

Abstract
In a rat heart allograft model, preventing T cell costimulation with CD40Ig leads to indefinite allograft survival, which is mediated by the induction of CD8+CD45RClo regulatory T cells (CD8+CD40Ig Tregs) interacting with plasmacytoid dendritic cells (pDCs). The role of TCR-MHC-peptide interaction in regulating Treg activity remains a topic of debate. Here, we identified a donor MHC class II–derived peptide (Du51) that is recognized by TCR-biased CD8+CD40Ig Tregs and activating CD8+CD40Ig Tregs in both its phenotype and suppression of antidonor alloreactive T cell responses. We generated a labeled tetramer (MHC-I RT1.Aa/Du51) to localize and quantify Du51-specific T cells within rat cardiac allografts and spleen. RT1.Aa/Du51-specific CD8+CD40Ig Tregs were the most suppressive subset of the total Treg population, were essential for in vivo tolerance induction, and expressed a biased, restricted Vβ11-TCR repertoire in the spleen and the graft. Finally, we demonstrated that treatment of transplant recipients with the Du51 peptide resulted in indefinite prolongation of allograft survival. These results show that CD8+CD40Ig Tregs recognize a dominant donor antigen, resulting in TCR repertoire alterations in the graft and periphery. Furthermore, this allopeptide has strong therapeutic activity and highlights the importance of TCR-peptide-MHC interaction for Treg generation and function.
Introduction
Allogeneic human-to-human transplantation remains the best treatment to replace organs that have failed following disease. The incompatibility between the MHC molecules of the recipient and donor cells is the main barrier to long-term success of organ transplantation. The induction of tolerance to the allograft has become a major objective, and certain tolerance strategies are beginning to be applied clinically (1). Different populations of Tregs have been described as being capable of inducing tolerance to allogeneic organs. Most of these Tregs are CD4+ Tregs, while CD8+ Tregs are less well defined (2).
We have previously described that costimulation blockade of CD40-CD40L interaction, one of the most efficient strategies to prolong organ allograft survival (3), induces CD8+CD45RClo Tregs (called CD8+CD40Ig Tregs) with potent suppressive capacity (2, 4–6). We showed that donor-specific CD8+CD40Ig Tregs, but not natural CD8+CD45RClo Tregs, transferred tolerance to naive transplant recipients. In addition, these cells acted in an unusual way, as allograft survival was dependent on their secretion of IFN-γ to enhance indoleamine 2,3-dioxygenase (IDO) expression by DCs and graft ECs (5). We also recently showed that the suppressive activity of CD8+CD40Ig Tregs mainly occurred in the presence of plasmacytoid DCs (pDCs) and that fibrinogen-like protein 2 (FGL2) was involved in the suppression (6).
The requirement for TCR interaction in shaping of the Treg population is an active and ongoing debate (2, 7). Some studies suggest that TCR specificity and diversity are critical for in vivo function and potency of CD8+ Tregs (2, 7–13). Different models for CD4+ Tregs have shown that antigen-specific Tregs are more potent suppressors than unrestricted Tregs (2, 14). It is also known that TCR diversity is critical for CD4+ Treg thymic selection and differentiation, and the TCR’s impact on Treg generation and function has been recently described (15). High-throughput sequencing has shown that naive Tregs with high TCR diversity expand more efficiently and are more adaptable and more efficient at suppressing graft versus host disease (GVHD) upon adoptive transfer than TCR-restricted Tregs (13, 16). Using an immunoscope, we previously demonstrated that CD8+CD40Ig Tregs accumulated a repertoire biased toward the Vβ11 element (5), suggesting the possibility of clonal expansion. To date, little is known about the recognition features of this Treg population or about CD8+ Treg populations in general.
In the present study, we investigated whether the TCR fine specificity of CD8+CD40Ig Tregs influences Treg function and allograft survival. Here, we have demonstrated for the first time, to our knowledge, in transplantation that induced CD8+CD40Ig Tregs recognize a dominant peptide derived from a polymorphic region of donor MHC class II molecules. This peptide expanded CD8+ Tregs in the presence of pDCs, at least ex vivo, and induced tolerance in naive transplanted recipients without additional treatment. In addition, we generated a specific tetramer and demonstrated both ex vivo and in vivo the dominant tolerance exerted by antigen-specific CD8+CD40Ig Tregs. Finally, we demonstrated that this peptide was recognized by Tregs expressing Vβ11- and Vβ18-specific TCRs. These TCRs comprised a private but restricted Vβ11 repertoire in the spleen and graft, but a private and diverse Vβ18 repertoire in the spleen and a more restricted Vβ18 repertoire in the graft that ensured efficient suppression of alloreactive immune responses.
Results
CD8+CD40Ig Treg activation in vitro.
In order to identify TCR recognition of allogeneic MHC-peptide complexes by CD8+CD40Ig Tregs and the subsequent activation of the function of these Tregs, we had to select a specific marker of activation allowing flow cytometric analysis after exposure to antigenic stimulation. Therefore, we screened molecules expressed at different time points by CD8+CD40Ig Tregs upon stimulation with polyclonal anti-CD3 and anti-CD28 antibodies. Expression of molecules on freshly isolated CD8+CD40Ig Tregs has been previously assessed by quantitative RT-PCR (qRT-PCR) (5) and demonstrated that among these molecules, CD25 and IFN-γ were markers distinguishing CD8+CD40Ig Tregs from other cell populations. On days 0, 1, 2, 3, and 6, we used flow cytometry to analyze Treg expression of CD71, CD25, and IFN-γ (Figure 1).
Figure 1. Screening of CD8+ Treg activation markers by flow cytometry.

(A) CD8+CD45RClo Tregs were isolated from the spleens of CD40Ig-treated rats by cell sorting and analyzed for their expression of T cell activation markers on day 0 and after 1 to 6 days of polyclonal stimulation with anti-CD3/CD28 antibodies. Gray-filled histograms represent the isotype control staining. (B) Data are summarized and plotted as the means ± SEM. n = 3.
We confirmed, on day 0, that CD8+CD40Ig Tregs expressed low levels of CD71 (0.83 ± 0.1%), CD25 (12.74 ± 6.1%), and IFN-γ (5.57 ± 3.3%). After polyclonal stimulation, CD71, CD25, and IFN-γ expression increased markedly from the first day and remained stable over time with, respectively, 82 ± 4.5%, 98.1 ± 1.9%, and 91.7 ± 7% positive cells on day 6.
In conclusion, we identified 3 markers of interest of CD8+CD40Ig Tregs with low basal expression and significant upregulation upon stimulation. Since CD25 was the most and earliest upregulated marker and since it was a marker previously described by us and others (5, 12), we selected this marker to assess CD8+CD40Ig Treg activation in the remaining aspects of this study.
CD8+CD40Ig Tregs recognized 2 donor MHC class II–derived peptides.
In the rat MHC–mismatched heart allograft model, donors (RT1u) and recipients (RT1a) are mismatched for all MHC molecules. We therefore aligned donor and recipient MHC I and II aa sequences and designed 82 overlapping 16-mer peptides matching the polymorphic domains of donor MHC I and II molecules (refs. 17–19 and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI71533DS1). Peptides were first grouped into pools of 6 to 8 peptides (30 μg/ml of each peptide) and tested in an in vitro assay in which immature or mature syngeneic recipient pDCs and sorted CD8+CD40Ig Tregs from CD40Ig-treated long-term allograft–bearing recipients were cocultured for 3 or 6 days. With immature pDCs, we observed no significant activation of CD8+CD40Ig Tregs on day 3 or day 6 with any of the allogeneic pools of peptides (data not shown). After stimulation with mature pDCs and pools of allogeneic peptides, we observed on day 3 a slight upregulation of CD25 expression in a small population of CD8+CD40Ig Tregs, and on day 6, a significant upregulation of CD25 expression following allogeneic stimulation (Supplemental Figure 2). These results suggest that some allogeneic peptides were efficiently recognized by CD8+CD40Ig Tregs and that this recognition led to increased CD25 expression. It also demonstrated that pDCs must be matured in our assay.
We next tested the stimulatory capacity of the 82 individual allopeptides in the presence of naive, matured syngeneic pDCs and CD8+CD40Ig Tregs purified from long-term survivors in a 6-day culture (Figure 2A). We observed that 2 peptides induced a highly significant upregulation of CD25 expression at the cell surface of CD8+CD40Ig Tregs: peptide 31 (called Bu31, 1.67-fold ± 0.09-fold vs. no peptide, P < 0.001), whose sequence overlaps with the peptide 32 sequence (P < 0.05), and peptide 51 (called Du51, 2.07-fold ± 0.18-fold vs. no peptide, P < 0.001). Du51 induced a stronger upregulation of CD25 expression than did Bu31, suggesting that Du51 is the dominant peptide recognized by CD8+CD40Ig Tregs, while Bu31 is subdominant. These results demonstrate that antigen-specific CD8+CD40Ig Tregs mainly recognized 2 peptides, Bu31 (YLRYDSDVGEYRAVTE) and Du51 (NREEYARFDSDVGEYR), derived respectively from the β1 chain of donor MHC class II RT1.Bu and RT1.Du molecules.
Figure 2. Analysis of CD8+CD45RClo Treg activation in response to donor-derived peptide stimulation.

(A) CD8+ Tregs were cocultured for 6 days with syngeneic CpG-matured pDCs in the presence of peptides. For each experiment, the percentage of CD25+ Tregs after 6 days of coculture with pDCs alone was given a value of 1. The mean value of 1 is equal to 32.85 ± 1.98%. Results are expressed as the ratio ± SEM between the percentage of CD25+ cells after peptide stimulation and the percentage of CD25+ cells in the control condition without peptide. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control condition (value 1.0). n = 4–18 for each peptide. (B) Analysis of Treg activation in response to Du51 shorter peptide derivatives. On the left, 18 Du51 derivatives are detailed and classified by aa sequence length, from 9 aa to 15 aa. The box highlights mismatched aa between the donor and recipient. On the right, Treg activation in response to Du51 derivatives was analyzed by CD25 expression. CD8+ Tregs were cocultured for 6 days with syngeneic CpG-matured pDCs in the presence of each peptide. Bars represent the ratio between the percentage of CD25+ cells after peptide stimulation and the percentage of CD25+ cells in the control condition without peptide. *P < 0.05, **P < 0.01, and ***P < 0.001 versus Du51 condition. n = 3–14 for each peptide.
CD8+CD40Ig Tregs recognized an unusually long allogeneic 15-mer peptide.
To determine the sequence of the natural dominant donor peptide recognized by antigen-specific CD8+CD40Ig Tregs, we used a library of degenerated peptides, ranging from 9-mer peptides with 1 aa lagging to 15-mer peptides with 2 or more aa lagging, derived from the dominant 16-mer Du51 (labeled 51-1 to 51-18) (Figure 2B). This library’s design was based on previous results and published reports (20–23) and was tested in the same in vitro assay described above. Interestingly, random peptide libraries have established that rat MHC class I RT1-Aa molecules showed a strong preference for 9- to 15-mer peptides bearing an arginine (R) at the C terminus (23).
None of the derivative 9-mer peptides 51-1 to 51-8 were able to induce activation of CD8+CD40Ig Tregs in a manner equivalent to that observed with the 16-mer Du51. However, we were able to induce a much stronger and substantial CD25 upregulation with a 15-mer derivative peptide (peptide 51-18) (2.04-fold ± 0.3-fold versus no peptide) missing the N-terminal asparagine (N). Contrary to other derivatives, CD25 upregulation induced by peptide 51-18 was not significantly different from that induced by Du51 (Figure 2B).
Altogether, these results show that a dominant MHC class II–derived 15-mer natural peptide (REEYARFDSDVGEYR) was presented to the CD8+CD40Ig Tregs and that such presentation induced activation of the specific cells.
Du51-activated CD8+CD40Ig Tregs displayed a modified phenotype and efficiently suppressed antigen-specific activated T cells.
The phenotype of Du51-activated CD8+CD40Ig Tregs was analyzed 6 days after stimulation (Figure 3, A and B). We previously demonstrated that CD8+CD40Ig Tregs acted through secretion of high levels of IFN-γ, which in turn induced IDO expression by DCs and graft ECs, and this action was necessary for tolerance induction in vivo (5). According to these results, we observed that stimulation of CD8+ Tregs by the peptide in the presence of pDCs led to significantly increased expression of IFN-γ, most likely by activated CD8+CD40Ig Tregs (Figure 3A). In the same culture supernatants, we observed decreased IL-12 production, likely of pDC origin, but no modification of IL-10 or TGF-β expression that could be produced by both CD8+CD40Ig Tregs and pDCs. We also observed an upregulation of CD71, CD28, and MHC class II, but no modification of Foxp3, expression after 6 days of peptide stimulation (Figure 3B).
Figure 3. CD8+ Treg phenotype following ex vivo allopeptide stimulation.

(A) Tregs and syngeneic CpG-matured pDCs were cultured for 6 days alone, with a control peptide, or with peptide Du51. Expression on CD8+ Tregs of the indicated markers was analyzed in the culture supernatant by ELISA (A) or by flow cytometry (B) after in vitro stimulation. Graphs represent the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. n = 4–7.
We previously demonstrated that CD8+CD40Ig Tregs, in the presence of allogeneic pDCs or syngeneic pDCs and a lysate of donor cells, could suppress the proliferation of syngeneic effector CD4+CD25– T cells, showing that CD8+CD40Ig Tregs acted through the direct and indirect pathway of allorecognition and that they are more efficient suppressor cells than naive CD8+CD45RClo Tregs (6). Here, we investigated whether Du51-stimulated CD8+CD40Ig Tregs could efficiently suppress effector T cell proliferation after a 6-day coculture and thus maintain their suppressive activity compared with CD8+CD40Ig Tregs stimulated with a nonactivating control peptide. We performed a mixed lymphocyte reaction (MLR) assay by stimulating CD8+CD40Ig Tregs for 6 days in the presence of syngeneic pDCs and Du51 or nonactivating peptide. Peptide-stimulated CD8+CD40Ig Tregs were then sorted using a FACSAria cell sorter and added in a direct MLR assay of sorted allogeneic pDCs and syngeneic CFSE-labeled CD4+CD25– effector T cells (Supplemental Figure 7). We hypothesized that expanded CD8+CD40Ig Tregs would exert bystander regulation of the proliferation of effector CD4+CD25– T cells stimulated by the direct allorecognition pathway (24, 25). To evaluate this, we measured the relative proportion of dividing CD4+CD25– T cells on day 6 of coculture (Supplemental Figure 3). In the absence of Tregs, we observed a relative proportion of 80.5% of dividing CD4+CD25– effector T cells on day 6, indicating that responding effector T cells had highly proliferated. The addition of Du51-stimulated Tregs resulted in a significant decrease in the relative proportion of dividing CD4+CD25– effector T cells, showing significant inhibition of the division of responding effector T cells compared with that seen in Tregs stimulated by a nonactivating peptide. Thus, Du51 antigen–specific CD8+CD40Ig Tregs maintained an efficient suppressive activity after in vitro activation, and Tregs that remained unstimulated for 6 days lost their suppressive activity and started to die from neglect.
Identification of Du51-specific CD8+CD45RClo Tregs using MHC class I tetramer revealed their enrichment in CD40Ig-treated recipients.
An important challenge in transplantation remains the identification of antigen-specific Tregs, as they represent a more potent suppressive population, and very few natural epitopes have been identified thus far. In addition, to date, in the rat and transplantation settings, no tools have been available to directly visualize and detect antigen-specific CD8+ Treg populations. To that end, we generated the MHC class I tetramer RT1.Aa/Du51, which was labeled with PE and allophycocyanin (APC), and stained specific populations in the spleen and graft (Figure 4, A and B).
Figure 4. Enrichment of Du51-specific Tregs in long-term CD40Ig-treated recipients.

T cell–enriched spleen (A) and total graft (B) from long-surviving recipients or from naive animals were incubated with RT1.Aa/Du51 tetramers labeled with PE- and APC-conjugated streptavidin. Top panels show representative plots. Bottom panels show results for the percentage of Tet+ cells among each population (left), the absolute number of cells (middle), and the percentage of total organ cells (right) ± SEM. *P < 0.05; **P < 0.01. n = 4–6.
Cells were first stained with a mixture of PE- and APC-conjugated RT1.Aa/Du51 tetramers, together with the control tetramer RT1.Aa/MTF-E, which was labeled with BV421 (Supplemental Figure 4). This dual fluorochrome strategy, previously described for antigen-specific CD8+ effector T cells, allows the discrimination of signal and noise staining, as specific CD8+ Tregs will bind equally to each version of RT1.Aa/Du51 tetramer, whereas random elements will not (26). Among double-positive stained events, nonpeptide-specific cells can be visualized and excluded, since they bind to a control tetramer bearing the same heavy-chain RT1.Aa, but with an irrelevant associated peptide (Supplemental Figure 4C). Cells were secondarily stained with CD8-PeCy7 and CD45RC-FITC to identify either CD8+CD45RClo Tregs or CD8+CD45RChi T cells. With such a strategy, we were able to identify Du51-specific cells among CD8+CD40Ig Tregs (2.19 ± 0.6% in the spleen and 1.16 ± 0.25% in the graft) (Figure 4, A and B). In the naive splenic CD8+CD45RClo Treg population, we found the precursor frequency to be 0.73 ± 0.2% (Figure 4A and Supplemental Figure 4C), demonstrating that even 120 days after transplantation and CD40Ig treatment, the frequency was still increased by approximately 3-fold and that a pool of donor-specific Tregs was identified in naive animals (Figure 4A and Supplemental Figure 4C). This difference was true when looking at the percentage and absolute number of tetramer-positive (Tet+) CD8+CD40Ig Tregs or naive CD8+CD45RClo T cells in the spleen (0.624 ± 0.128 vs. 0.173 ± 0.071, respectively, for percentage and 623,800 ± 127,700 vs. 172,600 ± 70,500 for absolute numbers in the spleen, P < 0.05). There were also significantly more Du51-specific cells among CD8+CD40Ig Tregs than among CD8+CD45RChi T cells from naive or CD40Ig-treated animals in terms of the percentage of positive cells in each population and the percentage or absolute number in the total spleen and the percentage in the graft (Figure 4, A and B). We observed no differences in percentages and absolute numbers between CD8+CD45RClo and CD8+CD45RChi T cells from the spleens of naive animals (Figure 4A and Supplemental Figure 4). Interestingly, Tet+ cells were localized both in the graft and the spleen within the CD8hi subset of CD8+CD45RClo Tregs (Figure 4, A and B).
Altogether, these results demonstrate that we were able to generate a functional RT1Aa/Du51 tetramer to detect alloantigen-specific CD8+ Tregs, a population that was significantly increased upon transplantation and CD40Ig treatment.
Superior suppressive capacity of Du51-specific CD8+CD45RClo Tregs mediated by direct and indirect pathways of allorecognition and requirement for in vivo tolerance induction.
We demonstrated the suppressive capacity of Du51-stimulated CD8+CD40Ig Tregs (Supplemental Figure 3). In this experiment, we wanted to study the suppressive capacity of freshly sorted RT1Aa/Du51 tetramer-specific CD8+CD40Ig Tregs (Figure 5A). Naive CD8+CD45RClo Tregs, total CD8+CD40Ig Tregs, RT1Aa/Du51 Tet– CD8+CD40Ig Tregs, and RT1Aa/Du51 Tet+ CD8+CD40Ig Tregs were sorted and incubated for 6 days with allogeneic (direct pathway) or alloantigen-loaded syngeneic pDCs (indirect pathway) and naive CFSE-labeled syngeneic CD4+CD25– T cells. In this assay, RT1Aa/Du51 tetramer–specific CD8+CD40Ig Tregs were activated by tetramer binding (data not shown and ref. 27). As previously described, total CD8+CD40Ig Tregs more efficiently suppressed the proliferation of effector CD4+CD25– T cells induced by both direct and indirect pathways of alloantigen presentation than did naive CD8+CD45RClo Tregs (Figure 5A). Interestingly, we observed a significant difference between the suppressive potential of Tet+ versus Tet– CD8+CD40Ig Tregs mediated by the direct pathway of allorecognition, with the Tet+ CD8+CD40Ig Tregs being the most potent suppressive cell subset, as shown by the decreased relative proportion of dividing CD4+CD25– T cells (Figure 5A). Although not significant, but with a statistical trend (P = 0.0571), we obtained the same difference regarding the indirect alloantigen presentation pathway (Figure 5A). Moreover, the suppression of CD4+CD25–-responding T cell proliferation was more effectively achieved by Du51-specific Tregs when induced by an indirect rather than a direct alloantigen presentation pathway. Interestingly, nonspecific Tet– CD8+CD40Ig Tregs tended to be less suppressive than total CD8+CD40Ig Tregs, highlighting the important contribution of Du51-specific CD8+ Tregs in the overall suppressive capacity of the total CD40Ig Treg pool. These results suggest that Du51 antigen–specific CD8+ Tregs are the most efficient Treg subpopulation of the total CD8+CD40Ig Treg pool.
Figure 5. In vitro and in vivo suppressive potential of Du51-specific CD40IgCD8+ Tregs.

(A) The relative proportion of CFSE-labeled LEW.1A dividing CD4+CD25– T cells stimulated with either donor LEW.1W (direct pathway) or alloantigen-loaded recipient LEW.1A (indirect pathway) pDCs was analyzed after 6 days of culture in the absence or presence of LEW.1A naive, total, Tet–, or Tet+ CD8+CD40Ig Tregs at a 1:1 effector/suppressor ratio. The proportion of dividing CD4+CD25– T cells in the control proliferation condition with pDCs only represented approximately 80% of the cells on day 6 and was given a value of 100 in each experiment. Graphs represent the mean ± SEM of the relative proportion of dividing CD4+CD25– T cells. *P < 0.05. n = 4. (B) 2.5 × 106 total or Tet– CD8+CD40Ig Tregs were injected i.v. into sublethally irradiated recipients (LEW.1A) the day before heart allotransplantation (LEW.1W). Graft survival was assessed by abdominal palpation of cardiac beating. **P < 0.01 for total (n = 4) versus Tet– CD8+CD40Ig Tregs (n = 3).
To study the in vivo relevance of the differential ex vivo suppressive effect observed for Du51-specific CD8+CD40Ig Tregs, we performed adoptive cell transfer experiments. Total CD8+CD40Ig or RT1Aa/Du51 Tet– CD8+CD40Ig Tregs were sorted and adoptively transferred into naive grafted irradiated recipients (Figure 5B). Unlike total CD8+CD40Ig Tregs, Tet– CD8+CD40Ig Tregs, which were depleted in Du51 antigen–specific cells, were unable to inhibit allograft rejection, demonstrating the crucial role of antigen-specific CD8+CD40Ig Tregs in the tolerogenic activity of the total CD8+CD40Ig Treg pool and in preventing allograft rejection and promoting infectious tolerance.
Site-specific accumulation of Vβ-biased CD8+CD45RClo T cells.
In naive T cells, the length distribution of the TCRβ chain CDR3 hypervariable region is Gaussian. Alteration of this Gaussian distribution is the hallmark of activation and selection of T cells in response to antigen. Using immunoscope analysis, we previously showed that CD8+CD40Ig Tregs expressed a specific altered Vβ11 repertoire (and a normal Gaussian-type distribution for all other Vβ families), with the same dominant CDR3β length in all animals (9 aa) (5). These previous results suggested the possibility of an oligoclonal expansion of CD8+CD40Ig T cells. We first confirmed the preferential expression of the Vβ11 chain by flow cytometry using an anti-mouse Vβ11 Ab that cross-reacts with rat (P < 0.01 compared with naive CD8+CD45RClo Tregs) (Supplemental Figure 5, A and B). We also investigated the repertoire of graft-infiltrated T cells (GITCs) from CD40Ig-treated recipients and observed an altered Vβ11 family with a peak of the same CDR3β length (9 aa) in all animals identical to that observed in splenocytes (ref. 5 and data not shown). Furthermore, GITCs displayed several other altered Vβ families (Vβ1, 7, 15, 17, and 18) with the same predominant CDR3β length for each Vβ family and for all animals (i.e., CDR3β length of 7 aa for the Vβ18 family). As compared with naive animals, the accumulation of Vβ11 family transcripts was increased 11-fold in splenic CD8+CD40Ig T cells (5) and 40-fold in the GITCs, while the second-most represented family (Vβ18) in GITCs showed an 11-fold increase and a CDR3β length of 7 aa for each animal (data not shown).
The fact that CD8+CD40Ig Tregs expressed a selected TCR Vβ11 gene family repertoire suggests an induced immune response, giving support to their memory phenotype and, furthermore, a biased TCR response to common antigens. The preferential accumulation of this TCR Vβ11 family repertoire and of other TCR Vβ families in the graft as compared with that in the spleen suggests a preferential migration of biased and expanded CD8+CD40Ig Tregs into the graft (6).
TCR repertoire sequencing reflected differential recruitment of CD8+CD40Ig Tregs to the tolerated organ versus spleen and a private restricted repertoire of Du51 antigen–specific Vβ11+CD8+CD40Ig Tregs.
To analyze the characteristics of the repertoire of Vβ11+ and Vβ18+ spleen and graft Tregs, cDNA obtained from naive, total CD40Ig–induced CD8+CD45RClo Tregs, or RT1Aa/Du51 tetramer–specific CD8+CD40Ig Tregs from 4 to 7 animals per group was amplified using Vβ- and Cβ-specific primers. PCR products were sequenced and TCR aa sequences were analyzed and summarized in Table 1.
Table 1.
Summary of the Vβ11 and Vβ18 repertoires of total or antigen-specific Tregs from spleen and graft from naive and CD40Ig-treated rats 120 days after transplantation
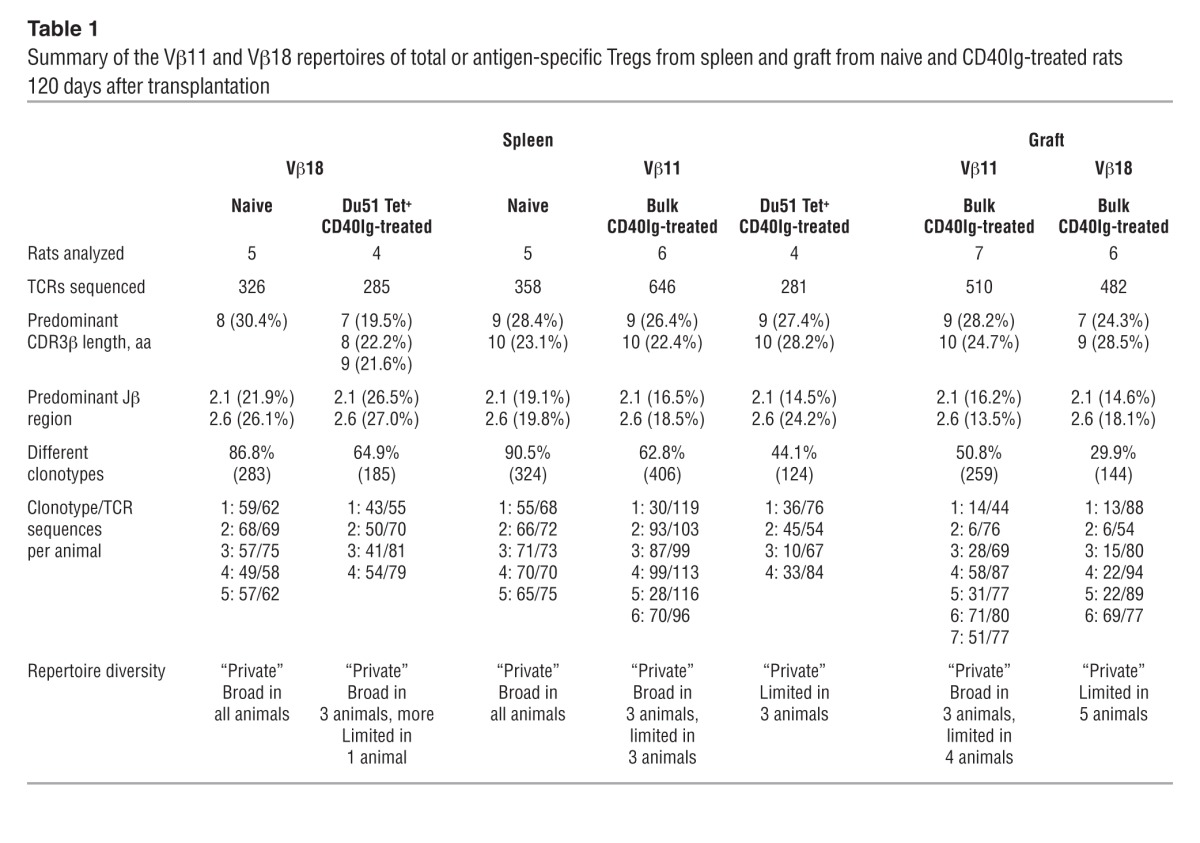
To assess the general TCR aa repertoire characteristics of each of the groups, we first considered the dominant features of the TCR aa clonotypes pooled across all animals in each group. The Vβ11 TCR clonotypes pooled across all animals in each group revealed predominant CDR3β lengths of 9 and 10 aa for CD8+ Tregs from naive spleens and CD40Ig-treated spleens and grafts (Table 1 and Supplemental Figure 6). Previous observations (5) and immunoscopic analysis (data not shown) of Vβ11 TCRs from naive/CD40Ig-treated spleens indicated a predominant CDR3β length of 9 aa. We therefore examined the CDR3 length distributions among the unique TCR clonotypes (Figure 6A and Figure 7A) and TCR repertoires (i.e., accounting for the number of copies of each clonotype; Figure 6B) for individual animals. The CDR3 length distributions for the TCR repertoires revealed substantial variation between treated animals and confirmed a predominant CDR3β length of 9 aa in many of the animals. In contrast, there was a difference in the predominant Vβ18 CDRβ lengths between naive spleens and treated grafts, with the CDR3 lengths of 8 aa prevalent among the Vβ18 TCR clonotypes pooled across all naive spleens and CDR3 lengths of 7 and 9 aa more common in the treated grafts (Table 1 and Supplemental Figure 6). However, as with the Vβ11-TCR repertoires, we observed substantial variation between treated animals in the CDR3 lengths dominating individual Vβ18 repertoires (Figure 7B).
Figure 6. Features and diversity of Vβ11 TCR aa repertoires.

Comparison of the features (A–D) and diversity (E and F) of Vβ11 TCR aa repertoires obtained from CD8+CD45RClo Tregs from naive spleens, bulk or Du51 Tet+ CD8+CD40Ig Tregs from spleens, and GITCs from CD40Ig-treated grafts. Shown are the percentages of the TCRβ aa clonotypes (A and C) and the TCRβ repertoires (i.e., including clonal dominance) (B and D) for each naive and CD40Ig-treated animal using a particular CDR3 length (A and B) and Jβ gene (C and D). The number of TCR aa clonotypes (E) and Simpson’s diversity indices (F) were estimated for a sample size of 44 TCR sequences per individual repertoire. The diversity of the TCR repertoires in CD40Ig-treated animals was compared with that of naive animals using a Kruskal-Wallis test and Dunn’s multiple comparison post tests. *P < 0.05.
Figure 7. Features and diversity of Vβ18 TCR aa repertoires.

Comparison of the features (A–D) and diversity (E and F) of Vβ18 TCR aa repertoires obtained from CD8+CD45RClo Tregs from naive spleens, GITCs from CD40Ig-treated grafts, and Du51 Tet+ CD8+CD40Ig Tregs from spleens. Shown are the percentages of the TCRβ aa clonotypes (A and C) and the TCRβ repertoires (i.e., including clonal dominance) (B and D) for each naive and CD40Ig-treated animal using a particular CDR3 length (A and B) and Jβ gene (C and D). The number of TCR aa clonotypes (E) and Simpson’s diversity indices (F) were estimated for a sample size of 44 TCR sequences per individual repertoire. The diversity of the TCR repertoires in CD40Ig-treated animals was compared with that of naive animals using a Kruskal-Wallis test and Dunn’s multiple comparison post tests. *P < 0.05; **P < 0.01.
Although the Vβ11 and Vβ18 chains of CD8+ Tregs recombined with a variety of Jβ genes (Figure 6C and Figure 7C), we found preferential usage of both the Jβ2.1 and Jβ2.6 TCR clonotypes pooled across all animals in each bulk naive spleen, CD40Ig-treated spleen, and in each CD40Ig-treated graft group (Table 1 and Supplemental Figure 6). However, the Jβs dominating individual TCR repertoires varied substantially between animals (Figure 6D and Figure 7D).
Another important feature of TCR repertoires is their clonotypic diversity. To statistically compare the diversity of the bulk Vβ11 and Vβ18 repertoires between the spleens of naive animals and the spleens and grafts of treated animals, we evaluated the number of different TCR aa clonotypes (Figure 6E and Figure 7E) and Simpson’s diversity index (Figure 6F and Figure 7F), with the latter reflecting the evenness of the clone size distribution across clonotypes. For both the Vβ11 and Vβ18 repertoires, we observed a significantly lower number of TCR clonotypes in the treated grafts compared with that in the naive spleens (Figure 6E and Figure 7E). Albeit not significant, there was also a trend toward reduced diversity of the Vβ11 repertoires from the bulk spleens of treated animals compared with those from naive animals (Figure 6, E and F). Substantially greater variation in TCR repertoire diversity in treated animals compared with that in naive animals was also evident. In particular, the large variation in Simpson’s diversity index between treated animals, indicating the prevalence of one or a few TCR clonotypes in some animals, was consistent with the interindividual variation we observed in CDR3 length and Jβ gene usage.
In order to more precisely analyze the repertoire of CD40Ig-induced antigen-specific Tregs, we studied the Vβ11 and Vβ18 repertoires of RT1Aa/Du51 tetramer–specific CD8+CD40Ig Tregs (Supplemental Tables 2 and 3). We observed considerable variation between individual animals in the preferential CDR3β length (Figure 6B and Figure 7B) and Jβ gene (Figure 6D and Figure 7D) usage of the Vβ11 repertoire. Interestingly, we found a significantly reduced number of clonotypes and overall diversity of the Vβ11, but not the Vβ18, repertoires from the Du51-specific splenic Tregs compared with those from the highly diverse naive Tregs from naive animals (Figure 6, E and F, Figure 7, E and F, and Table 1). The significantly lower Simpson’s diversity index for Vβ11 Du51 Tet+–treated spleen versus Vβ11 bulk naive spleen suggests that one or a few clonotypes tend to dominate several of these repertoires (as expected for an Ag-specific population) (Figure 6F). However, the Vβ18 Du51 Tet+ clonotypes from treated spleens had a diversity that was intermediate between the bulk Vβ18 repertoires from the naive spleens and those from the treated grafts (Figure 7, E and F). This observation was rather unexpected for an antigen-specific population and suggests that the Vβ18 clonotypes do not have a particularly high avidity for this peptide-MHC (pMHC).
Finally, we should point out that despite the greater interindividual variation in the Vβ11+ and Vβ18+ Du51-specific TCR repertoires, some clonotypes were found to be shared between Du51-specific TCR repertoires and were present in the bulk TCR repertoires from treated and naive animals (Supplemental Table 1).
Altogether, these results highlight (a) the largely private nature of both the Vβ11 and Vβ18 TCR repertoires; (b) the substantial clonotypic diversity of total TCR-biased CD8+CD40Ig Tregs in the spleen (Vβ11); (c) the more limited and distinct repertoires in the graft where the cells migrate and reside long term (Vβ11 and Vβ18); and (d) most importantly, the reduced clonotypic diversity of Du51-specific Vβ11+CD8+CD40Ig Tregs.
Tolerance induction by in vivo peptide treatment correlated with an increased proportion of CD8+CD45RClo Tregs and total inhibition of antidonor antibody responses.
To further determine the potential of the immunodominant peptide identified in the in vivo generation of CD8+CD45RClo Tregs and in allograft survival, animals were separately treated using two different protocols of peptide administration with no other treatment. In the first one, animals received 5 i.v injections of 500 μg of peptide (Figure 8A). We observed that injections of either control peptide or Du51 were not sufficient to induce a significant prolongation of allograft survival (respectively, 11 and 9.5 days; n = 4) (Figure 8A). In the second one, to improve the efficacy of the treatment and because such small peptides are rapidly eliminated from the recipient’s body, we tested mini-osmotic pumps with a constant i.p. delivery of 20.8 μg/hour of peptide for 28 days, starting on day 7 before transplantation. Interestingly, this protocol allowed significant prolongation of allograft survival (P < 0.01 compared with control peptide and no treatment), with 25% of indefinite allograft survival using Du51 compared with that achieved using control peptide (Figure 8A). To prove that tolerance induced by peptide infusion was dependent on CD8+ T cells and MHC class I presentation, we cotreated recipients with peptide infusion and either a depleting anti-CD8 mAb (OX8) or a blocking anti-MHC class I mAb (OX18) (Figure 8A), as previously described (5). Administration of both antibodies completely abolished allograft survival, indicating that recognition of MHC class I/antigen by CD8+ T cells was required in the establishment of the tolerance achieved with peptide infusion.
Figure 8. Tolerance induction after in vivo peptide Du51 infusion.

(A) Recipients were either untreated (n = 9), treated with 5 i.v injections of 0.5 mg peptide (n = 4), or treated with short-term continuous peptide infusion by an i.p mini-osmotic pump delivering either 0.5 mg/day alone (n = 8) or combined with a depleting anti-CD8 mAb (OX8) (n = 6) or an anti-MHC class I mAb (OX18) (n = 5), 1 mg/day in the LEW.1W/LEW.1A (n = 5) or BN/LEW.1A (n = 4) strain combination. **P < 0.01, 0.5 mg/day Du51 versus untreated animals and 0.5 mg/day of control peptide. *P < 0.05, 0.5 mg/day Du51 plus OX8 versus 0.5 mg/day Du51. ***P < 0.001, 1 mg/day Du51 versus untreated animals. **P < 0.01, 1 mg/day Du51 versus 1 mg/day Du51 in BN/1A. *P < 0.05 1 mg/day Du51 versus 0.5 mg/day Du51. (B) Anatomopatologic analysis of graft for signs of chronic rejection lesions comparing native heart with that of Du51-treated (20.83 μg/hour) long-term surviving recipients. A, adventitia; M, media. (C) IgG, IgG1, IgG2a, or IgG2b alloantibody production was evaluated in naive (n = 3), syngeneic (n = 3), untreated (n = 3), CD40Ig-treated (n = 3), long-term (n = 2), and Du51-treated animals with graft rejection (n = 4) less than 30 days after rejection or more than 120 days after transplantation. Graph represents MFI ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (D) Splenocytes were recovered after rejection or on day 120 and analyzed by flow cytometry for the absolute number of each subpopulation from naive (n = 12), long-term (n = 2), and Du51-treated recipients with graft rejection (n = 5). (E) Spleens from naive (n = 4), long-term Du51-treated (n = 2), and Du51-treated recipients with graft rejection (n = 4) were incubated with RT1.Aa/Du51 tetramers. Graphs represents the percentage of Tet+ cells among CD8+ Tregs and the absolute number of Tet+ CD8+ Tregs in spleen ± SEM.
Interestingly, an increased dose of Du51 administered by osmotic pump delivering 41.6 μg of peptide per hour induced an indefinite allograft survival in 80% of the recipients of Lewis 1W (LEW.1W) donor hearts (Figure 8A). However, Brown Norway (BN) third-party grafts were promptly rejected on day 7 after transplantation (Figure 8A), demonstrating that peptide Du51 infusion induces donor-specific tolerance mediated by CD8+ Tregs.
We analyzed grafted hearts and spleens of rejecting or long-term surviving recipients treated with peptide Du51 for signs of chronic rejection, presence of antidonor antibodies, proportion of total and Tet+ CD8+CD45RClo Tregs, and in vitro suppression of CD4+ effector T cells (Figure 8, B–E, and data not shown). Anatomopathologic analysis of the grafts of long-term recipients showed no signs of chronic rejection according to a previously established score (ref. 28 and Figure 8B). In addition, we observed a trend toward an increase in the percentage and absolute number of total CD8+ T cells, in particular of CD8+CD45RClo Tregs in the spleen (Figure 8D), as well as of Tet+ Du51–specific Tregs (Figure 8E) in long-term surviving peptide Du51–treated recipients compared with untreated or Du51-treated recipients that rejected their graft early. These results suggest that antigen-specific CD8+CD45RClo Tregs were induced or amplified by in vivo peptide treatment, while other subsets were not, and that these Tregs may be responsible for tolerance induction. We confirmed that these peptide-induced activated CD8+CD45RClo Tregs displayed suppressive activity ex vivo, since they efficiently inhibited effector CD4+ T cell proliferation in the same manner as freshly purified CD8+CD40Ig Tregs (data not shown). Finally, we observed complete inhibition of total IgG, IgG1, IgG2a, and IgG2b alloantibody production in long-term surviving Du51-treated recipients compared with that seen in untreated rats or Du51-treated recipients that rejected their graft early, results which could correlate with the absence of chronic rejection (ref. 28 and Figure 8C).
Discussion
Our current knowledge of how Tregs recognize peptides and the role of this recognition is very limited and mostly based on either transgenic mouse models using TCR gene transfer of CD4+ Tregs (8, 11) or on murine Qa-1–restricted CD8+ Tregs involved in autoimmune disease and cancer whose Qa-1 peptide repertoire has been described in the past few years (29). However, these studies suggest that antigen-specific Tregs crucially influence the outcome of long-term transplantation and contribute to the establishment of tolerance (30). Recent studies have characterized peptides recognized by CD8+ Tregs during cancer (a heme oxygenase-1–derived peptide), autoimmunity (Vβ-derived peptide), or even pregnancy (minor antigen–derived peptide), but not during transplantation (2). Transplantation represents a particular setting for the identification of antigen recognized by Tregs, since the presence of the graft is a continuous source of alloantigens and is most certainly essential for the function and maintenance of regulatory populations and thus the survival of the grafted organ (31). The recognition of alloantigens in the context of regulation has been shown by us and others to occur mainly by the indirect pathway of presentation, particularly that which is supported by pDCs (6, 30, 32).
In this report, we demonstrate for the first time to our knowledge that CD8+CD45RClo Tregs, through the indirect pathway of presentation, can recognize one dominant allopeptide, named Du51 (and one subdominant allopeptide), derived from the β1 domain of natural donor MHC class II molecules. These peptides share 80%–90% homology with human HLA class II molecules and thus could be used to detect specific CD8+ Tregs in humans. Using an MHC I–specific tetramer, we showed that Du51-specific CD8+CD45RClo Tregs were enriched in CD40Ig-treated long-term surviving recipients, expressed a biased restricted Vβ11 chain, displayed a strong suppressive activity ex vivo, and played a crucial role in tolerance induction upon adoptive transfer. Finally, peptide Du51 was shown to induce prolongation of allograft survival in vivo, inducing donor-specific CD8+ Tregs.
Here, we describe that the peptide Du51 displayed an unusual length of 15 aa and that shorter peptides tested failed to induce significant recognition by CD8+CD40Ig Tregs. Most of the literature has focused on short peptides (8–10 aa) bound to MHC class I, although it is known that 5%–10% of these are longer peptides (more than 10 aa) that can be presented by MHC class I molecules (33, 34). So far, such peptides have been identified for CTL models and are mostly derived from viral antigens. To the best of our knowledge, we provide the first description of a 15-aa peptide that can be structurally recognized by CD8+ Tregs, as shown with our RT1Aa/Du51 tetramers. Recent studies suggested that these long peptides drive a TCR recognition more focused on the peptide (35) and that TCRs recognize MHC class I peptides of a preferential length (36). The rat MHC class I molecule RT1.Aa has been known to accommodate particularly long peptides, with key-position residues such as Gln, Met, or Leu at position 2 (P2), Phe at P3, Pro at P4, and Arg at the C terminus (21, 22). Speir et al. demonstrated previously in a model of maternally transmitted minor histocompatibility antigen (MTF-E) of 13 residues that important anchor residues (in particular, arginine at P13) allowed binding with considerably bulged conformation (37). We also observed that Du51 displayed an Arg at the C terminus end and thus could help RT1.Aa accommodation of the peptide. Interestingly, some of the peptides that were tested by us were also tested by Ballet et al. on CD4+ and CD8+ T cells isolated from untreated animals with graft rejection (including the dominant peptide Du51) in the same mismatched cardiac allograft model (LEW.1W into LEW.1A) as ours (18). They found two immunodominant peptides referred to by us as peptides 47 and 55 that were derived from LEW.1W RT1.Du molecules and were involved in acute rejection of grafts from unmodified LEW.1A recipients. Importantly, peptide Du51 was not involved in acute rejection in their model, and the two immunodominant peptides identified by Ballet et al. were not recognized by our CD8+CD40Ig Tregs, suggesting that Tregs and non-Tregs did not recognize the same antigens.
We were able to produce an RT1.Aa/Du51 tetramer, which in addition to being a valuable tool to track antigen-specific cells, can also be used to determine TCR fine specificity and affinity. With this tetramer, we identified in naive animals a pool of antigen-specific Treg precursors of ~0.73%, which was expanded by about 3-fold 120 days after transplantation and CD40Ig treatment. This precursor frequency correlated with the observations of Leavenworth et al. made in a model of arthritis in mice, in which they analyzed the occurrence of Qa-1-Hsp60p216 and Qa-1.R72A-Qdm tetramer–specific CD8+ Tregs and described a naive frequency of approximately 1.65% and 0.46%, respectively, of positive CD8+ T cells for each (38). These observations suggest that the precursor frequency of antigen-specific cells in a Treg population are higher than the frequency of a given antigen-reactive non-Treg cell.
With the use of this MHC I tetramer, we compared the suppressive capacity of Du51-specific and nonspecific CD8+CD40Ig Tregs ex vivo in a coculture assay and in vivo through adoptive transfer of Tet– CD8+CD40Ig Tregs (depleted in Tet+ CD8+CD40Ig Tregs). We demonstrated ex vivo the superior suppressive potential of Tet+ over Tet– CD8+CD40Ig Tregs, which significantly inhibited CD4+ effector T cell proliferation stimulated by both the direct and indirect pathways of presentation, and did so most efficiently in the indirect allorecognition setting. These results are in agreement with several studies demonstrating the superior Treg suppressive potential of indirect specificity in vitro (39, 40). In vivo, we observed a rapid allograft rejection in naive grafted irradiated recipients transferred with Tet– CD8+CD40Ig Tregs compared with that in recipients adoptively transferred with total CD8+CD40Ig Tregs. In a similar manner, adoptive transfer of Tet– CD8αα+ Tregs (depleted in tetramer Qa-1/Hsp60p216–specific Tregs) could not prevent the development of autoimmune arthritis in a mouse model (38). Also, Tsang et al. showed a crucial role for CD4+ Tregs of indirect allospecificity in transplantation tolerance, demonstrating that only adoptive transfer of TCR-transduced CD4+ Tregs of both direct and indirect specificity, and not CD4+ Tregs of direct specificity only, can inhibit cardiac allograft rejection (11).
We have also analyzed different markers potentially expressed by antigen-specific Tregs. Foxp3 did not seem to be an interesting marker of CD8+CD40Ig Tregs unstimulated or stimulated with peptide, although upon several rounds of strong anti-CD3/anti-CD28/IL-2 stimulation, we were able to detect substantial levels of Foxp3 (data not shown). We also found increased IFN-γ production and decreased IL-12 expression after peptide-specific stimulation. Our model already showed IFN-γ to be a crucial cytokine (6).
Regarding the repertoire of induced CD8+CD40Ig Tregs, we previously showed that these cells preferentially used a TCR that recombined the Vβ11 chain and displayed a CDR3β of 9 aa in the spleen, suggesting the expansion of an oligoclonal population of Tregs (5). However, sequencing of approximately 700 CDR3β TCRs across 6 long-surviving animals demonstrated that total Vβ11+CD8+CD40Ig Tregs displayed a relatively diverse repertoire in the spleen. Nevertheless, we found a a more frequently repeated sequence in some animals. Analysis of the TCR repertoire in the graft revealed the predominance of two TCR Vβ chains: Vβ11 (as in the spleen) and Vβ18. Unlike the spleen, sequencing revealed biased and restricted repertoires for both chains, with some shared clonotypes for the Vβ18 TCR, as in the donor-specific blood transfusion model of induction of long-term allograft survival (41). However, the public sequence described by Douillard et al. was only found once in 1 animal in our study and did not represent the Vβ18 repertoire in our model. One interesting hypothesis would be that CD8+CD40Ig Tregs bearing such biased and restricted clonotypes are more potent suppressors and migrate early in the graft to exert their inhibitory activity and then stay localized in the tolerated transplant where immune regulation is required (42). In contrast, total CD8+CD40Ig Tregs that reside in the spleen but displayed a nonrestricted (Vβ18) or less-restricted (Vβ11) repertoire (and, consequently, closer to the naive Tregs’ repertoire) potentially identify distinct regulatory populations (that possibly do not express the same chemokine receptors) being recruited upon subsequent inflammation. Despite some shared clonotypes for both Vβ11 and Vβ18 (Supplemental Table 1), we could not find public CDR3 sequences. By more precisely analyzing the repertoire of antigen-specific CD8+CD40Ig Tregs in the spleen, which was achieved by focusing on tetramer Du51+ Tregs, we demonstrated reduced clonotypic diversity of Du51-specific Vβ11+CD8+CD40Ig Tregs (compared with the total CD8+CD40Ig Tregs from naive and CD40Ig-treated spleen and similar to the graft CD8+CD40Ig Tregs), but not of Du51-specific Vβ18+CD8+CD40Ig Tregs. Thus, the Vβ18 chain usage might not be optimal for recognition of this particular antigen. However, even this analysis of the antigen-specific population did not reveal public clonotypes for this Treg population. TCR sequences that are shared between individuals tend to be more efficiently produced by a process of convergent recombination (43) and thus present at a higher frequency in the naive T cell repertoire (44–46). However, interindividually shared TCR sequences are not necessarily dominant in the immune response to antigen (47). This appears to be the case in our model, suggesting that this process may be modified by either TCR expansion or TCRα chain pairing. More recent reports have revealed the important contribution of the α chain in specific pMHC recognition and how TCRαβ diversity should be taken into consideration, as some specific TCR Vα pairings dictate and alter MHC restriction (48, 49). In the literature, a consensus on TCR diversity of naive CD4+ Tregs was obtained, stating that more is better. Authors demonstrated that a high TCR diversity ensures optimal Treg expansion and function by increasing the probability of responding antigen-specific clones (13, 16, 50, 51). In our model, naive CD8+ Tregs displayed a highly diverse repertoire that was remodeled and biased by expansion of Du51-specific CD8+ Tregs after transplantation and CD40Ig treatment.
From a therapeutic point of view, the identification of natural peptide recognized by Tregs in transplantation is an important goal, as new strategies using amplified CD4+ Tregs are currently being tested in human transplantation (30, 52). The difficulty is that these human CD4+ Tregs amplified in a polyclonal fashion by nonspecific stimulus such as anti-CD3/CD28 antibodies are less efficient than antigen-specific Tregs (2, 53), display limited expansion capacity, and are usually outgrown by conventional effector T cells (54). The use of specific antigen to expand Tregs in short-term culture would surely improve clinical settings. As a proof of principle, we administered the dominant peptide Du51 in naive grafted recipients, without immunosuppressive treatment, and observed significant prolongation of allograft survival. This is the first time to our knowledge that this extended survival could be obtained with allopeptide alone (i.e., without immunosuppressive drugs) in rodents and clearly demonstrates both the efficiency of this peptide and the implication of the indirect pathway of presentation in tolerance induction. Some studies had described earlier immunomodulatory effects of HLA-derived peptides on alloimmune responses (55). In particular, an HLA-B7–derived peptide, named Allotrap, was shown to prolong skin and heart allograft survival when associated with ciclosporin administration in mice and rat, respectively (56, 57). Its effect was associated with modulation of heme oxygenase-1 activity (58). In our model, administration of either anti-CD8 or anti–MHC-I antibodies in combination with peptide Du51 completely abolished allograft survival. Thus, the in vivo therapeutic effect obtained with peptide Du51 infusion was directly linked to MHC class I presentation and CD8+ T cell induction. Moreover, the immunodominant peptide Du51 induced a donor-specific inhibition of alloimmune responses, as third-party grafts were promptly rejected. In addition, the Du51 peptide–induced allograft survival was accompanied by a total inhibition of antidonor antibodies that was probably related to the occurrence of antigen-specific CD8+ Tregs after infusion of the allopeptide. This result is important, as it shows the potency of peptide-induced Tregs in inhibiting acute and chronic allograft rejection occurrence and offers new possibilities in human transplantation. In addition, such expanded, highly suppressive Tregs could provide us with more fundamental information about new, poorly described genes overexpressed in such conditions that could be used as biomarkers (6). Finally, we could assume that the presence of this CD8+CD45RClo Treg population in some groups of patients is associated with a better prognosis during the course of disease (59, 60).
In conclusion, our study indicates that MHC class II donor antigens can be used to boost antigen-specific CD8+ Treg generation and/or function and that, in turn, these Tregs inhibit antidonor immune responses, allowing the establishment of a true tolerance. We also demonstrate that antigen-specific CD8+ Treg TCRs display a private and restricted repertoire that ensures efficient expansion and suppression of alloreactive immune responses. Altogether, these results highlight the importance of the TCR and its interaction with MHC/peptide and open new possibilities in the generation of this population that could be transferable to human settings.
Methods
Animals and cardiac transplantation models.
Allotransplantations of heart were performed between whole MHC–incompatible LEW.1W (RT1.Au as donors) and LEW.1A (RT1.Aa as recipients) male rats as previously described (5).
Adenovirus-mediated gene transfer.
The adenovirus (Ad) encoding for the extracellular portion of mouse CD40 fused to the constant domains of human IgG1 (AdCD40Ig) and the recombinant noncoding Addl324, as well as the procedure of intragraft delivery, have been described previously (5). Briefly, Ad particles (2.1010 infectious particles) were slowly injected at 3 points into the cardiac ventricular walls.
Peptides libraries.
Overlapping peptide libraries (16-mer) with 4 aa lagging were designed to cover the entire polymorphic sequences of MHC-I RT1.Au (α1, -2, and -3 domains), MHC II RT1.Bu (all domains), and MHC II RT1.Du (α2 and β1 domains), as previously published (17–19), and were manufactured by GL Biochem Ltd. Lyophilized peptides were dissolved in 0.4% sterile DMSO/sterile water and stored at –80°C. For control peptides, we used allogeneic nonactivating peptides 7, 26, and 39 in vitro and peptide 31 in vivo.
Degenerated 9- to 15-mer overlapping peptides with 1 to 2 aa lagging were designed to cover the sequence of a positively isolated 16-mer peptide and were synthesized by GL Biochem Ltd.
All peptides were shown to be greater than 95% homogeneous by analytical reverse-phase HPLC, and aa sequences were confirmed. Peptides were diluted in complete RMPI-1640 at a concentration of 120 μg/ml.
Cell purification.
T cells were purified as previously described and as shown in Supplemental Figure 8 (6). Briefly, total splenocytes were depleted with a cocktail of anti–γδ T cells (V65), anti-CD45RA cells (OX33), anti–CD161 NK cells (3.2.3), and anti–CD11b/c monocytes (OX42) using magnetic beads (Dynal). Enriched T cells were labeled with anti–CD45RC-biotin (OX22) and strepavidin-PE-Cy7, anti–CD8α-PE (OX8), anti–TCRαβ-Alexa 647 (R73), and anti–CD25-FITC (OX39) mAbs. CD8+CD45RClo T cells and CD4+CD25– T cells were sorted after gating of TCRαβ+ cells with a FACSAria cell sorter (BD Biosciences). The purity of sorted cell populations was greater than 99%.
pDCs were purified as previously described (6). Briefly, splenocytes recovered after collagenase digestion were further purified by negative depletion with anti-TCR (R73 and V65) T cells and anti-CD45RA (OX33) B cell mAbs. Enriched cells were labeled with anti–CD45R-PE (His24), anti–CD4-APC (OX35), anti–TCR-FITC (R73), and anti–CD45RA-FITC (OX33). pDCs, defined as CD45R+ and CD4+ cells, were sorted after gating on FITC– cells.
MLR.
For MLR coculture assays, pDCs from LEW.1A naive rats (1.25 × 104 cells), syngeneic CD8+CD40Ig Tregs (5 × 104 cells), and 120 μg/ml of allogeneic peptides were plated in triplicate in 200 μl of complete RPMI-1640 medium in round-bottom, 96-well plates for 6 days at 37°C and 5% CO2. pDCs were matured with 0.5 μM of CpG ODN 1826.
For direct MLR suppressive assays, sorted CFSE-labeled CD4+CD25– T cells from LEW.1A origin (5 × 104 cells) and allogeneic pDCs isolated from donor LEW.1W animals (1.25 × 104 cells) were plated in triplicate for 6 days in a final volume of 200 μl of completed RPMI-1640 medium in round-bottom, 96-well plates with FACS-sorted, freshly purified naive CD8+CD45RClo Tregs, peptide-expanded CD8+CD40Ig Tregs (5 × 104 cells), Du51 Tet+, or Tet– CD8+CD40Ig Tregs. For indirect MLR suppressive assays, splenocytes isolated from donor LEW.1W animals were frozen and thawed to induce apoptosis. Apoptotic cells were then cultured overnight with pDCs isolated from recipient LEW.1A animals (8:1 ratio) at 0.65 × 106 pDCs/ml. Alloantigen-loaded pDCs were finally washed and plated as described previously for the direct MLR.
For anti-CD3/anti-CD28 stimulation, round-bottom, 96-well plates were coated with anti-CD3 (1 μg/ml; BD Biosciences) mAb for 1 hour at 37°C, 5% CO2 and then washed, and 5.104 CD8+CD40Ig Treg cells were added to each well in 200 μl of completed RPMI-1640 plus anti-CD28 (10 μg/ml) for 1, 2, 3, and 6 days.
The proliferation of CFSE-labeled naive CD4+CD25– T cells and the phenotype of CD8+CD45RClo Tregs were analyzed by flow cytometry on a FACSCanto II cytometer (BD Biosciences) after gating on TCR+CD4+ cells or TCR+CD8+ cells among live cells (DAPI negative).
Extracellular and intracellular staining.
For extracellular staining, cells were stained with the following mAbs: anti-TCRαβ (R73, Alexa Fluor 647 conjugated), anti-CD8α (OX8, PE-Cy7 conjugated; eBioscience), anti-CD4 (W3.25, PE-Cy7 conjugated), anti-CD45RC (OX22, FITC conjugated), anti-CD28 (JJ319, biotin labeled), anti-CD71 (OX26, biotin labeled), anti-mouse Vβ11 (KT11, biotin labeled; AbD Serotec), anti-CD25 (OX39, biotin labeled), and anti-MHC-II (OX6, biotin labeled).
For intracellular staining, cells were then biotin labeled for Foxp3 (eBioscience) using a BD Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions.
All biotinylated mAbs were visualized using PerCP.Cy5.5 Streptavidin (BD Biosciences). A FACSCanto II cytofluorimeter (BD Biosciences) was used to measure fluorescence, and data were analyzed using FlowJo software, version 7.6.5 (Tree Star Inc.). Cells were first gated by their morphology, and dead cells were excluded by selecting DAPI-negative viable cells.
Cytokine assays.
IFN-γ and IL-10 were measured in coculture supernatants using OptEIA ELISA from BD Biosciences, and IL-12 and TGF-β were measured using ELISA from Invitrogen and R&D Systems, respectively.
Production of biotinylated RT1-Aa-peptide complexes.
Briefly, heavy-chain RT1-Aa and β2 microglobuline (β2m) were cloned in pET24 and produced in E. coli BL21-DE3. Recombinant proteins were produced as inclusion bodies, dissolved in 8 M urea, and refolded in vitro as previously described for human HLA-A2-peptide complexes (61). RT1-Aa, β2m, and Du51 peptides were refolded in 0.4 M L-arginine, 0.1 M Tris (pH 8), 2 mM EDTA, 5 mM reduced glutathione, and 0.5 mM oxidated glutathione for 5 days at 4°C. The solution was then concentrated, and the buffer was changed on a 10-kDa amicon membrane (Millipore). Folded MHC-peptide complexes were biotinylated with the BirA enzyme (Avidity) for 5 hours at 30°C and desalted on a Hiprep 26/10 desalting column (GE Healthcare). MHC-peptide complexes were then purified by anion exchange Q-sepharose chromatography. Biotinylation was tested by tetramerization with streptavidin (Sigma-Aldrich) at a molar ratio of 4:1.
Tetramerization and staining.
Tetramerization of RT1.Aa/Du51 was performed at room temperature by the addition of streptavidin-PE (Jackson ImmunoResearch) or streptavidin-APC (BD Biosciences) at a 4:1 molar ratio, in 4 equal aliquots added at 15-minute intervals. Likewise, the control tetramer RT1.Aa/MTF-E (ILFPSSERLISNR) was conjugated with streptavidin-BV421 (Biolegend) and represented 1.6 ± 0.7% of nonspecific staining among Du51-specific cells. These three reagents were mixed and added to plated cells at 10 μg/ml for 1 hour at 4°C. Cells were further stained for CD8 and CD45RC, and fluorescence was analyzed on a FACSCanto II cytometer (BD Biosciences). Cells were first gated by their morphology and dead cells excluded by selecting DAPI-negative cells.
PCR and sequencing.
cDNA was obtained as previously described (5). Briefly, total RNA was isolated and reverse transcribed using an MMLV Reverse-Transcriptase kit (Invitrogen) according to the manufacturer’s instructions. cDNA was PCR amplified using a Cβ primer and a Vβ11 primer, as described previously (41). PCR products were loaded onto a 2% agarose gel and electrophoresed. An ~300-bp band corresponding to PCR product was purified using a Nucleospin Extract II kit (MACHERY-NAGEL). Purified PCR products were further ligated into pCR 2.1-TOPO vector (TOPO TA cloning kit; Invitrogen) according to the manufacturer’s instructions, and single bacterial colonies were used for sequencing the TCR aa CDR3β regions.
TCR repertoire analysis.
The diversity of the TCR repertoires for each individual was evaluated using two different measures of diversity: the number of different TCR aa clonotypes and Simpson’s diversity index (62). Simpson’s diversity index accounts for the clonal dominance hierarchy and provides a relative measure of diversity that ranges in value from 0 (minimal diversity) to 1 (maximal diversity). To account for differences in the sizes of the TCR repertoire samples, the TCR repertoire diversity measures were estimated for each animal as the median value of 10,000 random draws of subsamples of 44 TCR sequences (i.e., the number of Vβ11 sequences obtained from the graft of treated animal 1, which was the minimum sample size across all animals) from the total TCR repertoire sample for each individual (62). The diversity measures were calculated using MATLAB (The MathWorks).
Peptide treatment in vivo.
Peptides (16-mer) were dissolved in 0.4% DMSO-PBS before injection. For the first protocol, single doses of peptide (500 μg/injection) were injected i.v. at different time points before and after transplantation on days –6, –3, 0, +3, and +7 into grafted LEW.1A recipients. In the second protocol, mini-osmotic pumps (ALZET) were implanted i.p. into recipients, and 16-mer peptides were delivered continuously at either 20.83 or 41.66 μg/hour for 14 days. A first pump was implanted on day –7 before transplantation and was replaced by a second one on day +7, allowing delivery of 14 or 28 mg of peptide per animal for 28 consecutive days. A depleting anti-CD8α mAb (OX8, IgG1, 3 mg/kg) or an anti–MHC class Ia and Ib mAb (OX18, 3 mg/kg) were injected i.p. twice a week from day –7 until rejection. Allografts were monitored daily by palpation, and allograft rejection was defined as complete cessation of a palpable heart beat.
Adoptive cell transfer.
Cell transfers were performed by i.v. injection of purified and sorted total or Du51 Tet– CD8+CD40Ig Tregs into LEW.1A recipients sublethally irradiated (4.5-Gy whole-body irradiation) on the day before transplantation.
Morphometric analysis of cardiac grafts.
The upper third of the graft was fixed in paraformaldehyde and embedded in paraffin. Five-micrometer coronal sections were stained with H&E-saffron. Tissues were analyzed by a pathologist (K. Renaudin) blinded to the groups, and chronic rejection was evaluated as previously described (63).
Donor-specific alloantibody detection.
Alloantibodies were analyzed by cytofluorimetry as described elsewhere (28). Briefly, after digestion by collagenase D and red blood cell lysis, allogeneic spleen cells were incubated with diluted (1/8) heat-inactivated serum and then with FITC-conjugated goat anti-rat IgG antibodies (H+L chain specific) (The Jackson Laboratories), a mouse anti-rat IgG1 mAb (MCA 194; Serotec), IgG2a (MCA 278; Serotec), or IgG2b (MCA 195; Serotec). Antibody binding was revealed using FITC-coupled F(ab)′2 goat anti-mouse IgG (The Jackson Laboratories). Cells were analyzed using a FACS Canto II cytofluorimeter (BD Biosciences), and the results were expressed as the mean channel fluorescence for each serum.
Statistics.
For the peptide activation test, a nonparametric Wilcoxon signed-rank test was performed, comparing column median to a hypothetical value of 1.0. Statistical significance for TCR Vβ11 expression, phenotype of activated cells, cytokine expression, proliferation assay, and tetramer staining was evaluated by a 2-tailed Mann-Whitney U test. Graft survival was analyzed by a Kaplan-Meier log-rank test. Two-way ANOVA and Bonferroni’s post tests were used for donor-specific antibody analysis and splenocyte phenotypic characterization. Analyses were made with GraphPad Prism software, version 5.04 (GraphPad Software). For diversity analysis, Kruskal-Wallis and Dunn’s multiple comparison post tests were performed using GraphPad Prism, version 6.0c. A P value less than 0.05 was considered significant.
Study approval.
All animal experiments were approved by the ethics committee for animal care and use of the French Pays de la Loire region.
Supplementary Material
Acknowledgments
We thank F-Xavier Hubert for review of the manuscript, Claire Usal for technical assistance with animal models, and Bernard Vanhove and Bernard Martinet for purification of OX18 and OX8 antibodies. We thank the Vector Core of the University Hospital of Nantes, which is supported by the Association Française Contre les Myopathies, for producing the adenoviral vectors. We thank the Fondation Progreffe for financial support and Crédit Agricole for the donation of the FACSAria cell sorter. This work was supported by a Marie Curie fellowship from the 6th Framework Programme (FP) of the European Union, an ESOT Junior Basic Science Grant awarded to C. Guillonneau and an INSERM-Région Pays de la Loire Fellowship awarded to E. Picarda, and an Australian Research Council Future Fellowship awarded to V. Venturi. This work has partially benefited from state support managed by the National Research Agency under the “Investissements d’avenir” program (ANR-11-LABX-0016-01).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(6):2497–2512. doi:10.1172/JCI71533.
References
- 1.Bluestone JA, Auchincloss H, Nepom GT, Rotrosen D, St Clair EW, Turka LA. The Immune Tolerance Network at 10 years: tolerance research at the bedside. Nat Rev Immunol. 2010;10(11):797–803. doi: 10.1038/nri2869. [DOI] [PubMed] [Google Scholar]
- 2.Guillonneau C, Picarda E, Anegon I. CD8+ regulatory T cells in solid organ transplantation. Curr Opin Organ Transplant. 2010;15(6):751–756. doi: 10.1097/MOT.0b013e32834016d1. [DOI] [PubMed] [Google Scholar]
- 3.Li XL, Menoret S, Le Mauff B, Angin M, Anegon I. Promises and obstacles for the blockade of CD40-CD40L interactions in allotransplantation. Transplantation. 2008;86(1):10–15. doi: 10.1097/TP.0b013e31817c4b97. [DOI] [PubMed] [Google Scholar]
- 4.Guillot C, et al. Prolonged blockade of CD40-CD40 ligand interactions by gene transfer of CD40Ig results in long-term heart allograft survival and donor-specific hyporesponsiveness, but does not prevent chronic rejection. J Immunol. 2002;168(4):1600–1609. doi: 10.4049/jimmunol.168.4.1600. [DOI] [PubMed] [Google Scholar]
- 5.Guillonneau C, et al. CD40Ig treatment results in allograft acceptance mediated by CD8CD45RC T cells, IFN-γ, and indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117(4):1096–1106. doi: 10.1172/JCI28801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li XL, et al. Mechanism and localization of CD8 regulatory T cells in a heart transplant model of tolerance. J Immunol. 2010;185(2):823–833. doi: 10.4049/jimmunol.1000120. [DOI] [PubMed] [Google Scholar]
- 7.Picarda E, Anegon I, Guillonneau C. T-cell receptor specificity of CD8(+) Tregs in allotransplantation. Immunotherapy. 2011;3(4 suppl):35–37. doi: 10.2217/imt.11.37. [DOI] [PubMed] [Google Scholar]
- 8.Tsang JY, et al. The potency of allospecific Tregs cells appears to correlate with T cell receptor functional avidity. Am J Transplant. 2011;11(8):1610–1620. doi: 10.1111/j.1600-6143.2011.03650.x. [DOI] [PubMed] [Google Scholar]
- 9.Joffre O, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med. 2008;14(1):88–92. doi: 10.1038/nm1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang Q, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199(11):1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsang JY, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest. 2008;118(11):3619–3628. doi: 10.1172/JCI33185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. 2011;3(83):83ra42. doi: 10.1126/scitranslmed.3002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wing JB, Sakaguchi S. TCR diversity and Treg cells, sometimes more is more. Eur J Immunol. 2011;41(11):3097–3100. doi: 10.1002/eji.201142115. [DOI] [PubMed] [Google Scholar]
- 14.Masteller EL, Tang Q, Bluestone JA. Antigen-specific regulatory T cells — ex vivo expansion and therapeutic potential. Semin Immunol. 2006;18(2):103–110. doi: 10.1016/j.smim.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Ohkura N, Sakaguchi S. Regulatory T cells: roles of T cell receptor for their development and function. Semin Immunopathol. 2010;32(2):95–106. doi: 10.1007/s00281-010-0200-5. [DOI] [PubMed] [Google Scholar]
- 16.Fohse L, et al. High TCR diversity ensures optimal function and homeostasis of Foxp3+ regulatory T cells. Eur J Immunol. 2011;41(11):3101–3113. doi: 10.1002/eji.201141986. [DOI] [PubMed] [Google Scholar]
- 17.van Denderen B, Peche H, Gagne K, Usal C, Cuturi MC, Soulillou JP. Identification of immunodominant donor MHC peptides following rejection and donor strain transfusion-induced tolerance of heart allografts in adult rats. Eur J Immunol. 2001;31(5):1333–1339. doi: 10.1002/1521-4141(200105)31:5<1333::AID-IMMU1333>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 18.Ballet C, et al. Indirect CD4+ TH1 response, anti-donor antibodies and diffuse C4d graft deposits in long term recipients conditioned by donor antigens priming. Am J Transplant. 2008;9(4):697–708. doi: 10.1111/j.1600-6143.2009.02556.x. [DOI] [PubMed] [Google Scholar]
- 19.Ettinger RA, Moustakas AK, Lobaton SD. Open reading frame sequencing and structure-based alignment of polypeptides encoded by RT1-Bb, RT1-Ba, RT1-Db, and RT1-Da alleles. Immunogenetics. 2004;56(8):585–596. doi: 10.1007/s00251-004-0725-7. [DOI] [PubMed] [Google Scholar]
- 20.Thorpe CJ, Moss DS, Powis SJ, Howard JC, Butcher GW, Travers PJ. An analysis of the antigen binding site of RT1.Aa suggests an allele-specific motif. Immunogenetics. 1995;41(5):329–331. doi: 10.1007/BF00172160. [DOI] [PubMed] [Google Scholar]
- 21.Powis SJ, et al. The rat cim effect: TAP allele-dependent changes in a class I MHC anchor motif and evidence against C-terminal trimming of peptides in the ER. Immunity. 1996;4(2):159–165. doi: 10.1016/S1074-7613(00)80680-9. [DOI] [PubMed] [Google Scholar]
- 22.Stevens J, Wiesmuller KH, Barker PJ, Walden P, Butcher GW, Joly E. Efficient generation of major histocompatibility complex class I-peptide complexes using synthetic peptide libraries. J Biol Chem. 1998;273(5):2874–2884. doi: 10.1074/jbc.273.5.2874. [DOI] [PubMed] [Google Scholar]
- 23.Stevens J, Wiesmuller KH, Walden P, Joly E. Peptide length preferences for rat and mouse MHC class I molecules using random peptide libraries. Eur J Immunol. 1998;28(4):1272–1279. doi: 10.1002/(SICI)1521-4141(199804)28:04<1272::AID-IMMU1272>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 24.Karim M, Feng G, Wood KJ, Bushell AR. CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood. 2005;105(12):4871–4877. doi: 10.1182/blood-2004-10-3888. [DOI] [PubMed] [Google Scholar]
- 25.Wan Q, et al. Probing the effector and suppressive functions of human T cell subsets using antigen-specific engineered T cell receptors. PLoS One. 2013;8(2):e56302. doi: 10.1371/journal.pone.0056302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chattopadhyay PK, et al. Techniques to improve the direct ex vivo detection of low frequency antigen-specific CD8+ T cells with peptide-major histocompatibility complex class I tetramers. Cytometry A. 2008;73(11):1001–1009. doi: 10.1002/cyto.a.20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wooldridge L, Lissina A, Cole DK, van den Berg HA, Price DA, Sewell AK. Tricks with tetramers: how to get the most from multimeric peptide-MHC. Immunology. 2009;126(2):147–164. doi: 10.1111/j.1365-2567.2008.02848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guillonneau C, et al. Anti-CD28 antibodies modify regulatory mechanisms and reinforce tolerance in CD40Ig-treated heart allograft recipients. J Immunol. 2007;179(12):8164–8171. doi: 10.4049/jimmunol.179.12.8164. [DOI] [PubMed] [Google Scholar]
- 29.Kim HJ, Cantor H. Regulation of self-tolerance by Qa-1-restricted CD8(+) regulatory T cells. Semin Immunol. 2011;23(6):446–452. doi: 10.1016/j.smim.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol. 2012;12(6):417–430. doi: 10.1038/nri3227. [DOI] [PubMed] [Google Scholar]
- 31.Hamano K, Rawsthorne MA, Bushell AR, Morris PJ, Wood KJ. Evidence that the continued presence of the organ graft and not peripheral donor microchimerism is essential for maintenance of tolerance to alloantigen in vivo in anti-CD4 treated recipients. Transplantation. 1996;62(6):856–860. doi: 10.1097/00007890-199609270-00026. [DOI] [PubMed] [Google Scholar]
- 32.Gokmen MR, Lombardi G, Lechler RI. The importance of the indirect pathway of allorecognition in clinical transplantation. Curr Opin Immunol. 2008;20(5):568–574. doi: 10.1016/j.coi.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 33.Burrows SR, Rossjohn J, McCluskey J. Have we cut ourselves too short in mapping CTL epitopes? Trends Immunol. 2006;27(1):11–16. doi: 10.1016/j.it.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Hickman HD, et al. Toward a definition of self: proteomic evaluation of the class I peptide repertoire. J Immunol. 2004;172(5):2944–2952. doi: 10.4049/jimmunol.172.5.2944. [DOI] [PubMed] [Google Scholar]
- 35.Liu YC, et al. The energetic basis underpinning T-cell receptor recognition of a super-bulged peptide bound to a major histocompatibility complex class I molecule. J Biol Chem. 2012;287(15):12267–12276. doi: 10.1074/jbc.M112.344689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ekeruche-Makinde J, et al. Peptide length determines the outcome of TCR/peptide-MHCI engagement. Blood. 2013;121(7):1112–1123. doi: 10.1182/blood-2012-06-437202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Speir JA, Stevens J, Joly E, Butcher GW, Wilson IA. Two different, highly exposed, bulged structures for an unusually long peptide bound to rat MHC class I RT1-Aa. Immunity. 2001;14(1):81–92. doi: 10.1016/S1074-7613(01)00091-7. [DOI] [PubMed] [Google Scholar]
- 38.Leavenworth JW, Tang X, Kim HJ, Wang X, Cantor H. Amelioration of arthritis through mobilization of peptide-specific CD8+ regulatory T cells. J Clin Invest. 2013;123(3):1382–1389. doi: 10.1172/JCI66938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hara M, et al. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol. 2001;166(6):3789–3796. doi: 10.4049/jimmunol.166.6.3789. [DOI] [PubMed] [Google Scholar]
- 40.Jiang S, Tsang J, Game DS, Stevenson S, Lombardi G, Lechler RI. Generation and expansion of human CD4+ CD25+ regulatory T cells with indirect allospecificity: Potential reagents to promote donor-specific transplantation tolerance. Transplantation. 2006;82(12):1738–1743. doi: 10.1097/01.tp.0000244932.29542.9e. [DOI] [PubMed] [Google Scholar]
- 41.Douillard P, et al. Donor-specific blood transfusion-induced tolerance in adult rats with a dominant TCR-Vβ rearrangement in heart allografts. J Immunol. 1996;157(3):1250–1260. [PubMed] [Google Scholar]
- 42.Hayashi Y, Tsukumo S, Shiota H, Kishihara K, Yasutomo K. Antigen-specific T cell repertoire modification of CD4+CD25+ regulatory T cells. J Immunol. 2004;172(9):5240–5248. doi: 10.4049/jimmunol.172.9.5240. [DOI] [PubMed] [Google Scholar]
- 43.Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T-cell responses? Nat Rev Immunol. 2008;8(3):231–238. doi: 10.1038/nri2260. [DOI] [PubMed] [Google Scholar]
- 44.Quigley MF, et al. Convergent recombination shapes the clonotypic landscape of the naive T-cell repertoire. Proc Natl Acad Sci U S A. 2010;107(45):19414–19419. doi: 10.1073/pnas.1010586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robins HS, et al. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2010;2(47):47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venturi V, et al. A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J Immunol. 2011;186(7):4285–4294. doi: 10.4049/jimmunol.1003898. [DOI] [PubMed] [Google Scholar]
- 47.Thomas PG, Handel A, Doherty PC, La Gruta NL. Ecological analysis of antigen-specific CTL repertoires defines the relationship between naive and immune T-cell populations. Proc Natl Acad Sci U S A. 2013;110(5):1839–1844. doi: 10.1073/pnas.1222149110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Day EB, et al. Structural basis for enabling T-cell receptor diversity within biased virus-specific CD8+ T-cell responses. Proc Natl Acad Sci U S A. 2011;108(23):9536–9541. doi: 10.1073/pnas.1106851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turner SJ, Rossjohn J. αβ T cell receptors come out swinging. Immunity. 2011;35(5):660–662. doi: 10.1016/j.immuni.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Haribhai D, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35(1):109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adeegbe D, Matsutani T, Yang J, Altman NH, Malek TR. CD4(+) CD25(+) Foxp3(+) T regulatory cells with limited TCR diversity in control of autoimmunity. J Immunol. 2010;184(1):56–66. doi: 10.4049/jimmunol.0902379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Safinia N, Leech J, Hernandez-Fuentes M, Lechler R, Lombardi G. Promoting transplantation tolerance; adoptive regulatory T cell therapy. Clin Exp Immunol. 2013;172(2):158–168. doi: 10.1111/cei.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Renner P, et al. Antigen-specific recognition is critical for the function of regulatory CD8(+)CD28(–) T cells. Transpl Immunol. 2010;22(3–4):144–149. doi: 10.1016/j.trim.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Tang Q, Bluestone JA, Kang SM. CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. J Mol Cell Biol. 2012;4(1):11–21. doi: 10.1093/jmcb/mjr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zang W, Murphy B. Peptide-mediated immunosuppression. Am J Ther. 2005;12(6):592–599. doi: 10.1097/01.mjt.0000178766.60234.e2. [DOI] [PubMed] [Google Scholar]
- 56.Buelow R, Veyron P, Clayberger C, Pouletty P, Touraine JL. Prolongation of skin allograft survival in mice following administration of ALLOTRAP. Transplantation. 1995;59(4):455–460. doi: 10.1097/00007890-199502270-00001. [DOI] [PubMed] [Google Scholar]
- 57.Murphy B, Kim KS, Buelow R, Sayegh MH, Hancock WW. Synthetic MHC class I peptide prolongs cardiac survival and attenuates transplant arteriosclerosis in the Lewis→Fischer 344 model of chronic allograft rejection. Transplantation. 1997;64(1):14–19. doi: 10.1097/00007890-199707150-00004. [DOI] [PubMed] [Google Scholar]
- 58.Iyer S, et al. Characterization and biological significance of immunosuppressive peptide D2702.75-84(E → V) binding protein. Isolation of heme oxygenase-1. J Biol Chem. 1998;273(5):2692–2697. doi: 10.1074/jbc.273.5.2692. [DOI] [PubMed] [Google Scholar]
- 59.Ordonez L, Bernard I, L’Faqihi-Olive FE, Tervaert JW, Damoiseaux J, Saoudi A. CD45RC isoform expression identifies functionally distinct T cell subsets differentially distributed between healthy individuals and AAV patients. PLoS One. 2009;4(4):e5287. doi: 10.1371/journal.pone.0005287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ordonez L, et al. A higher risk of acute rejection of human kidney allografts can be predicted from the level of CD45RC expressed by the recipients’ CD8 T cells. PLoS One. 2013;8(7):e69791. doi: 10.1371/journal.pone.0069791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci U S A. 1992;89(8):3429–3433. doi: 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Venturi V, Kedzierska K, Turner SJ, Doherty PC, Davenport MP. Methods for comparing the diversity of samples of the T cell receptor repertoire. J Immunol Methods. 2007;321(1–2):182–195. doi: 10.1016/j.jim.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 63.Guillonneau C, et al. The role of TNF-related activation-induced cytokine-receptor activating NF-κB interaction in acute allograft rejection and CD40L-independent chronic allograft rejection. J Immunol. 2004;172(3):1619–1629. doi: 10.4049/jimmunol.172.3.1619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.