(A) Echocardiographic long-axis view of 16-month-old control and atrogin-1 KO hearts. Ao, aorta. (B) Quantification of LV internal diameter (LVID), LV posterior wall (LVPW) thickness, LV length, LA anterior-posterior wall (LA ant-post wall), ejection fraction, and E wave deceleration time (EdT) in 16-month-old control and atrogin-1 KO mice. Error bars indicate SEM (*0.01 < P < 0.05; **P < 0.01; controls, n = 5 mice; atrogin-1 KO, n = 7 mice). (C) Quantification of heart rate (bpm) in freely moving aged control and atrogin-1 KO mice. Error bars indicate SEM (n = 4 mice for each group). (D) ECG recording in freely moving aged control (bottom) and atrogin-1 KO (top, 2 examples) mice. Atrogin-1 KO mice show runs of ventricular tachycardia. Estimation of heart rate is reported in the first ECG trace from an atrogin-1 KO mouse. (E) Quantification of ventricular ectopies per minute in aged control and atrogin-1 KO mice. Error bars indicate SEM (**P < 0.01; n = 4 mice for each group). PVC, premature ventricular complexes.
Figure 3. Atrogin-1 KO mice develop myocardial interstitial fibrosis during aging.

(A–F) Hematoxylin-eosin staining on ventricular cryosections from (A and D) 6-, (B and E) 9-, and (C and F) 16-month-old atrogin-1 KO mice, showing the presence of interstitial fibrosis in 9-month-old and 16-month-old atrogin-1 KO hearts. Scale bar: 1 mm (A–C); 50 μm (D–F). (G and H) Confocal immunofluorescence analysis of ventricular cryosections from 16-month-old (G) control and (H) atrogin-1 KO mice costained with antibodies against collagen I (red signal) and sarcomeric actinin (α-actinin, green signal). The right images in G and H show high-magnification views of the white boxes in the composite image (left images). White arrows indicate collagen I accumulation in the LV interstitium of atrogin-1 KO hearts. Scale bar: 1 mm (left images); 50 μm (right images). (I) Survival probability of male control (black line) and atrogin-1 KO (red line) mice, evaluated by using Kaplan-Meier method. Comparison of control and atrogin-1 KO survival curves was performed by both Mantel-Cox and Gehan-Breslow-Wilcoxon tests. Atrogin-1 KO mice show a significant reduction in the life span, as compared with that of controls (P < 0.05).
Increased apoptosis and ER stress in atrogin-1–null hearts.
The presence of interstitial fibrosis in atrogin-1 KO hearts prompted us to investigate whether increased apoptosis rates were detectable. The number of apoptotic cardiomyocytes, assessed either using detection of the cleaved form of caspase-3 or performing TUNEL assay, was increased already in 9-month-old hearts, in which myocardial fibrotic remodeling was still moderate (Supplemental Figure 2A). In aged KO mice, apoptotic nuclei were mainly detected within the LV and in the IVS (TUNEL: LV, atrogin-1 KO vs. control: 11.37 ± 5.50 vs. 0.13 ± 0.47 nuclei per field; IVS, atrogin-1 KO vs. control: 7.22 ± 3.58 vs. 0.36 ± 0.60 nuclei per field) (Figure 4, A–C), both regions characterized by the highest degree of fibrosis (Figure 3, G and H). We thus used electron microscopy to investigate cardiomyocyte ultrastructure and gain insight into the cellular mechanisms leading to apoptosis in the atrogin-1 KO hearts. This analysis revealed cardiomyocyte areas with diffuse sarcomeric disarray and expansion of ER cisternae. Moreover, within the region of ER proliferation, we found small electron-pale mitochondria (Figure 4D and Supplemental Figure 2B). These ultrastructural features are consistent with activation of the ER stress response, which was evaluated by assessing UPR genes (e.g., ATF4 and its downstream targets CHOP and GADD34) and protein markers (e.g., eIF2α, CHOP, and the ER stress chaperone BIP). Interestingly, upregulation of UPR genes, phosphorylation of eIF2α, and BIP and CHOP accumulation were detected in atrogin-1 KO hearts and were already present in 9-month-old animals, prior to the onset of overt myocardial alterations ultimately leading to cardiomyopathy (Figure 4, E–G), strongly supporting that activation of ER stress preceded apoptosis and subsequent myocardial fibrosis.
Figure 4. Atrogin-1 KO hearts show increased cardiomyocyte apoptosis and ER stress.

(A) Confocal immunofluorescence analysis of ventricular cryosections from 16-month-old control and atrogin-1 KO mice costained with wheat germ agglutinin (wga, green signal) and an antibody against the cleaved isoform of caspase-3 (red signal). Arrows indicate apoptotic cardiomyocytes in atrogin-1 KO hearts. Scale bar: 25 μm. (B) Immunofluorescence and bright-field images of heart cryosections from aged control and atrogin-1 KO mice simultaneously stained with an antibody to dystrophin and used for TUNEL assay (brown signal). Nuclei were counterstained with DAPI (blue signal). The asterisks indicate an apoptotic cardiomyocyte in an atrogin-1 KO heart. Scale bar: 10 μm. (C) Quantification of TUNEL-positive nuclei in aged control and atrogin-1 KO hearts. Error bars indicate SEM (***P < 0.001; n = 6 hearts for each group). (D) Electron micrographs of ventricular thin sections from aged control and atrogin-1 KO mice. The inset shows abnormal ER cisternae in atrogin-1 KO cardiomyocytes. Scale bar: 1 μm; 1.5 μm (inset). (E) Real-time quantitative PCR analysis of ventricular extracts from 9- and 16-month-old control and atrogin-1 KO mice demonstrating upregulation of genes involved in ER stress. Error bars indicate SEM (*0.01 < P < 0.05; **P < 0.01; n = 5 hearts for each group). (F and G) Western blotting on ventricular extracts from (F) 9- and (G) 16-month-old control and atrogin-1 KO mice, showing a marked increase of ER stress activation markers in atrogin-1 KO hearts.
The autophagy/lysosome machinery is impaired in atrogin-1–null hearts.
We have demonstrated previously that, in skeletal muscles, impairment of autophagy leads to intracellular protein accumulation and increased apoptosis (31, 32), and skeletal muscle–specific inhibition of autophagy causes UPR activation (33). Moreover, interplay between UPS and autophagy has been proposed recently in the setting of neurodegenerative diseases (34), indicating that impairment of UPS impinges on the correct function of the autophagy/lysosome system. Interestingly, atrogin-1 KO hearts were characterized by ultrastructural features similar to those of autophagy-defective muscles, such as sarcomeric disarray and changes in mitochondrial morphology (33). Therefore, we assessed the state of the autophagy/lysosome system in adult and aged atrogin-1 KO hearts. Accumulation of Bnip3 (data not shown) and beclin-1 proteins (Figure 5A) indicated that autophagy was induced in the atrogin-1 KO hearts. However, we detected increased levels of p62 and lipidated LC3 proteins (Figure 5, A and B), a finding consistent with autophagy impairment (35). In line with this, immunofluorescence revealed that those atrogin-1 KO cardiomyocytes that showed sarcomeric disruption had abundant p62 aggregates and LC3 puncta (Figure 5, C and D, and Supplemental Figure 3A). Importantly, LC3 and p62 staining did not colocalize (Figure 5E and Supplemental Figure 3, B–D), suggesting that LC3-positive puncta are truly autophagosomes and are not due to LC3 sequestration into p62-positive protein aggregates. Western blot and immunofluorescence analyses revealed an increase in the amount of the lysosomal marker LAMP2 and in lysosome number in atrogin-1–null cardiomyocytes (Figure 5, F and G). We thus sought to determine whether autophagic and lysosomal markers colocalized, using coimmunofluorescence for LC3 and LAMP2, which demonstrated that the majority of autophagosomes were juxtaposed to lysosomes but did not colocalize (Figure 5H and Supplemental Figure 4). To further confirm that ablation of atrogin-1 causes a block of the autophagy flux, we treated adult control and atrogin-1 KO mice with bafilomycin, an inhibitor of autophagosome/lysosome fusion. Consistent with our hypothesis, bafilomycin increased the LC3-II band in control mice, while it did not elicit any significant change in atrogin-1 KO hearts (Figure 5I). Altogether, these results suggest that, in atrogin-1 KO cardiomyocytes, despite normal autophagy induction, autophagosome/lysosome fusion is compromised, culminating in the block of the autophagic flux.
Figure 5. Atrogin-1 ablation leads to impairment in the autophagy flux.

(A and B) Western blotting on ventricular extracts from aged control, atrogin-1 KO, and starved control (starv) mice. The LC3-II/β-tubulin ratio was evaluated in control and atrogin-1 KO hearts. Error bars indicate SEM (*P < 0.01; n = 6 for each group). (C and D) Confocal immunofluorescence on ventricular cryosections from 9- and 16-month-old control and atrogin-1 KO mice costained with antibodies against sarcomeric actinin (α-actinin) and (C) p62 or (D) LC3, showing p62 and LC3 aggregates in both adult and aged atrogin-1 KO cardiomyocytes (arrows). Scale bar: 20 μm (inset in C); 25 μm (C, 9- and 16-month-old control and 9-month-old atrogin-1 KO, and D); 50 μm (C, 16-month-old atrogin-1 KO). (E) Confocal 3D reconstruction of a ventricular cardiomyocyte from an aged atrogin-1 KO mouse costained with antibodies against p62 and LC3. Scale bar: 20 μm (left image); 10 μm (right image). (F) Western blotting on ventricular extracts from 9- and 16-month-old control and atrogin-1 KO hearts, showing LAMP2 accumulation in atrogin-1 KO hearts. (G) Confocal immunofluorescence on ventricular cryosections from aged control and atrogin-1 KO mice costained with antibodies against LAMP2 and dystrophin. Scale bar: 50 μm. (H) Confocal 3D reconstruction of a ventricular cardiomyocyte from aged atrogin-1 KO mice costained with antibodies against LAMP2 and LC3. Scale bar: 30 μm; 5 μm (inset). (I) Western blotting on ventricular extracts from aged control and atrogin-1 KO mice treated either with DMSO or the lysosome inhibitor bafilomycin (BFL). The LC3-II/β-tubulin ratio was evaluated in DMSO- and BLF-treated control and atrogin-1 KO hearts. Error bars indicate SEM (**P < 0.01; n = 6 for each group).
The ESCRT-III protein CHMP2B accumulates in atrogin-1–null hearts.
In order to explain how the lack of atrogin-1, a ubiquitin ligase so far associated with the proteasomal function, leads to inhibition of the autophagy/lysosome system, we considered the possibility that atrogin-1 regulates the turnover of proteins involved in the autophagy/lysosome machinery. To address this issue, we used in vivo stable isotope labeling of amino acids in cell culture (SILAC) mass spectrometry analysis to compare heart protein turnover rates in the 9-month-old atrogin-1 KO and control mice. Metabolic protein profiling with labeled amino acids (pulsed SILAC) is a straightforward approach to assess changes in protein turnover. Here, we fed the mice for 2 weeks with a diet containing a labeled isotope of the essential amino acid lysine (13C6 lysine [Lys-6]). Subsequent mass spectrometry in heart tissue extracts allowed, for every protein detected, quantification of both its absolute amount and the Lys-6–incorporating fraction (newly synthesized within the diet period), thus inferring the protein turnover rate. Consistent with the reduced efficiency of autophagy/lysosome system in atrogin-1 KO hearts, pulsed SILAC experiments demonstrated that substrates of lysosomes (36), including Tau, filamin C, GAPDH, and BAG3, had decrease protein turnover and/or increased protein concentration (Supplemental Tables 2 and 3).
Given that our results indicate that the maturation and fusion of autophagosome/lysosome vesicle is impaired in KO hearts, we selected among the proteins accumulated in atrogin-1–null hearts 32 candidate proteins involved in vesicular trafficking (Table 1 and Supplemental Table 4). We further sorted these hits for biological function and focused on the presence of 2 ESCRT proteins, VPS25 and CHMP2B, that belong to ESCRT-II and ESCRT-III complexes, respectively. ESCRT proteins are necessary for autophagosome formation, and they have been shown to have a role in autophagosome/lysosome fusion and/or in sealing the membrane of the autophagosome (37). Interestingly, a mutation in CHMP2B causing its altered turnover has been linked to the block of autophagy and subsequent neurodegeneration (38), and we thus focused on this protein. Western blotting and immunofluorescence demonstrated that CHMP2B was increased in atrogin-1 KO hearts already at 9 months of age and accumulated further at 16 months (Figure 6, A and B). Whereas in control hearts CHMP2B distributed homogenously throughout the cardiomyocyte, in atrogin-1 KO hearts, it was found only in large aggregates in areas depleted of sarcomeric proteins (Figure 6B and Supplemental Figure 5, A and B). Compared with the controls, CHMP2B staining only minimally overlapped with the lysosomal marker LAMP2 in the KO cardiomyocytes (Figure 6C and Supplemental Figure 5C), suggesting that impaired CHMP2B clearance may result in incomplete autophagic flux.
Table 1.
Proteins involved in intracellular vesicle trafficking and maturation
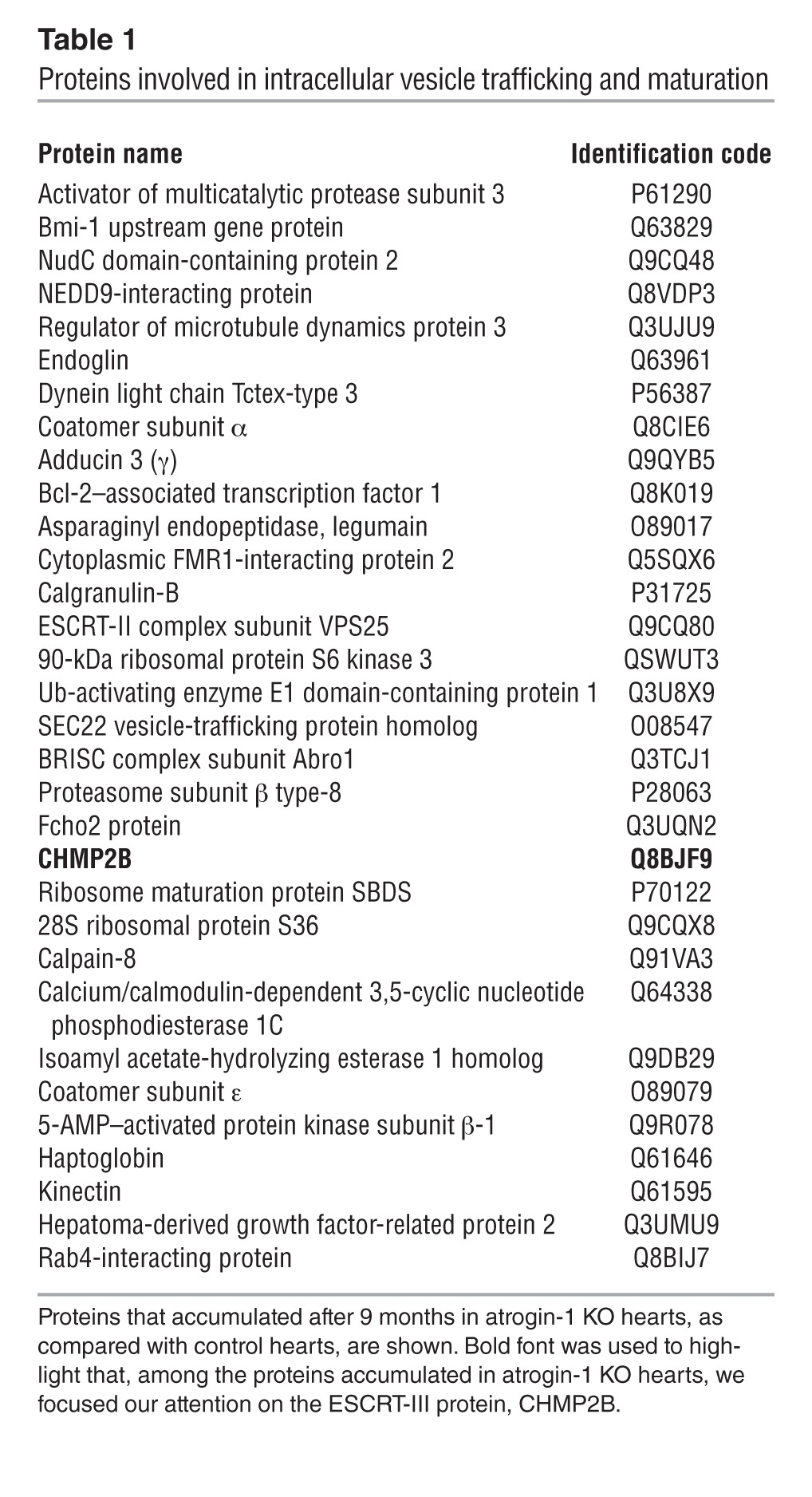
Figure 6. The ESCRT-III protein CHMP2B is a novel target of atrogin-1.

(A) Western blotting on ventricular extracts from 9- and 16-month-old control and atrogin-1 KO mice. (B) Confocal immunofluorescence on ventricular cryosections from 9- and 16-month-old control and atrogin-1 KO mice costained with antibodies against sarcomeric actinin (α-actinin) and CHMP2B. White arrows show CHMP2B accumulation. Scale bar: 20 μm; 5 μm (inset). (C) Confocal 3D reconstruction of a ventricular cardiomyocyte costained with antibodies to LAMP2 and CHMP2B. Note no coexpression of LAMP2 and CHMP2B. Scale bar: 2 μm. (D) Immunoprecipitation with an anti-CHMP2B antibody on protein extracts from control and MG132-treated cultured cardiomyocytes, showing CHMP2B polyubiquitination upon UPS block. (E) Western blotting on protein extracts from control and MG132-treated cultured cardiomyocytes, showing CHMP2B accumulation upon UPS inhibition. (F) Confocal immunofluorescence on control and MG132-treated cultured cardiomyocytes costained with antibodies to cardiac troponin I (cTnI) and CHMP2B. Note CHMP2B accumulation in cardiomyocyte areas with sarcomeric disruption. Scale bar: 50 μm; 2 μm (insets). (G) Cultured cardiomyocytes were cotransfected with myc-tag atrogin-1 and flag-tag CHMP2B, proteins were isolated and used for coimmunoprecipitation analysis. Note that CHMP2B coimmunoprecipitates with atrogin-1, indicating a direct interaction between CHMP2B and atrogin-1. (H and I) Immunoprecipitation with an antibody specific for flag-tag (CHMP2B) on extracts from the tibialis anterior of control mice transfected by electroporation with vectors encoding for CHMP2B, SKP1, CUL1, and ROC in the presence or the absence of atrogin-1. The Western blots show increased (H) higher molecular weights and (I) ubiquitination of CHMP2B in the presence of atrogin-1, as compared with controls.
CHMP2B is a target of atrogin-1.
We used in vitro experiments to directly test the hypothesis that CHMP2B is degraded by the UPS upon interaction with atrogin-1. WT rat neonatal cardiomyocytes were treated with the proteasome inhibitor MG132, which caused CHMP2B polyubiquitination (Figure 6D) and accumulation (Figure 6E) that occurred predominantly in areas depleted of sarcomeric proteins (Figure 6F and Supplemental Figure 6). In addition, flag-tagged CHMP2B coimmunoprecipitated with myc-tagged atrogin-1 in cells cotransfected with atrogin-1 and CHMP2B (Figure 6G). Atrogin-1 is a ubiquitin ligase belonging to the SCF family that binds the substrate and forms a complex with SKP1/CUL1/ROC to transfer ubiquitin from the E2 enzyme to the substrate. Importantly, when atrogin-1 was coexpressed with the SKP1/CUL1/ROC complex in skeletal muscles, it enhanced CHMP2B ubiquitination (Figure 6H). Altogether, these data indicate that atrogin-1 interacts directly with CHMP2B and mediates its targeting to the UPS for degradation.
Since our in vivo results also suggest that accumulation of CHMP2B in atrogin-1 KO hearts may result in autophagy impairment, we used CHMP2B overexpression to test the hypothesis that accumulation of this ESCRT-III protein impairs autophagic flux in normal cardiomyocytes. Cells were transiently transfected with YFP-LC3 to label autophagic vesicles, thus enabling quantification of the cell fraction occupied by autophagosomes with confocal microscopy. Consistent with our hypothesis, CHMP2B overexpression was sufficient to increase the number of YFP-LC3 vesicles (Figure 7, A–C). Importantly, bafilomycin treatment did not increase LC3-positive puncta in CHMP2B-overexpressing cardiomyocytes, further supporting the concept that CHMP2B accumulation negatively regulates the autophagic flux (Figure 7, C and D).
Figure 7. CHMP2B overexpression causes an impairment in the autophagy flux per se.

(A and B) Confocal immunofluorescence analysis of neonatal WT cardiomyocytes transfected either with (A) LC3-YFP (green signal) or (B) LC3-YFP/CHMP2B and stained with an antibody against CHMP2B (red signal). (C) Cultured neonatal cardiomyocytes were transfected either with YFP-LC3 (green signal, –CHMPB) or cotransfected with YFP-LC3– and CHMP2B-expressing vectors (+CHMP2B). Autophagosomes were analyzed by confocal microscopy. The same analysis was performed also in the presence of the autophagy inhibitor bafilomycin (+BFL). The right-most images in A–C show high-magnification views of the white boxes in the left images. Scale bar: 10 μm (left and middle images); 1 μm (right images). (D) Quantification of the total number of YFP-LC3 vesicles in cardiomyocytes both at baseline and upon CHMP2B overexpression both in presence and absence of bafilomycin. Error bars indicate SEM (**P < 0.01; n = 50 cardiomyocytes for each group).
Collectively, our in vitro data corroborate the in vivo findings and demonstrate that CHMP2B is a molecular target of atrogin-1 and support the concept that the failure to degrade CHMP2B results in its accumulation and consequent block of autophagy.
In vivo silencing of CHMP2B in atrogin-1 KO cardiomyocytes removes the block of autophagy and decreases apoptosis.
Based on our observations, we hypothesized that CHMP2B downregulation in atrogin-1 KO hearts would reduce its accumulation and minimize its inhibitory effect on autophagy, thus reducing UPR activation and protecting cardiomyocytes from apoptosis. To this purpose, 9-month-old atrogin-1 KO mice were transduced with an adeno-associated viral vector (AAV9) encoding both small hairpin RNA (shRNA) against the CHMP2B gene and GFP to label infected cells. Four weeks after infection, about 50% of the cardiomyocytes expressed GFP and had reduced immunoreactivity for anti-CHMP2B antibody (mean CHMP2B fluorescence intensity: GFP-positive cardiomyocytes, 108 ± 8 AU; non-GFP-expressing cardiomyocytes, 44 ± 2 AU), indicating that CHMP2B was significantly knocked down in infected cells (Figure 8A and Supplemental Figure 7A). No decrease in CHMP2B fluorescence was observed in cardiomyocytes from hearts transduced with a control adeno-associated viral vector (AAV9) encoding GFP (mean CHMP2B fluorescence intensity: GFP-positive cardiomyocytes, 108 ± 8 AU; non-GFP-expressing cardiomyocytes: 108 ± 0.3 AU) (Figure 8A and Supplemental Figure 7B). Remarkably, in the shRNA-treated mice, most of the nontransduced cells had intracellular CHMP2B aggregates (Figure 8B, white arrow), which were never observed in GFP-positive cardiomyocytes. Furthermore, LC3 and p62 aggregates, which were frequently detected in cardiomyocytes of 9-month-old atrogin-1 KO mice (Figure 8, C–E), were present in cardiomyocytes that did not express shRNAs against CHMP2B but were never observed in cells that expressed oligos against CHMP2B (Figure 8, C–E), suggesting that knockdown of CHMP2B restored normal autophagy. That CHMP2B knockdown restored the autophagic flux in atrogin-1 KO mice was further supported by the colocalization of autophagosomes and lysosomes in CHMP2B-downregulated cardiomyocytes (Figure 8F). Moreover, while a marked fraction of GFP-negative cardiomyocytes expressed cleaved caspase-3, apoptosis markers were never expressed in cardiomyocytes in which CHMP2B was knocked down (Figure 8G). These results validate the notion that the reduction of CHMP2B protein level protects atrogin-1 KO cardiomyocytes from the block of autophagy and induction of cell death resulting from CHMP2B proteotoxicity.
Figure 8. CHMP2B knockdown protects atrogin-1 KO hearts from block of autophagy and cardiomyocyte apoptosis.

(A) Evaluation of mean CHMP2B fluorescence intensity in cryosections from AAV9-GFP-U6-CHMP2Bsh– and AAV9-GFP–treated atrogin-1 KO mice, costained with antibodies to CHMP2B and GFP. Both control (GFP–) and silenced GFP+ cardiomyocytes were analyzed. Error bars represent SEM (**P < 0.01; n = 800 cardiomyocytes from 4 hearts). CHMP2Bsh cardiomyocytes show a significant decrease in CHMP2B fluorescence, as compared with that of controls. No decrease in CHMP2B fluorescence was observed in cardiomyocytes transduced with control viral vector. (B) 3D reconstruction of a cryosection from an AAV9-GFP-U6-CHMP2Bsh–treated atrogin-1 KO mouse costained with antibodies to CHMP2B and GFP, showing no CHMP2B accumulation in GFP+ cells. The white arrow indicates GFP-negative, nonsilenced cells with CHMP2B aggregates visible. Scale bar: 25 μm. (C and D) Confocal immunofluorescence on cryosections from AAV9-GFP-U6-CHMP2Bsh–treated atrogin-1 KO mice, costained with antibodies to GFP and (C) LC3 or (D) p62, showing no accumulation of LC3 and p62 in GFP+ CHMP2Bsh cardiomyocytes (arrows). Scale bar: 25 μm; 10 μm (inset in C). (E) Evaluation of percentage of p62-accumulating cardiomyocytes in GFP– noninfected and GFP+ CHMP2Bsh cells, showing that CHMP2B silencing prevents p62 accumulation. Error bars represent SEM (*0.01 < P < 0.05; n = 450 cardiomyocytes from 4 hearts). (F) Confocal immunofluorescence on cryosections from AAV9-GFP-U6-CHMP2Bsh–treated atrogin-1 KO mice, costained with antibodies to GFP (blue signal), LC3 (green signal), and LAMP2 (red signal), showing colocalization of LC3 and LAMP2 in GFP+ CHMP2Bsh cardiomyocytes. Asterisks indicate GFP-positive, CHMP2Bsh cardiomyocytes. Section symbols indicate GFP-negative, nonsilenced cardiomyocytes. Higher-magnification views of boxed images appears to the right. Scale bar: 25 μm (costaining images); 5 μm (right colocalization images); 10 μm (left colocalization images). (G) Confocal immunofluorescence on cryosections from AAV9-GFP-U6-CHMP2Bsh–treated atrogin-1 KO mice costained with antibodies to GFP and cleaved caspase-3. Higher-magnification views of boxed images appears to the right. Arrows indicate GFP-negative, nonsilenced cardiomyocytes showing cleaved caspase-3 expression. Scale bar: 50 μm (left 3 images); 20 μm (right-most image).
Discussion
Tight regulation of proteostasis plays a housekeeping cellular function in postmitotic tissues, such as the myocardium and the nervous tissue, that rely critically on mechanisms to control both the quality of newly synthesized proteins and the efficient removal of damaged and unfolded/misfolded proteins. The coordinated work of the UPS and the autophagy/lysosome system (7–9) is necessary, i.e., when UPS targets are too large/too numerous to be degraded by the proteasome, autophagy must intervene. Here, we identify a novel link between UPS and autophagy and show that the muscle-specific ubiquitin ligase atrogin-1 controls turnover of the ESCRT-III family protein CHMP2B, which controls the autophagy system (39). In atrogin-1 KO hearts, the primary failure to regulate CHMP2B turnover causes its intracellular aggregation (Figure 6B and Supplemental Figure 5B), contributing to the secondary impairment of autophagy that, by amplifying unfolded/misfolded proteotoxicity, ensues in cardiomyopathy. These results highlight the importance of regulated proteolysis in the heart and show that the loss of atrogin-1 per se is sufficient to cause cardiac damage, which leads to cardiomyopathy when protein quality control becomes less efficient, as occurring in aging (ref. 25 and Figure 9).
Figure 9. Role of atrogin-1 in the cross-talk between cardiomyocyte UPS and autophagy/lysosome system.

(A) The muscle-specific ubiquitin ligase atrogin-1 regulates the turnover of CHMP2B, an ESCRT-III protein essential for lysosome biogenesis and autophagosome/lysosome fusion to form the autophagosome/lysosome. Ablation of atrogin-1 reduces CHMP2B turnover, leading to CHMP2B accumulation into cardiomyocyte areas characterized by sarcomeric depletion. (B) The impaired turnover of CHMP2B compromises endosome maturation and autophagosome/lysosome fusion, resulting in the block of the autophagic flux (i.e., lysosome and autophagosome accumulation), activation of markers of ER stress, expansion of ER cisternae, and modifications of mitochondria morphology, all of which lead to cardiomyocyte apoptosis, resulting in a diffuse myocardial interstitial fibrosis (Figures 3 and 4).
Proteotoxicity cardiomyopathy in the atrogin-1 KO mice.
Initially, atrogin-1 was identified as a mediator of skeletal muscle atrophy and being involved in the degradation of calcineurin and the activation of FoxO1 and FoxO3 transcription factors (18, 19, 40, 41); more recently, a role in the proteolysis of the NF-κB inhibitor IkB (20) has been reported. Our systematic morphological, functional, and biochemical analyses of the atrogin-1 KO hearts at different ages, ranging from 6 to 16 months, demonstrated aging-related myocardial remodeling, which was characterized by the coexistence of hypertrophic and atrophic cells (42) in the LV (Figure 1, E and F). When looked at at the ultrastructural level, abnormalities observed included ER proliferation, disappearance of contractile elements, and abnormal mitochondria, all of which are consistent with activated ER stress and impairment of autophagy (Figures 4 and 5). Such reduction of the autophagy flux resulted in the accumulation of p62- and LC3-positive autophagosomes and was further supported by the blunted increase in the lipidated form of LC3 upon lysosome inhibition (Figure 5I). The resulting defect caused the formation of p62-positive aggregates and increased cardiomyocyte apoptosis, which was followed by fibrotic myocardial replacement, eventually causing diastolic dysfunction (Figures 2 and 3).
Defective control of proteostasis has been implicated in cardiomyopathies due to either accumulation of incorrectly folded proteins (e.g., the product of mutant cMyBP-C and CryAB genes) (43, 44) or acquired dysfunction of protein quality control, as occurring in heart failure (45–47). In the former scenario, the myocardium features abundant fibrotic replacement, and these cardiomyopathies have a restrictive pattern, similar to that observed in the atrogin-1 KO mice (Figure 2, A and B).
In other models, the genetic ablation of Atg5, a critical gene for autophagosome formation, causes cardiomyopathy and heart failure, supporting the concept that autophagy inhibition is detrimental for cardiac function (48). Conversely, it has been demonstrated recently that enhancement of autophagy ameliorates cardiac function and prevents accumulation of intracellular aggregates in a murine model of proteotoxic cardiomyopathy caused by mutant CryABR120G (49).
When we explored autophagy at higher detail in the atrogin-1 KO mice, autophagosomes and lysosomes appeared increased in number and size but segregated (Figure 5, F–H), suggesting a failure in the late steps of autophagy. This feature resembles that of myopathies with lysosomal defect (e.g., Danon disease) (50, 51) and prompted us to search whether atrogin-1 targets included proteins involved in the endosome/lysosome maturation and fusion with autophagosomes. The use of unbiased pulsed labeled proteomics in vivo (SILAC) allowed us to identify, among the proteins with increased turnover in the KO hearts, the ESCRT-III family protein CHMP2B, a previously unrecognized target of atrogin-1.
CHMP2B is a novel target of atrogin-1.
The combined molecular, biochemical, proteomic, and in vivo approaches in the KO murine model identified CHMP2B as a novel target of atrogin-1. CHMP2B is a component of the heteromeric ESCRT-III complex essential for endosome trafficking, virus budding, cytokinesis, and the autophagy process (26). CHMP2B is necessary for cell viability, and its ablation impairs autophagy, causing inhibition of autophagosome/lysosome fusion and, therefore, accumulation of autophagosomes. Mutations in CHMP2B have been found in familial neurodegenerative diseases (e.g., frontotemporal dementia and amyotrophic lateral sclerosis), in which, consistent with a modulatory role in autophagy, brain pathology shows intracellular accumulation of p62-positive protein aggregates (22, 39).
Since the turnover of CHMP2B is defective in the atrogin-1 KO hearts, the clearance of the unfolded/misfolded forms of the protein is compromised. The progressive build up of the aggregate-prone CHMP2B would inevitably increase the possibility of aggregation and inclusion formation. Interestingly, when CHMP2B is expressed in WT-cultured cardiomyocytes, intracellular aggregates develop in a time-dependent manner (our unpublished data), suggesting that, above a given concentration threshold, the protein has the tendency to form amyloid-like complexes. In support of this, bioinformatics prediction (Protein Aggregation Prediction Server; http://www.abl.ku.edu/ProA/) (52) shows 3 aggregation-prone domains in CHMP2B, resulting in a high aggregation score. It may be speculated that formation of intracellular CHMP2B aggregates impairs autophagy either by sequestering the functional form of the protein or other regulatory factors (e.g., interacting components of the ESCRT-III or ESCRT-II complexes), with subsequent inhibition of ESCRT complex function that ultimately affects lysosomes maturation and the autophagy system.
During aging, the reduced efficiency of autophagy in the atrogin-1 KO mice adds to the decreased protein quality control, aggravating the proteotoxic load and triggering activation of UPR and ER stress, which eventually lead to increased cardiomyocyte apoptosis. Among the proteins with impaired turnover in the atrogin-1 KO heart, our proteomics analysis identified other putative targets of the ubiquitin ligase, e.g., ribosomal S6 kinase 2 (RSK2), that might contribute to the pathological events that lead to the cardiomyopathy in addition to CHMP2B. It is thus likely that the atrogin-1 KO cardiomyopathy results from the impairment of multiple signaling pathways converging in ER stress and apoptosis. However, the downregulation of CHMP2B is sufficient to significantly reduce cardiomyocyte death, indicating that its accumulation has a dominant role in the pathogenesis of myocardial remodeling in the absence of atrogin-1.
Proteasome inhibition: friend or foe?
Usui et al. demonstrated recently that atrogin-1 KO mice are protected from developing myocardial hypertrophy upon transverse aortic coarctation through sustained IkB-mediated NF-κB inhibition (20), suggesting that proteasome inhibition might be beneficial to prevent hypertrophy in pressure overload (53, 54). Our findings support the concept that sustained inhibition of atrogin-1 is deleterious for cardiac muscle and should be avoided because it affects basic cellular functions, including autophagy regulation and ER function. In addition, our results are of immediate clinical relevance given the increasing use of proteasome inhibitors as antitumor treatment (55). Recent case reports provide alarming signals that treatment with bortezomib, a proteasome inhibitor approved by the FDA for multiple myeloma, might be associated with cardiotoxicity (56–58), and in experimental models, bortezomib treatment caused protein accumulation, ER stress, and ultrastructural abnormalities in both cardiac sarcomeres and mitochondria (59).
Conclusions.
In conclusion, these data highlight the importance of regulated proteolysis in the heart and show that the loss of atrogin-1 per se is sufficient to cause cardiac damage, which evolves into cardiomyopathy when protein quality control becomes less efficient, as occurring in aging. Such cardiomyopathy represents a novel model of proteotoxic myocardial remodeling, which will be useful to determine the mechanism of impaired proteostasis and its contribution to cardiomyocyte damage. Further studies will aim to determine whether mutations in the genes connected to the atrogin-1 signaling cascade may be responsible for human familial cardiomyopathies of undetermined genetic cause.
Methods
Mice.
Atrogin-1 KO mice were provided by David Glass (Novartis Institutes for Biomedical Research, Cambridge, Massachusetts, USA). Atrogin-1+/– mice were backcrossed with the inbred C57BL/6J strain (Charles River) for 7 generations. Atrogin-1 KO mice were obtained by crossing heterozygous atrogin-1+/– mice and identified by PCR analysis. All experiments were conduced in 6-, 9-, and 16-month-old atrogin-1+/–, atrogin-1, and C57BL/6J WT male mice.
Echocardiography and echo-Doppler.
Echocardiography was performed both in adult and aged atrogin-1 KO mice and age- and sex-matched littermate controls, using a Vevo 2100 (VisualSonics) system equipped with a 30-MHz transducer. Anesthesia was induced with 3% isoflurane and maintained with 1.5% isoflurane during constant monitoring of temperature, respiration rate, and ECG. Two-dimensional cine loops with frame rates of 200 frames per second of a long-axis view and a short-axis view at proximal, mid, and apical level of the LV were recorded. IVS and LV posterior wall thicknesses, LV internal diameter, and maximal LV length were measured in systole and in diastole from the long-axis B-mode image, according to standard procedures. Ejection fraction (EF) was determined by the following formula, based on the Simple method (Simp): %EF = 100 × systolic LV volume/diastolic LV volume.
To assess changes in LV shape, the sphericity index was calculated at end-diastole and end-systole as the volume of the LV divided by the volume of a sphere with a diameter equal to the LV longest axis (measured in the apical view) using the following formulas (4, 5):
End-diastolic sphericity index = end-diastolic LV volume/(3/4 × π × [end-diastolic Simp length]3)
(Equation 1)
End-systolic sphericity index = end-systolic LV volume/(3/4 × π × [end-systolic Simp length]3)
(Equation 2)
As this ratio increases, the ventricle becomes more spherical. The Doppler frequency images of ascending aorta and mitral flow were acquired from the long-axis B-mode image. The Doppler frequency image of pulmonary artery flow was acquired from a cranial long-axis view. Doppler analysis of mitral valve flow was performed from the long-axis B-mode image, placing the sample volume in the LV below the mitral annulus. Echocardiographic image acquisition and analysis were performed by a single operator, blinded to the mouse genotype.
Telemetry implantation and ECG recording.
Telemeters (Data Sciences International) were implanted in 15-month-old WT and atrogin-1 KO male mice. Animals were anesthetized with 1.5% isoflurane (v/v O2), and implantation was performed according to the manufacturer’s procedure. After 10 days to allow complete recovery from surgery, ECG recording was started and carried on for 2 months.
Tissue samples and immunofluorescence analysis.
The hearts were harvested from C57BL/6J WT, atrogin-1+/–, and atrogin-1 KO mice; fixed in 1% paraformaldehyde (Sigma-Aldrich) at room temperature for 15 minutes; equilibrated in sucrose gradient; frozen in liquid nitrogen; sectioned; and processed for histological and immunofluorescence analyses. Ten-micron myocardial sections were obtained with a cryostat (Leica CM1850, Leica Microsystems GmbH) and incubated with primary antibodies diluted in PBS supplemented with 1% BSA and 0.5% Triton X-100 overnight at 4°C. Antibodies used in this study are listed in Supplemental Table 5. FITC- and TRITC-conjugated secondary antibodies, all from Jackson lab, were used to detect primary antibodies.
TUNEL assay.
TUNEL assay was performed in 9- and 16-month-old control and atrogin-1 KO heart cryosections, by using the ApopTag peroxidase in situ apoptosis detection system, following the procedure suggested by the manufacturer (Chemicon). A total of 6 nonconsecutive 10-μm cryosections from the mid-portion of the ventricles for each heart were analyzed. Cryosections were stained with an antibody to dystrophin, and nuclei were counterstained with DAPI. The number of apoptotic nuclei was estimated in 18 randomly selected fields from the RV (n = 6) and the LV (n = 6) walls and the IVS (n = 6) using ImageJ software.
Morphometric quantification of cardiomyocyte cross-sectional area.
Cardiomyocyte cross-sectional area was quantified in both adult and aged control and atrogin-1 KO mouse hearts. Five nonconsecutive cryosections from the mid-portion of the ventricles per heart were stained with an antibody against dystrophin and analyzed with fluorescence microscopy. The images of 12 randomly chosen fields, area of 69,000 μm2 from the RV (n = 6) and the LV (n = 6), were acquired with a fluorescence microscope (Leica DC130, Leica Microscopes), and cardiomyocyte cross-sectional area was evaluated using a custom-developed routine running in Matlab (MathWorks).
Electron microscopy analysis.
We used conventional fixation-embedding procedures based on glutaraldehyde-osmium fixation and Epon embedding (33).
Western blotting.
For details, see the Supplemental Methods.
Real-time quantitative PCR.
For details, see the Supplemental Methods and Supplemental Table 6.
Immunoprecipitation.
While cells were lysed as described for Western blot, tissues were ground in Nitrogen and then added to lysis buffer (50 mM Tris-HCl pH 7.5, 1 mM EGTA, 10% glycerol, 0.1% Triton, 50 mM MgCl2) and further homogenized with a Polytron (Kinematica). Lysates were then centrifuged for 5 minutes at 5,000 g, supernatant was collected, and protein concentration was quantified. 500 μg of and 1 mg of total proteins, respectively from cells and muscles, were used for every immunoprecipitation. After a preclearing with A/G+ beads (Santa Cruz), lysates were cleaned from beads and incubated overnight with the selected antibody. The following day, A/G+ beads were added to lysate to bind the antibody-protein complex and left 2 hours in agitation at 4°C. Then, precipitated proteins were washed with ice-cold PBS and loaded in a denaturating gel.
Mass spectrometry–based proteomics.
For details, see the Supplemental Methods.
Bafilomycin mouse treatment.
Bafilomycin (Sigma-Aldrich) was dissolved in DMSO and intraperitoneally injected in adult (9-month-old) control and atrogin-1 KO mice at a dosage of 2 mg/kg. Mice were maintained in absence of food. Four hours after injection, mice were sacrificed and hearts were removed and processed for molecular and biochemical analyses. A group of control mice injected with the same amount of the vehicle was also tested.
In vitro UPS blockage.
Cardiomyocytes cultured for 4 days were treated with 20 μM MG132 for either 12 or 24 hours. At the end of MG132 treatment, cells were either fixed for immunofluorescence analyses or used for protein isolation, as described in the Supplemental Methods.
In vivo CHMP2B silencing.
Fifty μl of either 1 × 1013 per ml AAV9-GFP-U6-CHMP32Bsh or 1 × 1013 per ml AAV9-GFP (both from VECTOR BIOLABS) were injected via tail vein in 10-month-old atrogin-1 KO mice. Mice were sacrificed 3 and 4 weeks upon infection, and hearts were harvested and processed for immunofluorescence and molecular analyses.
Statistics.
All data are expressed as mean ± SEM. Comparison between the experimental groups was performed by using 2-tailed Student’s t test. P < 0.05 was considered statistically significant.
Study approval.
All experimental procedures were performed according to the European Commission guidelines and have been approved by the local ethical committee and the relevant Italian authority (Ministero della Salute, Ufficio VI), in compliance of Italian Animal Welfare Law (law no. 116/1992 and subsequent modifications) and complying with the Directive 2010/63/EU of the European Parliament.
Supplementary Material
Acknowledgments
This work was supported by the Foundation Leducq, Telethon-Italy (TCP04009); the European Community Seventh Framework Program of FP7 (MYOAGE, contract 223576); the European Research Council (MyoPHAGY, contract 282310); the Italian Ministry of Education to M. Sandri; University of Padova (Progetto Ateneo 2010, contract CPDA0977); the European Community Seventh Framework Program FP7/2007-2013 under grant agreement HEALTH-F2-2009-241526, EUTrigTreat, and Telethon-Italy (GGP11224) to M. Mongillo; and University of Padova (Progetto Giovani Studiosi 2010, contract: GRIC101133) to T. Zaglia. We thank David J. Glass for providing atrogin-1 KO mice; Stefano Schiaffino for critical reading; Domenico Corrado for interpretation of ECG data; Giulia Borile, Emilio Bigon, Eleonora Piasentini, and Silvia Gaiani for technical assistance.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(6):2410–2424. doi:10.1172/JCI66339.
References
- 1.Weeks KL, McMullen JR. The athlete’s heart vs. the failing heart: can signaling explain the two distinct outcomes? Physiology (Bethesda). 2011;26(2):97–105. doi: 10.1152/physiol.00043.2010. [DOI] [PubMed] [Google Scholar]
- 2.Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res. 2011;90(2):234–242. doi: 10.1093/cvr/cvr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baskin KK, Taegtmeyer H. Taking pressure off the heart: the ins and outs of atrophic remodelling. Cardiovasc Res. 2011;90(2):243–250. doi: 10.1093/cvr/cvr060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlossarek S, Carrier L. The ubiquitin-proteasome system in cardiomyopathies. Curr Opin Cardiol. 2011;26(3):190–195. doi: 10.1097/HCO.0b013e32834598fe. [DOI] [PubMed] [Google Scholar]
- 5.Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106(3):463–478. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willis MS, Patterson C. Into the heart: the emerging role of the ubiquitin-proteasome system. J Mol Cell Cardiol. 2006;41(4):567–579. doi: 10.1016/j.yjmcc.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Zheng Q, Li J, Wang X. Interplay between the ubiquitin-proteasome system and autophagy in proteinopathies. Int J Physiol Pathophysiol Pharmacol. 2009;1(2):127–142. [PMC free article] [PubMed] [Google Scholar]
- 8.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584(7):1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 9.Korolchuk VI, Menzies FM, Rubinsztein DC. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy. 2009;5(6):862–863. doi: 10.4161/auto.8840. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99(12):1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 11.Gomes AV, Zong C, Ping P. Protein degradation by the 26S proteasome system in the normal and stressed myocardium. Antioxid Redox Signal. 2006;8(9–10):1677–1691. doi: 10.1089/ars.2006.8.1677. [DOI] [PubMed] [Google Scholar]
- 12.Bodine SC, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294(5547):1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 13.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98(25):14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandri M, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412. doi: 10.1016/S0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stitt TN, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14(3):395–403. doi: 10.1016/S1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 16.Zaglia T, et al. Cardiac sympathetic neurons provide trophic signal to the heart via beta2-adrenoceptor-dependent regulation of proteolysis. Cardiovasc Res. 2013;97(2):240–250. doi: 10.1093/cvr/cvs320. [DOI] [PubMed] [Google Scholar]
- 17.Skurk C, et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280(21):20814–20823. doi: 10.1074/jbc.M500528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li HH, et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114(8):1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li HH, et al. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117(11):3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Usui S, et al. Endogenous muscle atrophy F-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-κB. Circ Res. 2011;109(2):161–171. doi: 10.1161/CIRCRESAHA.110.238717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Portbury AL, Willis MS, Patterson C. Tearin’ up my heart: proteolysis in the cardiac sarcomere. J Biol Chem. 2011;286(12):9929–9934. doi: 10.1074/jbc.R110.170571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filimonenko M, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179(3):485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17(18):1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 24.Rusten TE, et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol. 2007;17(20):1817–1825. doi: 10.1016/j.cub.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 25.Powell SR. The ubiquitin-proteasome system in cardiac physiology and pathology. Am J Physiol Heart Circ Physiol. 2006;291(1):H1–H19. doi: 10.1152/ajpheart.00062.2006. [DOI] [PubMed] [Google Scholar]
- 26.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458(7237):445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 27.Higami Y, Shimokawa I. Apoptosis in the aging process. Cell Tissue Res. 2000;301(1):125–132. doi: 10.1007/s004419900156. [DOI] [PubMed] [Google Scholar]
- 28.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109(13):1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 29.Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122(25):2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336(4):267–276. doi: 10.1056/NEJM199701233360407. [DOI] [PubMed] [Google Scholar]
- 31.Grumati P, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16(11):1313–1320. doi: 10.1038/nm.2247. [DOI] [PubMed] [Google Scholar]
- 32.Yu Z, et al. Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet. 2011;7(10):e1002321. doi: 10.1371/journal.pgen.1002321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masiero E, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10(6):507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Cecarini V, et al. Crosstalk between the ubiquitin-proteasome system and autophagy in a human cellular model of Alzheimer’s disease. Biochim Biophys Acta. 2012;1822(11):1741–1751. doi: 10.1016/j.bbadis.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 35.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larsen KB, Lamark T, Overvatn A, Harneshaug I, Johansen T, Bjorkoy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy. 2010;6(6):784–793. doi: 10.4161/auto.6.6.12510. [DOI] [PubMed] [Google Scholar]
- 37.Metcalf D, Isaacs AM. The role of ESCRT proteins in fusion events involving lysosomes, endosomes and autophagosomes. Biochem Soc Trans. 2010;38(6):1469–1473. doi: 10.1042/BST0381469. [DOI] [PubMed] [Google Scholar]
- 38.Lee JA, Liu L, Gao FB. Autophagy defects contribute to neurodegeneration induced by dysfunctional ESCRT-III. Autophagy. 2009;5(7):1070–1072. doi: 10.4161/auto.5.7.9823. [DOI] [PubMed] [Google Scholar]
- 39.Lee JA, Gao FB. Roles of ESCRT in autophagy-associated neurodegeneration. Autophagy. 2008;4(2):230–232. doi: 10.4161/auto.5384. [DOI] [PubMed] [Google Scholar]
- 40.Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006;20(10):1660–1670. doi: 10.1096/fj.05-5560com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heineke J, et al. Calcineurin protects the heart in a murine model of dilated cardiomyopathy. J Mol Cell Cardiol. 2010;48(6):1080–1087. doi: 10.1016/j.yjmcc.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maron BJ, Ferrans VJ, Roberts WC. Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am J Pathol. 1975;79(3):387–434. [PMC free article] [PubMed] [Google Scholar]
- 43.Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109(2):151–160. doi: 10.1161/CIRCRESAHA.110.237339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlossarek S, Mearini G, Carrier L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J Mol Cell Cardiol. 2011;50(4):613–620. doi: 10.1016/j.yjmcc.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 45.Tresse E, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6(2):217–227. doi: 10.4161/auto.6.2.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Predmore JM, et al. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121(8):997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caleshu C, et al. Furthering the link between the sarcomere and primary cardiomyopathies: restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am J Med Genet A. 2011;155A(9):2229–2235. doi: 10.1002/ajmg.a.34097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taneike M, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6(5):600–606. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 49.Bhuiyan MS, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123(12):5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malicdan MC, Noguchi S, Nonaka I, Saftig P, Nishino I. Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromuscul Disord. 2008;18(7):521–529. doi: 10.1016/j.nmd.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 51.Sugimoto S. A novel vacuolar myopathy with dilated cardiomyopathy. Autophagy. 2007;3(6):638–639. doi: 10.4161/auto.4931. [DOI] [PubMed] [Google Scholar]
- 52.Fang Y, Gao S, Tai D, Middaugh CR, Fang J. Identification of properties important to protein aggregation using feature selection. BMC Bioinformatics. 2013;14:314. doi: 10.1186/1471-2105-14-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Depre C, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114(17):1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 54.Stansfield WE, Moss NC, Willis MS, Tang R, Selzman CH. Proteasome inhibition attenuates infarct size and preserves cardiac function in a murine model of myocardial ischemia-reperfusion injury. Ann Thorac Surg. 2007;84(1):120–125. doi: 10.1016/j.athoracsur.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 55.Micel LN, Tentler JJ, Smith PG, Eckhardt GS. Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. J Clin Oncol. 2013;31(9):1231–1238. doi: 10.1200/JCO.2012.44.0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Enrico O, et al. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br J Haematol. 2007;138(3):396–397. doi: 10.1111/j.1365-2141.2007.06659.x. [DOI] [PubMed] [Google Scholar]
- 57.Hacihanefioglu A, Tarkun P, Gonullu E. Acute severe cardiac failure in a myeloma patient due to proteasome inhibitor bortezomib. Int J Hematol. 2008;88(2):219–222. doi: 10.1007/s12185-008-0139-7. [DOI] [PubMed] [Google Scholar]
- 58.Voortman J, Giaccone G. Severe reversible cardiac failure after bortezomib treatment combined with chemotherapy in a non-small cell lung cancer patient: a case report. BMC Cancer. 2006;6:129. doi: 10.1186/1471-2407-6-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nowis D, et al. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am J Pathol. 2010;176(6):2658–2668. doi: 10.2353/ajpath.2010.090690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.