

Abstract
Epithelial tumor cells that have undergone epithelial-to-mesenchymal transition (EMT) are typically prone to metastasis and drug resistance and contribute to a poor clinical outcome. The transcription factor ZEB1 is a known driver of EMT, and mediators of ZEB1 represent potential therapeutic targets for metastasis suppression. Here, we have shown that phosphatidylinositol 3-kinase–targeted (PI3K-targeted) therapy suppresses metastasis in a mouse model of Kras/Tp53-mutant lung adenocarcinoma that develops metastatic disease due to high expression of ZEB1. In lung adenocarcinoma cells from Kras/Tp53-mutant animals and human lung cancer cell lines, ZEB1 activated PI3K by derepressing miR-200 targets, including amphiregulin (AREG), betacellulin (BTC), and the transcription factor GATA6, which stimulated an EGFR/ERBB2 autocrine loop. Additionally, ZEB1-dependent derepression of the miR-200 and miR-183 target friend of GATA 2 (FOG2) enhanced GATA3-induced expression of the p110α catalytic subunit of PI3K. Knockdown of FOG2, p110α, and RHEB ameliorated invasive and metastatic propensities of tumor cells. Surprisingly, FOG2 was not required for mesenchymal differentiation, suggesting that mesenchymal differentiation and invasion are distinct and separable processes. Together, these results indicate that ZEB1 sensitizes lung adenocarcinoma cells to metastasis suppression by PI3K-targeted therapy and suggest that treatments to selectively modify the metastatic behavior of mesenchymal tumor cells are feasible and may be of clinical value.
Introduction
Lung cancer is the foremost cause of cancer-related mortality, with high incidence rates, robust metastatic propensity, and frequent de novo and acquired resistance to therapy (1). Even in the setting of “addiction” to mutant receptor tyrosine kinases (RTKs) that are pharmacologically targetable, tumors undergo only transient responses to targeted therapy and subsequently develop drug resistance that frequently results in death (2). The incidence of KRAS mutations exceeds that of RTK mutations in lung adenocarcinoma, and currently available therapeutic strategies do not effectively target mutant KRAS (3). KRAS-mutant lung adenocarcinomas commonly harbor oncogenic mutations in TP53 and STK11 that confer metastatic capacity (4). Thus, inhibition of mutant oncoproteins reduces tumor burden but does not eradicate disease in patients with lung cancer, warranting the development of other treatment strategies that complement the beneficial effects of tumor cytoreduction.
In the “migrating stem cell hypothesis,” metastases arise from a small population of tumor cells with the capacity to undergo epithelial-mesenchymal transition (EMT), a reversible process characterized by a loss of polarized features, detachment from neighboring cells, increased motility and invasion, and resistance to standard cytotoxic chemotherapy (5). EMT is induced by several families of transcriptional repressors, including ZEB, SNAIL, and basic helix-loop-helix factors (6). ZEB1 is highly expressed in epithelial cancers (e.g., prostate, lung, and pancreatic cancers), and its expression is correlated with a poor prognosis (7). ZEB1 represses the expression of epithelial genes and certain microRNAs (miRs), including miR-183, miR-203, and miR-200 family members (i.e., miR-200a, miR-200b, miR-200c, miR-141, and miR-429), which function not only as strong inducers of epithelial differentiation but also as inhibitors of stem cell properties (8–10). Reciprocally, miR-200 family members directly target the ZEB1 3′-untranslated region (3′-UTR), creating a double-negative feedback loop that regulates ZEB1 and miR-200 expression (11).
In mice that develop metastatic lung adenocarcinoma from the expression of a latent KrasG12D allele and a knock-in Trp53R172HΔG allele (“KP” mice) (12), the ZEB1/miR-200 axis plays a central role in metastasis regulation. When injected into syngeneic, immunocompetent mice, lung adenocarcinoma cell lines derived from KP mice (KP cells) are uniformly tumorigenic, but have variable metastatic potential (high or low) (13). In monolayer culture, highly metastatic KP cells have a mesenchymal phenotype, high ZEB1 levels, and low miR-200 levels, whereas poorly metastatic KP cells have an epithelial phenotype, low ZEB1 levels, and high miR-200 levels (13). Highly metastatic KP cells exhibit plasticity in 3-dimensional cultures, forming polarized epithelial spheres that undergo EMT in response to treatment with transforming growth factor-β (TGF-β), whereas poorly metastatic KP cells do not form such spheres or undergo EMT (13). The ability of highly metastatic KP cells to undergo EMT, invade, and metastasize is abrogated by the ectopic expression of the miR-200b/200a/429 genomic cluster (13).
In the present study, we used KP mice to identify downstream mediators of ZEB1 that are pharmacologically actionable for the purpose of developing therapeutic strategies to suppress metastasis. We focused our attention on phosphatidylinositol 3-kinase (PI3K), because it has been implicated in the expansion of various normal stem cell populations and tumor-initiating cells at the bronchoalveolar duct junction in KRAS-driven lung adenocarcinoma models (14–20). Our findings suggest that ZEB1 activates PI3K in lung adenocarcinoma cells, thereby sensitizing mesenchymal tumor cells to metastasis suppression by PI3K-targeted therapy. We conclude that treatments to selectively modify the metastatic behavior of mesenchymal tumor cells are feasible and may be of clinical value.
Results
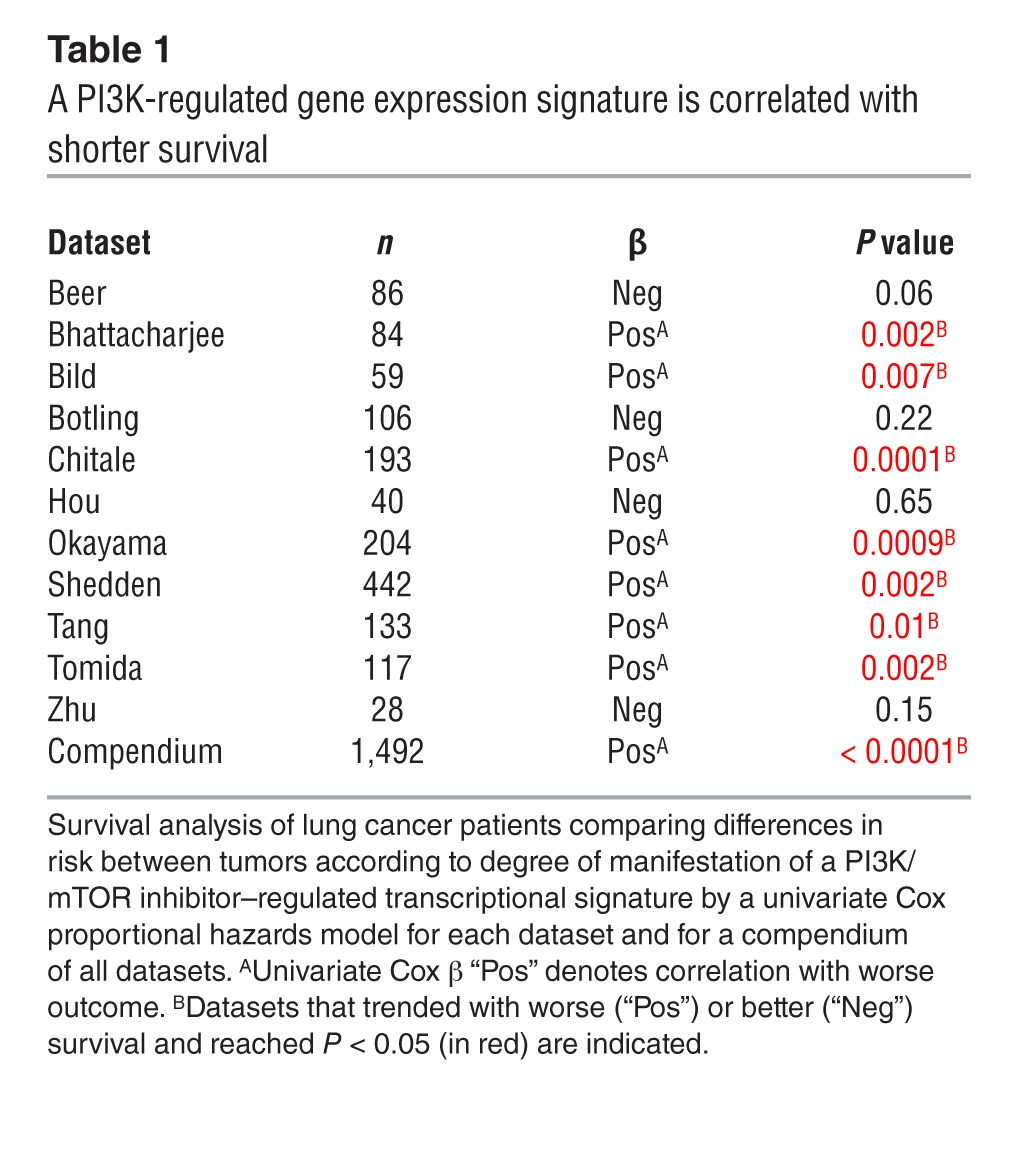
PI3K-regulated gene expression signature is correlated with shorter survival.
To determine whether PI3K activation is a determinant of clinical outcome in patients with early-stage lung adenocarcinoma, we probed tumors from 11 independent cohorts (n = 1,492 patients) using a gene expression signature consisting of 1,801 genes that were up- or downregulated in cancer cells treated with small-molecule inhibitors of PI3K or its downstream mediator, mTOR (21). After grouping the tumors on the basis of their relative gene signature score values (upper, middle, and lower third), we found that patients with the strongest manifestation of the expression signature had a shorter overall survival duration, both in 9 cohorts analyzed individually and in a compendium of all 11 cohorts; the 5-year overall survival rates were 48%, 61%, and 71% for the upper, middle, and lower thirds of PI3K signature scores, respectively (Figure 1A and Table 1). In one dataset for which KRAS mutation status was available (22), there was no correlation between KRAS mutation status and clinical outcome or the presence of the gene signature; however, the gene signature was prognostic in the KRAS-mutant and wild-type groups. Overall, these findings suggest that the transcriptional programs associated with PI3K-dependent pathways are more active in aggressive human lung adenocarcinomas, but are not activated exclusively in KRAS-mutant tumors.
Figure 1. Mesenchymal lung adenocarcinoma cells are sensitive to PX-866.

(A) Survival analysis of lung cancer patients comparing differences in risk between tumors according to degree of manifestation of a PI3K/mTOR inhibitor–regulated transcriptional signature. Kaplan-Meier plot compares the top, bottom, and middle thirds of the T-score values. (B) H&E staining of metastatic tumors (arrows) in KP mice. Scale bars: 200 μm. (C) Numbers (n) of visible lung tumors (scatter plot) and mice with (w/) or without (w/o) histologically documented metastases (Met) (bar graph). Metastasis incidence P value (Fisher’s exact test). (D) Invasion assays. In a single experiment, invasive KP cells were quantified after a 24-hour incubation with PX-866 or vehicle. Mean ± SD from triplicate samples. (E) Scatter plots of primary tumor volume and numbers of visible lung metastases in a single experiment in which syngeneic mice were injected with 344SQ cells (5 mice per cohort) or 531LN2 cells (9 mice per cohort) and then treated with PX-866 or vehicle. Mean ± SD was calculated for each cohort. (F and G) Invasion assays. Invasive KP cells (F) and H1299 cells (G) were quantified in the presence or absence of PX-866. H1299 cells were treated with (+) or without (–) doxycycline (Dox) (1 μg/ml) to induce expression of the miR-200b/a/429 construct. Mean ± SD from triplicate samples. vec, vector.
Table 1.
A PI3K-regulated gene expression signature is correlated with shorter survival
A PI3K inhibitor suppresses metastasis.
To directly determine whether PI3K drives metastasis in lung adenocarcinoma, we treated KP mice with PX-866, a class I PI3K antagonist (23), or vehicle beginning at 4 to 5 months of age (when they had developed multifocal lung adenomas) and ending at 8 to 9 months of age (when they had invasive lung adenocarcinomas and distant metastases) (12). PX-866 treatment inhibited PI3K activity on the basis of reduced AKT Ser473 phosphorylation levels in tumor-bearing lung tissues (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI72171DS1). Although lung tumor multiplicity did not differ between the 2 cohorts, the incidence of distant metastasis was much lower in PX-866–treated mice (Figure 1, B and C). A histologic analysis of tumor tissues revealed no significant difference between the cohorts in the frequencies of lung adenocarcinomas or their precursors (atypical alveolar hyperplasia, adenoma, or bronchoalveolar cell carcinoma-like lesions) (Table 2), suggesting that PX-866 mitigated metastasis without suppressing the histological progression of lung tumors.
Table 2.
Histologic analysis of lung tumors in KP mice

To determine whether PX-866 inhibits the metastatic properties of established lung adenocarcinoma cells, we used a panel of KP cells and human lung cancer cell lines. These cell lines had epithelial or mesenchymal features on the basis of their levels of epithelial markers (miR-200 family members and E-cadherin) and mesenchymal markers (ZEB1, Snail2, and vimentin), but E-cadherin levels were not different in KP cells (Supplemental Figure 2, A–C), consistent with partial EMT in mesenchymal KP cells, as reported previously (13). Unlike epithelial KP cells, mesenchymal KP cells generate highly metastatic tumors in syngeneic, immunocompetent mice and form polarized epithelial spheres in 3-dimensional cultures that undergo EMT in response to extracellular cues, whereas epithelial KP cells are poorly metastatic and do not form such spheres (13). Our in vitro studies of mesenchymal KP cell lines revealed that PX-866 markedly reduced cell invasion in Boyden chambers (Figure 1D and Supplemental Figure 2D). In contrast, PX-866 did not suppress the invasion of epithelial KP cell lines (Figure 1D and Supplemental Figure 2D) or affect the proliferation or colony formation of any tested cell lines (Supplemental Figure 3, A and B). In the human lung cancer cell lines, we found that PX-866 treatment did not detectably change cell proliferation or colony formation (Supplemental Figure 3, C and D), but did inhibit the migration of mesenchymal cells, the basal activity of which was higher than that of epithelial cells (Supplemental Figure 4A); it also decreased the invasion of the 2 cell lines (H1299 and H157) that had measurable invasive activity in Boyden chambers (Supplemental Figure 4B).
To determine whether PX-866 has an antimetastatic effect on invasive lung adenocarcinoma cells, we injected syngeneic, immunocompetent mice subcutaneously with highly metastatic KP cells (344SQ or 531LN2) 1 day after the start of PX-866 treatment, which potently decreased intratumoral AKT Ser473 phosphorylation (Supplemental Figure 5A). After a 6-week course of treatment with PX-866, mice bearing 344SQ tumors had diminished numbers of lung metastases, but no change in primary tumor size, whereas the same schedule of PX-866 treatment markedly reduced primary tumor size and the incidence of lung metastases in mice injected with 531LN2 cells (Figure 1E). PX-866 had no significant effect on 531LN2 tumor growth or metastasis when treatment was delayed until 7 days after 531LN2 cell injection (Supplemental Figure 5B), suggesting that PX-866 decreased the attachment of 531LN2 tumors.
On the basis of the evidence described above that epithelial and mesenchymal KP cells are differentially sensitive to invasion suppression by PX-866, we postulated that sensitivity to PX-866 is regulated by the ZEB1/miR-200 axis, which controls bidirectional transitions between epithelial and mesenchymal states (24). Compared with its effects on control transfectants (393P_vector), PX-866 more potently inhibited the invasion of an epithelial KP cell line (393P) that was stably transfected with ZEB1, which induces EMT and increases the invasive and metastatic properties of 393P cells (ref. 25, Figure 1F, and Supplemental Figure 6A). Conversely, sensitivity to PX-866 was suppressed by ectopic expression of the miR-200b/200a/429 cluster in a highly invasive, mesenchymal KP cell line (344SQ) that underwent mesenchymal-epithelial transition (MET) and lost metastatic properties after ectopic miR-200b/200a/429 expression (ref. 13, Figure 1F, and Supplemental Figure 6A). Similarly, ectopic miR-200b/200a/429 expression desensitized a mesenchymal human lung adenocarcinoma cell line (H1299) to PX-866 treatment (Figure 1G and Supplemental Figure 6B). The inhibitory effect of PX-866 on PI3K-dependent signaling, as measured by suppression of AKT phosphorylation, was enhanced by ectopic ZEB1 and attenuated by ectopic miR-200b/200a/429 (Supplemental Figure 6C). Thus, the ZEB1/miR-200 axis regulated cell sensitivity to PI3K antagonism.
ZEB1 increases p110α catalytic activity by stimulating an EGFR/ERBB2 autocrine loop.
We reasoned that the increased sensitivity of mesenchymal KP cells to PX-866 may result from higher basal PI3K enzymatic activity in those cells. To examine this possibility, we first performed an ELISA to quantify the levels of phosphatidylinositol (3,4,5)-triphosphate (PIP3) produced in vitro by p110α, p110β, p110γ, or p110δ immunoprecipitated from a panel of epithelial and mesenchymal KP cells. Mesenchymal KP cells had significantly higher p110α activity, whereas the activity levels of the other p110 family members did not significantly differ between mesenchymal and epithelial cells (Figure 2A).
Figure 2. ZEB1 activates p110α through an EGFR/ERBB2 autocrine loop.

(A) ELISA of PIP3 levels (picomoles) catalyzed in vitro by p110 isozymes immunoprecipitated from epithelial (307P, 393P, 412P, and 531P1) and mesenchymal (344P, 344SQ, 531P2, and 531LN2) KP cells. (B) ELISA. p110α-specific enzymatic activity in KP cells. Mean ± SD from triplicate samples. (C) Array-based detection of phosphorylated RTKs (p-RTK) in cell lysates. Duplicate phosphorylated EGFR (p-EGFR) and ERBB2 (p-ERBB2) are indicated (arrows); positive control proteins are in the top left, top right, and lower right corners. Densitometric analysis (bar graphs) of p-EGFR and p-ERBB2 expressed as mean values of duplicate spots in each transfectant relative to those of 393P_vector cells, which were set at 1.0. (D) Western blot analysis using phospho-specific (p-) and total protein antibodies. Controls include blotting for actin. (E) qPCR analysis of mRNA levels of ERBB ligands. Mean ± SD from triplicate samples. *P < 0.01 compared with 393P_vector; #P < 0.01 compared with 344SQ_vector. (F) ELISA. β-cellulin (BTC) and amphiregulin (AREG) levels in conditioned media. Mean ± SD from triplicate samples. (G and H) Western blot analysis using phospho-specific and total protein antibodies (G) and ELISA of p110α enzymatic activity (H). 393P_ZEB1 cells were treated with neutralizing antibodies against AREG and/or BTC for 24 hours. IgG was used as a negative control. Mean ± SD from triplicate samples. (I) Western blot analysis. Cells were treated with drug or vehicle (0) for 24 hours. Controls include blotting for actin. (J) ELISA. p110α enzymatic activity in 393P_ZEB1 cells treated with the indicated doses of drug or vehicle (0) for 24 hours. Mean ± SD from triplicate samples.
We sought to understand why p110α enzymatic activity was higher in mesenchymal KP cells than in epithelial KP cells. Expression of the PTEN lipid phosphatase, a suppressor of PI3K activity, was not diminished in mesenchymal KP cells (Supplemental Figure 7, A and B), and genomic sequencing studies revealed no activating Pik3ca somatic mutations in mesenchymal KP cells (data not shown). However, p110α catalytic activity was increased by ectopic ZEB1 expression and diminished by ectopic miR-200b/200a/429 expression (Figure 2B), leading us to postulate that the ZEB1/miR-200 axis targets an upstream regulator of PI3K. STAT3 Tyr705 phosphorylation was similar in epithelial and mesenchymal KP cells grown in a monolayer culture or as primary tumors in syngeneic, immunocompetent mice (Supplemental Figure 7, A and C), excluding cytokine receptor signaling as a major contributor to the differential p110α activity in epithelial and mesenchymal KP cells. An array-based analysis of total phosphotyrosine levels on 39 RTKs, many of which activate PI3K (26), revealed that phosphorylation of EGFR and ERBB2 was increased by ectopic ZEB1 and diminished by ectopic miR-200b/200a/429 (Figure 2C), which was confirmed by Western blot analysis using phosphorylation site–specific antibodies against EGFR and ERBB2 (Supplemental Figure 7D). Phosphorylated EGFR and ERBB2 were detected in 5 of 7 mesenchymal KP cell lines and in 0 of 5 epithelial KP cells (Figure 2D). The mRNA levels of certain EGFR ligands (Hbegf, Areg, Btc, and Ereg) were higher in 344SQ than in 393P cells, upregulated by ectopic ZEB1 in 393P cells, and minimally decreased by ectopic miR-200b/200a/429 in 344SQ cells (Figure 2E), which was confirmed by ELISAs of amphiregulin and β-cellulin protein levels in conditioned media samples (Figure 2F). The mRNAs of other ligands for EGFR (Egf and Tgfa) or ERBB3 and ERBB4 (Nrg1, Nrg2, Nrg3, and Nrg4) were not detectable. We observed that EGFR phosphorylation and PI3K activity were suppressed by treatment with neutralizing antibodies against amphiregulin and β-cellulin, administered in combination but not singly (Figure 2, G and H); treatment with tyrosine kinase inhibitors that were selective for EGFR (erlotinib) or EGFR and ERBB2 (afatinib) prominently decreased the phosphorylation of EGFR and ERBB2 (Figure 2I) and their downstream effectors (ERK, AKT, and mTOR) (Supplemental Figure 7E) and diminished p110α enzymatic activity (Figure 2J) but not invasion (data not shown). Although the reason for the lack of anti-invasive activity is not clear, feedback loops activated by EGFR/ERBB2 antagonists (27) may have contributed. These findings suggest that ZEB1 increased p110α catalytic activity by stimulating an EGFR/ERBB2 autocrine loop.
Next, we examined the mechanisms by which ZEB1 increased the expression of amphiregulin and β-cellulin. The activities of luciferase reporters containing the Areg or Btc 3′-UTRs were repressed by cotransfection of miR-200 pre-miRs, and this suppression was mitigated by site-directed mutagenesis of the predicted miR-200 binding sites in the Areg and Btc 3′-UTRs (www.targetscan.org) (Figure 3A). Furthermore, ectopic ZEB1 expression increased RNA polymerase II binding to the Areg and Btc promoters (Figure 3B), suggesting that ZEB1 upregulated the expression of these genes at the transcriptional level. Given that ZEB1 typically functions as a transcriptional repressor, we postulated that it relieves the repression of a transcriptional activator of Areg and Btc. On the basis of the presence of predicted binding sites in the Areg and Btc gene promoters (http://jaspar.genereg.net), we performed cotransfection assays to determine whether ZEB1, ETS1, GATA3, GATA4, or GATA6 can activate luciferase reporters containing Areg or Btc promoter elements. GATA6 activated both promoters (Figure 3C) and bound to the Areg and Btc gene promoters in ChIP assays (Figure 3D). We found that GATA6 levels were increased by ectopic ZEB1 expression and suppressed by ectopic miR-200b/200a/429 expression (Figure 3E). Cotransfection of miR-200a pre-miRs suppressed the activity of a Gata6 3′-UTR luciferase reporter, and this suppression was mitigated by site-directed mutagenesis of a predicted miR-200 binding site in the Gata6 3′-UTR (www.targetscan.org) (Figure 3F). In conclusion, ZEB1 increased the expression of amphiregulin and β-cellulin by derepressing miR-200 targets (Areg, Btc, and Gata6).
Figure 3. BTC and AREG are transcriptionally or post-transcriptionally regulated by the ZEB1/miR-200 axis.

(A) 3′-UTR reporter assays. 344SQ cells were transiently cotransfected with pre–miR-200s and Btc or Areg 3′-UTR luciferase reporters in which predicted miR-200 binding sites were wild-type (WT) or mutated (Mut). Results were normalized using a dual firefly/Renilla luciferase system. Mean ± SD from triplicate samples. (B) RNA polymerase II (Pol II) ChIP assays of Areg, Btc, and Hbegf promoters. Mean ± SD from triplicate samples. (C) GATA6 ChIP assays on Btc and Areg promoters. IgG was used as a negative control. (D) Promoter reporter assays. 393P cells were transiently cotransfected with Btc or Areg promoter luciferase reporters (500 ng/well in 24-well plates) and control or a GATA6 expression vector (500 ng). Mean ± SD from triplicate samples. (E) qPCR analysis (bar graph) and Western blot analysis (gels) of GATA6 expression. qPCR values, normalized on the basis of ribosomal protein L32 mRNA levels, represent the mean ± SD from triplicate samples and were expressed relative to 393P_vector cells (Vec), which were set at 1.0. Actin was included as a loading control. (F) 3′-UTR reporter assays. 344SQ cells were transiently cotransfected with pre–miR-200a and Gata6 3′-UTR luciferase reporters, in which predicted miR-200 binding sites were wild-type or mutated. Results were normalized using a dual firefly/Renilla luciferase system. ZEB1 3′-UTR was used as a positive control. Mean ± SD from triplicate samples. Results are expressed relative to the normalized luciferase activity in hRL-transfected (empty vector–transfected) cells.
ZEB1 regulates the expression of PI3K pathway signaling components.
Given that the amplification of certain PI3K signaling components is oncogenic (28–30), we hypothesized that these genes are transcriptional targets of the ZEB1/miR-200 axis. The effects of ectopic ZEB1 on the levels of numerous PI3K signaling components are opposite those of ectopic miR-200b/200a/429 (Figure 4A). The expression of 2 genes with large fold changes, Pik3ca and Rheb, was confirmed by immunoblotting of KP and human lung cancer cells (Figure 4B and Supplemental Figure 8A). To determine whether p110α promotes metastasis, we performed shRNA-mediated depletion of p110α, which reduced p110α protein levels by 90% and 60% in Pik3ca shRNA #1 and #2 transfectants, respectively (Figure 4C). p110α depletion had no detectable effect on cell proliferation in monolayer culture (Figure 4D), led to decreased soft agar colony formation (Figure 4E) and Boyden chamber invasion (Figure 4F), and caused decreased lung metastasis incidence without significantly changing the size of primary tumors in syngeneic, immunocompetent mice (Figure 4G). To investigate the prometastatic role of RHEB, we rendered 344SQ cells RHEB deficient by transfecting them with either of 2 different shRNAs (Supplemental Figure 8B); this reduced the phosphorylation of S6 ribosomal protein (Supplemental Figure 8C), decreased invasion in Boyden chambers (Figure 5A), and reduced the numbers of lung metastases without consistently decreasing the size of primary tumors in syngeneic, immunocompetent mice (Figure 5B).
Figure 4. p110α mediates invasion and metastasis.

(A) qPCR analysis of the mRNA levels of genes in the PI3K pathway. Values were normalized on the basis of ribosomal protein L32 mRNA levels and are expressed as ratios (393P_ZEB1 to 393P_vector; 344SQ_miR-200 to 344SQ_vector). (B) Western blot analysis of KP cells. (C) qPCR assays (bar graph) and Western blot analysis (gels) of 344SQ_Pik3ca shRNA cells (shRNA #1 and #2) and 344SQ_scr shRNA cells (Scr). Controls include 344SQ_scr shRNA cells and blotting for actin. qPCR values were normalized on the basis of ribosomal protein L32 mRNA levels and represent the mean ± SD from triplicate samples. (D) Density of 344SQ_Pik3ca shRNA cells and 344SQ_scr shRNA cells in monolayer culture was measured by MTT assay at the indicated time points. Values represent the mean ± SD from triplicate samples. (E) Soft agar colony formation. Colonies were photographed (images) and counted (bar graph) after 14 days of incubation. Original magnification, ×4. Values represent the mean ± SD from triplicate wells. (F) Invasion assays. Cells were photographed (images) and quantified (bar graph). Scale bars: 100 μm. (G) Scatter plots of primary tumor volume (left scatter plot) and numbers of visible lung metastases (right scatter plot) in syngeneic mice injected in the flanks with 344SQ_Pik3ca shRNA (n = 5) or 344SQ_scr cells (n = 5). Each mouse is indicated with a dot. Mean ± SD calculated for each cohort.
Figure 5. p110α and RHEB are downstream mediators of ZEB1.

(A) Invasion assays. Invasive 344SQ_Rheb shRNA cells (#3 and #5) and 344SQ_scr shRNA cells were photographed (images) and quantified (bar graph) after 24 hours of incubation. Mean ± SD from triplicate samples. Scale bars: 100 μm. (B) Scatter plots of primary tumor weight and numbers of visible lung metastases in syngeneic mice injected with 344SQ_Rheb shRNA or 344SQ_scr cells. Mean ± SD for each cohort. (C) RNA polymerase II ChIP assays of Pik3ca (top) and Rheb (bottom) promoters. (D and E) Luciferase assays of PIK3CA (D) and Rheb (E) promoter activity. 344SQ cells were cotransfected with GATA3 expression and reporter plasmids. Results were normalized using a dual firefly/Renilla luciferase system. Mean ± SD from triplicate samples. Results are expressed relative to the normalized luciferase activity in pGL3 basic vector–transfected cells. (F and G) GATA3 ChIP assays on the PIK3CA (F) and RHEB (G) promoters in 293T cells and H322 cells, respectively. Controls include whole-cell lysates (input), ChIP using IgG, and amplification of an upstream region in the PIK3CA promoter (PCR #1) and intron 7 of RHEB (PCR #2) containing no predicted GATA-binding sites. Sizes of the PCR products generated from regions containing predicted GATA-binding sites in PIK3CA promoter (PCR #2 and PCR #3) and RHEB promoter (PCR #1) are indicated.
To gain insight into the mechanisms by which PI3K-dependent signaling promotes invasion, RNA samples from 344SQ_Pik3ca shRNA cells and 344SQ_scrambled (344SQ_scr) cells were subjected to global transcriptional profiling. Using 344SQ_scr cells as the reference, 697 Affymetrix probe sets (538 unique genes) were differentially expressed (353 upregulated and 185 downregulated by a factor of at least 2.0; P < 0.01; Supplemental Figure 8D and Supplemental Table 1). Of 18 highly up- or downregulated genes from the arrays sampled, 15 were differentially expressed by quantitative RT-PCR (qRT-PCR) analysis (Supplemental Figure 8E). We performed an enrichment analysis (Fisher’s exact test using gene ontology terms [www.ingenuity.com]) to examine the predominant gene functions in the expression signature, which revealed enrichment in “cellular movement” (114 genes, P = 4.73 × 10–8 to 6.20 × 10–3), “cellular development” (135 genes, P = 1.61 × 10–9 to 6.40 × 10–3), “cellular assembly and organization” (78 genes, P = 1.91 × 10–7 to 6.20 × 10–3), “cellular function and maintenance” (129 genes, P = 1.91 × 10–7 to 6.20 × 10–3), and “protein synthesis” (37 genes, P = 5.34 × 10–7 to 5.63 × 10–3). Of particular interest among the downregulated genes were promigratory genes that encode cell surface receptors (Flt1, Itgb5, and Fzd6), secreted ligands (Nrg1 and Wnt9), collagens (Col4a4 and Col4a5), and regulators of the actin cytoskeleton (Tns1, Myh10, and Six4) (31–36), several of which were confirmed by qRT-PCR analysis (Supplemental Figure 8F). Flt1 encodes a cell surface receptor (VEGFR1) that is required for invasion and metastasis in KP cells (36), but reconstitution of FLT1 expression in 344SQ_Pik3ca shRNA cells (Supplemental Figure 8G) was not sufficient to enhance invasion in Boyden chamber assays (Supplemental Figure 8H). We conclude that PI3K upregulates the expression of multiple promigratory genes that act cooperatively to promote cell invasion.
ZEB1 upregulates the expression of Pik3ca and Rheb through a friend of GATA 2/GATA3–dependent mechanism.
We queried the mechanisms by which ZEB1 increased the expression of Pik3ca and Rheb. Despite having a predicted miR-200 binding site (www.targetscan.org), a Rheb 3′-UTR reporter was not suppressed by cotransfection of miR-200 pre-miRs (Supplemental Figure 9A). Ectopic miR-200b/200a/429 expression decreased RNA polymerase II binding to the Pik3ca and Rheb gene promoters (Figure 5C), suggesting that these genes were regulated at the transcriptional level. On the basis of the presence of predicted binding sites in the PIK3CA and Rheb gene promoters (http://jaspar.genereg.net), we performed cotransfection assays to determine whether GATA3, GATA4, GATA6, p53, ΔNp63, ZEB1, or ETS1 can activate luciferase reporters containing PIK3CA or Rheb promoter elements. GATA3 activated both promoters, whereas a mutant GATA3 that was deficient in DNA-binding activity did not have this effect (Figure 5, D and E, and Supplemental Figure 9, B and C), and GATA3 bound to the promoter regions of PIK3CA (Figure 5F) and RHEB (Figure 5G) in ChIP assays. These are noteworthy findings, given that GATA3 is a driver of invasion and metastasis in KP cells (37).
Although GATA3 levels did not change in response to ectopic ZEB1 expression in 393P cells (data not shown), the levels of friend of GATA 2 (FOG2), a GATA3 coactivator that formed complexes with GATA3 in 344SQ cells (Figure 6A), were increased by ectopic ZEB1 in 393P cells (Figure 6B), and FOG2 was expressed at higher levels in mesenchymal than in epithelial KP cells (Figure 4B). The Fog2 3′-UTR has predicted binding sites for miR-200c and miR-183 (www.targetscan.org), which are transcriptional targets of ZEB1 in 393P cells (25). The activity of a Fog2 3′-UTR luciferase reporter was repressed by cotransfection of miR-200c or miR-183 pre-miRs, which was mitigated by site-directed mutagenesis of the predicted miR-200 and miR-183 binding sites in the Fog2 3′-UTR (Figure 6C), and FOG2 levels were downregulated by transfection of miR-200c pre-miRs (Figure 6D), leading us to conclude that miR-200c and miR-183 serve as intermediates in ZEB1-induced upregulation of FOG2 expression. Introduction of Fog2 shRNAs into 344SQ cells and HCC827_ZEB1 cells decreased p110α expression (Figure 6, E and F and Supplemental Figure 10). FOG2 knockdown in 344SQ cells reduced invasion in Boyden chambers (Figure 6G) and diminished lung metastases, but not primary tumor size, in syngeneic, immunocompetent mice (Figure 6H). Thus, we conclude that ZEB1 upregulates p110α expression and promotes metastasis by derepressing FOG2, a miR-200 and miR-183 target.
Figure 6. FOG2 is a mediator of ZEB1.

(A) Coimmunoprecipitation of FOG2 and GATA3 in 344SQ cells. A GFP-tagged GATA3 expression vector was generated to discriminate between exogenous GATA3 and IgG heavy chain on the basis of size. Extracts were subjected to immunoprecipitation with IgG, anti-FOG2, or anti-GATA3 antibodies. Total extracts and immunoprecipitates were then blotted with anti-FOG2 or -GATA3. (B) qPCR analysis (bar graph) and Western blot analysis (gels) of FOG2 expression. qPCR values, normalized on the basis of ribosomal protein L32 mRNA levels, represented the mean ± SD from triplicate samples and were expressed relative to 393P_vector cells (Vec), which were set at 1.0. (C) 3′-UTR reporter assays. 344SQ cells were transiently cotransfected with Fog2 3′-UTR reporters, in which the predicted miR binding sites were wild-type or mutated (mut) and control, miR-183, or miR-200c precursors. Mean ± SD from triplicate samples. (D) Western blot analysis of FOG2 expression in 344SQ cells that had been transiently transfected with indicated miRNA precursors. (E and F) Western blot analysis of FOG2 (E) and p110α (F) in 344SQ_scr shRNA cells (Scr) and 344SQ_Fog2 shRNA cells. (G) Invasion assays. Invasive 344SQ_Fog2 shRNA (#3 and #4) and 344SQ_scr shRNA cells were photographed (images) and quantified (bar graph). Mean ± SD from triplicate samples (bar graph). Scale bars: 100 εm. (H) Scatter plots of primary tumor weight (left) and numbers of visible lung metastases (right) in syngeneic mice injected with 344SQ_Fog2 shRNA (#3 and #4) or 344SQ_scr cells. Mean ± SD for each cohort. Actin was included as a loading control for Western blots.
FOG2 is not required for ZEB1-induced mesenchymal differentiation.
On the basis of its proinvasive role, we postulated that FOG2 mediates ZEB1-induced mesenchymal differentiation, a process that is temporally linked with EMT-associated cell invasion (6). An analysis of the transcriptional profiles of human lung and breast tumors classified as mesenchymal or epithelial on the basis of known EMT markers revealed that FOG2 (ZFPM2) expression was not correlated with clinical outcome, but was correlated strongly with the presence of an EMT signature (Table 3) and ZEB1 expression (Figure 7A). However, FOG2 knockdown in 344SQ cells did not change cell morphologic characteristics (Supplemental Figure 11A) or the expression of epithelial or mesenchymal markers (Supplemental Figure 11B). Similarly, treatment with PX-866 did not change the basal or TGF-β–induced expression of epithelial and mesenchymal markers in KP cells (Supplemental Figure 11, C and D) or human lung cancer cells (Supplemental Figure 11E). On the basis of these findings, we conclude that ZEB1-induced mesenchymal differentiation and invasion are separable processes.
Table 3.
Correlation of selected genes with EMT status in breast and lung tumors
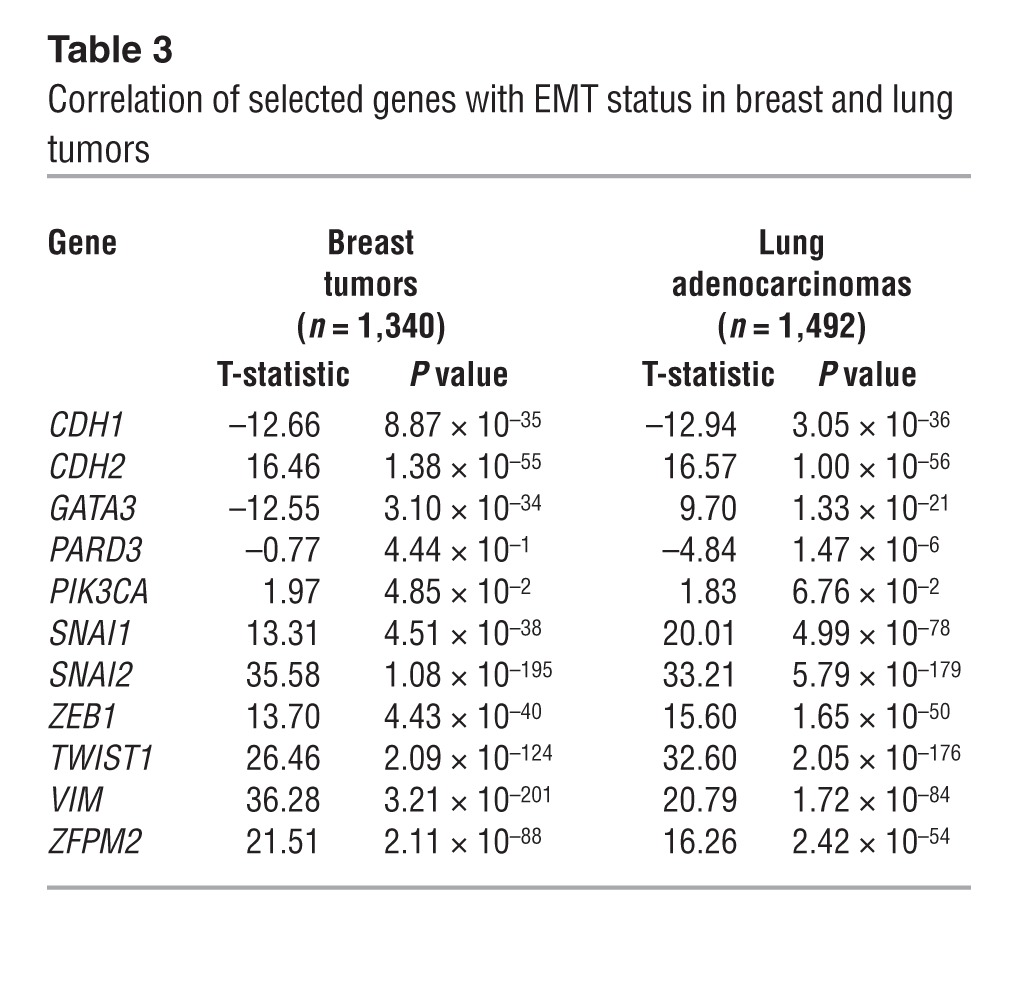
Figure 7. FOG2 expression was correlated with EMT.

(A) Heatmap depiction of correlations between selected genes and EMT status in breast and lung tumors. Two gene expression array datasets were examined, one from human breast cancer (n = 1340 tumors) and one from human lung adenocarcinoma (n = 1492 tumors). Each tumor profile was scored for the degree of EMT manifestation on the basis of a 16-gene signature of canonical EMT markers (CDH1, CDH2, DSP, FOXC2, FN1, ITGB6, MMP2, MMP3, MMP9, OCLN, SNAI2, SNAI1, SOX10, TWIST1, VIM, and GSC) (21). Tumors were scored by subtracting the sum of the normalized values for the epithelial markers from the sum of the values for the mesenchymal markers. Pearson’s correlations of genes with EMT scores are indicated. Red denotes a significant positive correlation; blue denotes a negative correlation. (B) Schematic illustration of the pathway described herein.
Discussion
In this study, we explored the feasibility of metastasis inhibition in a mouse model driven by expression of mutant KRAS and p53 that closely mimics the features of poor-prognosis human lung adenocarcinomas (12). Treatment with a PI3K antagonist suppressed distant metastasis without affecting spontaneous lung tumor multiplicity, which is consistent with previous findings that PI3K inhibition does not inhibit the growth of spontaneous Kras-mutant lung adenocarcinomas in mice (38). In the context of recent reports that EMT sensitizes tumor cells to AXL RTK inhibitors (39), these findings suggest that therapeutic approaches to target mesenchymal tumor cells are feasible and of clinical value.
PI3K is a critical mediator of oncogenic KRAS and is activated through multiple mechanisms in KRAS-mutant cells. KRAS binds directly to the p110α catalytic subunit, and abrogating this interaction suppresses tumor development in mouse models of lung cancer (16). RTKs can also function as intermediaries. In KRAS-mutant colon carcinoma cells, IGF-1R is highly expressed, binds to PI3K, and is required for PI3K activation (40). High expression of RON in KRAS-mutant lung and pancreatic tumors is part of a “KRAS addiction” gene expression signature that activates PI3K, maintains epithelial differentiation, and promotes tumor cell viability (4). In mice that develop KRAS-mutant human pancreatic tumors, EGFR activates PI3K and is required for tumor outgrowth (41). Here, ZEB1 activated PI3K by derepressing miR-200 targets (AREG, BTC, and GATA6) that stimulated an EGFR/ERBB2 autocrine loop and by derepressing a miR-200 and miR-183 target, FOG2, that enhanced GATA3-induced expression of p110α (Figure 7B). Collectively, these findings suggest that the pathways through which KRAS-mutant cancer cells activate PI3K become more numerous as the cells acquire additional driver mutations, implying that sustained PI3K activity is essential for the malignant progression of KRAS-mutant tumors.
Central to the “migrating stem cell hypothesis” is the belief that highly metastatic tumor cells can transit reversibly between epithelial and mesenchymal states and acquire stem cell properties in response to extracellular cues (24). These dynamic events are regulated by a double-negative feedback loop between ZEB1 and miR-200. The link between TP53 gain of function, which underlies this cellular plasticity, and ZEB1 upregulation is unclear. One candidate is miR-130b, which is a transcriptional target of mutant p53 that binds directly to the ZEB1 3′-UTR (42). Targets of the miR-200 family include SOX2, KLF4, BMI1, and SUZ12, which function as master regulators of cell specification during embryonic development (10, 43). Similar to these master regulators, GATA3 plays a fundamental role in development and tumorigenesis (44). In the breast, GATA3 is required for luminal epithelial differentiation and commitment, and its expression is lost as breast epithelial cells progress to fully transformed adenocarcinomas (45). Reconstitution of GATA3 expression in undifferentiated breast carcinoma cells is sufficient to induce epithelial differentiation and suppress metastasis (45). In contrast to its suppressive effect on EMT and metastasis in the breast, GATA3 induces EMT and promotes metastasis in lung cancer cells through transcriptional repression of the miR-200b/200a/429 cluster (37). We evaluated large human tumor datasets and found that, consistent with its opposing roles in breast and lung cancer cells, GATA3 expression was correlated with an epithelial state in breast cancer and a mesenchymal state in lung cancer. These findings suggest that the biologic role of GATA3 varies dramatically in different tumor cell types.
The complexity of GATA3 as a regulator of tumor cell biology depends partly on its transcriptional cofactors. GATA3 associates with FOG1 or FOG2, which can function as coactivators or corepressors of target genes (46). FOG2 is critical for normal cardiac development in mice, and germline FOG2 mutations are associated with congenital cardiac defects in humans (47, 48). FOG2-deficient epicardial cells can undergo EMT (48), as we observed in KP cells, but GATA3 deficiency shifts KP cells into an epithelial state that is refractory to TGF-β treatment (37), suggesting that FOG2 mediates its actions in part through GATA factors other than GATA3. Unlike FOG1, which binds preferentially to GATA1, GATA2, and GATA3 and is expressed predominantly in hematopoietic cells, FOG2 binds to GATA3, GATA4, GATA5, and GATA6 and is expressed broadly in epithelial, myocardial, neuronal, and hepatic cells (46). GATA factors share varying degrees of homologic features and have functions that are partially interchangeable. For example, the erythrocytic phenotype of Gata1-null mice can be partially rescued by knocking Gata3 into the Gata1 locus, presumably because of common target genes regulated by these GATA factors (44). However, GATA2 and GATA3 have nonoverlapping biologic roles in KRAS-mutant lung adenocarcinoma cells; GATA2 maintains the survival of KRAS-mutant human lung cancer cells (49), whereas the viability of KP cells is not reduced by GATA3 depletion (37). In sum, FOG2 may mediate its actions through multiple GATA factors with overlapping and nonoverlapping functions.
Despite reports demonstrating a temporal linkage between EMT and metastasis (6, 50), the role of EMT in metastasis is unclear. The findings reported here are consistent with a growing body of evidence that EMT is neither sufficient nor required for metastasis. Expression of the polarity proteins PARD3 and SCRIB is silenced in advanced cancers, and targeted deletion of PARD3 or SCRIB promotes tumorigenesis and metastasis and compromises cell-cell cohesion without inducing EMT (51, 52). While the expression of an EMT activator (TWIST1 or PRRX1) is sufficient to induce tumor cell invasion and dissemination, this expression must be downregulated and EMT must be reversed to allow colonization at distant sites (50, 53). Similarly, miR-200 family members can inhibit invasion in primary tumors while promoting colonization at distant sites (13, 54). Collectively, these findings suggest that EMT is part of a larger and more complex transcriptional program that is driven by EMT activators like ZEB that confer specific biologic properties needed to complete each stage of the metastasis cascade.
Methods
Mouse experiments.
For the syngeneic tumor experiments, 1 × 106 KP cells were subcutaneously injected into the flanks of 10- to 16-week-old wild-type littermates of KP mice bred in our mouse colony. These mice were killed 6 weeks after injection or at the first signs of morbidity. For the PX-866 treatment experiments, PX-866 (2 mg/kg/d) or its vehicle (5% alcohol in drinking water) was orally administered 3 times a week. The drug was administered for 6 weeks in 8- to 12-week-old wild-type mice bearing syngeneic tumors or for 16 weeks in 16- to 20-week-old KP mice bearing spontaneous lung tumors. Necropsies were performed on KP mice to quantify the number of primary lung tumors; to diagnose the histological features of H&E-stained, formalin-fixed, paraffin-embedded lung tumor tissues, as described previously (55); and to determine the presence or absence of metastases to regional mediastinal lymph nodes and distant extrathoracic sites. Necropsies were performed on wild-type mice that had been injected with syngeneic tumor cells to measure the diameters and weights of primary flank tumors and to determine whether the tumor cells had spread to distant sites on the basis of a visual examination of the lungs and histologic evaluation of H&E-stained, formalin-fixed, paraffin-embedded lung tissue.
Antibodies.
We purchased anti-p110α (sc-7174), anti-p110β (sc-7175), anti-p110γ (sc-7177), anti-p110δ (sc-7176), anti-FOG2 (sc-10755), anti-GATA3 (sc-9009), anti-GATA6 (sc-2768), anti-ZEB1 (sc-10572), anti-actin (sc-1616), normal rabbit IgG (sc-2027), normal goat IgG (sc-2028), and HRP-conjugated anti-goat IgG (sc-2768) antibodies from Santa Cruz Biotechnology Inc.; anti–p-AKT (pS473: 4060, pT308: 9275), anti-AKT (9272), anti-RHEB (4935), anti-total EGFR (2232), anti–p-EGFR (pY1068: 2243; pY1173: 2237), anti–total ERBB2 (2248), anti–p-ERBB2 (pY877: 2241, pY1248: 2244), anti-ZEB1 (3396), anti-vimentin (5741), anti-Snail2 (9585), anti–E-cadherin (3195), anti-pS6 (2211), anti-S6 (2212), anti-PTEN (9552), anti–p-STAT3 (9131), anti-STAT3 (9132), anti–p-ERK1/2 (9101), anti-ERK1/2 (9102), anti–p-mTOR (5536), anti-mTOR (2983), HRP-conjugated anti-mouse IgG (7076), and HRP-conjugated anti-rabbit IgG (7074) antibodies from Cell Signaling Technology; and anti-FLT1 (MAB471), AREG (AF989), and BTC (AF1025) neutralizing antibodies from R&D Systems.
Cell culture and generation of shRNA transfectants.
The isolation and culture conditions for murine KP cell lines, transfectants derived from them, and human lung cancer cell lines were described previously (13, 25). Human lung cancer cell lines that express ectopic miR-200b/200a/429 under the control of a doxycycline-inducible promoter (H1299) or that constitutively express ectopic ZEB1 (HCC827) will be described elsewhere. Unless indicated otherwise, cells were treated with 25 nM PX-866 or vehicle. We purchased sets of shRNA constructs for murine Rheb (Open Biosystems) and Fog2 (OriGene). shRNA constructs for Pik3ca were generated by inserting the following sequences into the pLKO.1 shRNA expression vector (8453; Addgene): for shRNA #1, CCGGTGTGAAACAAGACGACTTTGTGACCTTCGGCTTCAAGAGAGCCGAAGGTCACAAAGTCGTCTTGTTTCATTTTTG, and for shRNA #2, CCGGTGAGAGGTTTCAGGAGATGTGTTACAAGGCTTTCAAGAGAAGCCTTGTAACACATCTCCTGAAACCTCTTTTTTG. Two micrograms of each shRNA construct was transfected into cells using lipofectamine and PLUS reagent (Invitrogen). Stable transfectants were generated by culturing the cells in the presence of puromycin (15 μg/ml) for 4 to 6 weeks.
Cell invasion assay.
The upper chambers of growth factor–reduced Matrigel invasion chambers (354483; BD Biosciences) were seeded with cells (1 × 105 cells per well) in triplicate wells. Medium (RPMI 1640), with or without 10% FBS, was placed in the lower and upper chambers, respectively. Drugs were added to the lower chambers at the time the medium was added. Cells were incubated at 37°C for 24 hours. The invaded cells were visualized with Diff-Quik Stain (Dade Behring). Three microscopic fields (original magnification, ×10) were photographed and counted per chamber, and the results were expressed as the mean ± SD of invaded cells from the replicate wells.
Cell proliferation assay.
Cells (1 × 103 cells/well) were plated in quadruplicate in RPMI 1640 supplemented with 10% FBS on 96-well cell culture plates and treated with or without PX-866 for 3 days. Cells were then subjected to a 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay (M5655; Sigma-Aldrich) for 3 hours at 37°C before being lysed in dimethyl sulfoxide (200 μl/well). Cell proliferation was quantified at an optical density of 570 nm on a BenchMark microplate reader (BioRad).
Colony formation assay.
Cells in monolayer culture were harvested by being briefly digested with 0.05% Trypsin-EDTA (25300-054; Invitrogen), resuspended in DMEM containing 10% FBS, and aspirated through a 25-gauge needle to generate single-cell suspensions. Cells (1 × 104) were mixed with 0.3% agar and plated on 0.6% agar in DMEM supplemented with 20% FBS in 6-well plates. Cultures were maintained for 14 days to allow the formation of cell colonies, which were visualized by MTT staining (M5655; Sigma-Aldrich), manually counted, and expressed as the mean ± SD of the number of colonies from replicate (triplicate) wells.
Protein extraction, immunoprecipitation, and Western blotting.
Cells were lysed in lysis buffer containing 10 mM Tris (pH 7.4), 1 mM EDTA, 0.5 mM EGTA, 150 mM NaCl, 1% Triton X-100, 50 mM NaF, 10 mM Na4P2O7, 1 mM PMSF, and protease inhibitor cocktail (P8340; Sigma-Aldrich). The lysates were quantified for protein concentrations with BCA protein assay reagents (Pierce Biotechnology). For FOG2/GATA3 coimmunoprecipitation studies, 393P_ZEB1 cells were cotransfected with FOG2/FLAG-pCK (a gift from V. N. Kim, Seoul National University, Seoul, Republic of Korea) and GATA3/pEGFP-C3 (subcloned from GATA3/pcDNA3.1; 1332; Addgene) plasmids. Cell lysates were then immunoprecipitated with anti-FOG2 or anti-GATA3 antibody, and the immunoprecipitates were subjected to Western blotting using anti-GATA3 or anti-FOG2 antibody, respectively. For Western blotting, proteins (30 μg per sample) were electrophoresed in SDS-polyacrylamide gel and transferred onto nitrocellulose membranes (Amersham Hybond ECL; GE Healthcare). The membranes were blocked with 5% skim milk in Tris-buffered saline and Tween-20 and then incubated with primary antibodies overnight at 4°C. After being washed with Tris-buffered saline and Tween-20 for 30 minutes at ambient temperature, the membrane was further incubated with HRP-conjugated secondary antibodies for 1.5 hours, followed by 30 minutes of washing with Tris-buffered saline and Tween-20. Protein bands were visualized with ECL substrates (Pierce Biotechnology).
PI3K activity assay. p110 isozyme–specific activity was measured using an ELISA kit (K1000s; Echelon).
In brief, p110 isozymes were immunoprecipitated from total cell lysates (500 μg) using anti-p110α, -β, -γ, or -δ–specific antibodies, and the immunoprecipitates were isolated with Protein A/G Plus Agarose (sc-2003; Santa Cruz Biotechnology Inc.). PI3K reactions were run using the class I PI3K physiologic substrate PI(4,5)P2 (PIP2). The enzyme reactions, PIP3 standards, and controls were then mixed and incubated with immunoprecipitated p110 protein. This mixture was then transferred to a PIP3-coated microplate for competitive binding. A peroxidase-linked secondary detector and colorimetric detection were used to quantify the amount of PIP3 (picomoles) produced by PI3K by comparing the enzyme reactions with a PIP3 standard curve.
Phosphorylated RTK arrays.
Using the Proteome Profiler Mouse Phospho-RTK array kit (R&D Systems), array membranes were incubated overnight with 300 μg of total protein lysates, and phosphorylated RTKs were detected using an anti-phosphotyrosine HRP detection antibody and ECL reaction. Pixel densities of phosphorylated RTK spots were analyzed using Quantity One (Bio-Rad).
ELISA.
BTC (MB0323) and AREG (MA0623) ELISA kits were purchased from NeoBioLab. The assay was performed on conditioned media samples collected from cell cultures according to the manufacturer’s protocol.
RNA extraction and qPCR assays.
Cells were grown to 80% confluence in RPMI 1640 supplemented with 10% FBS on 6-well plates, washed with ice-cold PBS, and subjected to RNA extraction using 1 ml of TRIzol (Invitrogen). mRNA was reverse transcribed using SuperScript III (Invitrogen). For qPCR reactions, 1:10 dilutions of cDNA products were amplified using SYBR Green PCR Master Mix and analyzed using the 7500 Real-Time PCR System (both from Applied Biosystems). For the analysis of miR-200 family members, TaqMan assays (Applied Biosystems) were performed using 10 ng of total RNA. mRNA and miRNA expression values were normalized on the basis of ribosomal protein L32 mRNA and snoRNA-135 levels, respectively. The qPCR primers used in this study are listed in Supplemental Table 2.
mRNA expression profiling.
Snap-frozen samples were shipped on dry ice to Asuragen for processing and analysis using GeneChip Mouse Genome 430A 2.0 Array Chips (Affymetrix). Total RNA was isolated and quantified, and gene expression data were generated as described previously (13); arrays were quantile normalized. Array data have been made available through the Gene Expression Omnibus (GEO GSE46869). Two-sided Student’s t tests and fold changes with log-transformed data were used to determine any significant differences between groups of samples in mean gene mRNA levels, as described previously (56). Expression patterns were visualized as color maps using Java TreeView software (57). Gene ontology annotation terms were searched for within gene sets using SigTerms (58).
ChIP assay.
Cells were cross-linked with 1% formaldehyde and incubated in lysis buffer (50 mM Tris-HCl [pH 8.1], 1% SDS, 10 mM EDTA, and protease inhibitor cocktail) on ice for 10 minutes and sonicated (Cole-Parmer GEX-130 sonicator; pulse mode for 10 seconds, followed by no pulsing for 10 seconds for 20 cycles at 50% power). For RNA polymerase II ChIP assays, 344SQ_vector and 344SQ_miR-200 cells were immunoprecipitated with anti-RNA polymerase II antibody (Millipore) or control mouse IgG. DNA was eluted and purified with a PCR purification kit (QIAGEN), and qPCR analysis was carried out using specific primers to amplify Pik3ca or Rheb promoters. To detect binding of GATA3 to the PIK3CA and RHEB gene promoters, anti-GATA3 antibodies were used to immunoprecipitate GATA3/chromatin complexes from H322 cells and 293T cells (after transiently transfecting the latter with a GATA3 expression vector), as previously described (37). Anti-GATA6 antibodies were used to immunoprecipitate GATA6/chromatin complexes from 393P_ZEB1 cells. The PCR primers for ChIP assays are listed in Supplemental Table 3.
Luciferase assay.
Murine Rheb promoter (–2,251 to +749 bp from the transcription start site) was isolated by PCR amplification from a murine BAC clone (RPCI-23 64J21; BACPAC Resource Center) and ligated into the pGL3-Basic vector (Promega). Murine Btc (–2,081 to +674 bp) and Areg (–2,122 to +626 bp) promoters were isolated by PCR amplification from genomic DNA of TC-1 mouse embryonic stem cells. Human PIK3CA promoter (–829 to +105 bp) in a pGL3-Basic vector was a gift from Arezoo Astanehe (University of British Columbia, Vancouver, British Columbia, Canada). 344SQ cells were seeded on 24-well plates (1 × 105 cells/well) 1 day before transfection. Cells were cotransfected with promoter reporters (500 ng) and wild-type or mutant GATA3 (Addgene plasmids #1332 and #1334, respectively) or GATA6 (a gift from David Berger, the Michael E. DeBakey VA Medical Center, Houston, Texas, USA). A pRL-TK vector (50 ng, Promega) was cotransfected as an internal control. After 48 hours of transfection, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. The ZEB1 3′-UTR (1.9 kb; a gift from Gregory Goodall, University of Adelaide, Adelaide, Australia) was subcloned into the pCI-neo-hRL vector. Btc (2,069 bp), Areg (253 bp), and Gata6 (1,153 bp) 3′-UTRs were isolated by genomic PCR and ligated into the same vector. The reporters (500 ng) were cotransfected with synthetic miR-200 precursors (5 nM; Ambion) into 344SQ cells (1 × 105 cells/well). pGL3-Control (50 ng; Promega) was used as an internal control. After 48 hours, luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega). Results were normalized on the basis of firefly luciferase.
Immunohistochemical analysis.
Tissue sections were stained with anti–p-STAT3 antibody (Y705, 9145; Cell Signaling Technology) and HRP-conjugated anti-rabbit secondary antibody (8114, SignalStain Boost immunohistochemical detection reagent). SignalSlide Phospho-Stat1/3/5 (8105) immunohistochemical control was used as a control. Staining was performed using the Autostainer Plus automated stainer machine (Dako). Staining intensity was evaluated in a blinded manner to avoid bias.
Statistics.
To probe human lung adenocarcinomas with the CMap PI3K gene signature (21), we assembled a “compendium” dataset of 11 published datasets of expression profiles of human lung adenocarcinomas (n = 1,492 tumors) for which survival data were available (22, 59–68). The genes in each dataset were first normalized to standard deviations from the median; if multiple human array probesets referenced the same gene, the probeset with the highest variation was used to represent the gene. To score each tumor in the compendium dataset for similarity to the CMap PI3K gene signature, we derived a “T-score” metric in relation to the experimental signature, similar to that in previous analyses (69); in brief, the T-score was defined for each external profile as a 2-sided T-statistic that compared, within the profile, the mean of the upregulated genes in the signature with the mean of the downregulated genes in the signature. For human breast cancer, we used another compendium array dataset, described previously (70). Unless otherwise indicated, comparisons between 2 mean values ± SD (experimental versus control) were performed using a 2-tailed Student’s t test. P values less than 0.05 were considered statistically significant.
Study approval.
All animal experiments were reviewed and approved by the IACUC of The University of Texas MD Anderson Cancer Center.
Supplementary Material
Acknowledgments
This work was supported by R01 grants CA157450 (to J.M. Kurie) and P50 CA70907 from The University of Texas Southwestern/MD Anderson Cancer Center Lung Specialized Program of Research Excellence (to J.M. Kurie). J.M. Kurie is the Elza and Ina A. Shackelford Endowed Professor in Lung Cancer Research. Y. Yang was supported by the Mayo Clinic Foundation. D.L. Gibbons was supported by NCI K08 grant CA151651 and by an International Association for the Study of Lung Cancer Fellow Grant and received financial support from Waun Ki Hong (MD Anderson Cancer Center). C.J. Creighton was supported by P30 grant CA125123. We thank Fengju Chen, Yiqun Zhang, and David R. Jones for technical assistance.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(6):2696–2708. doi:10.1172/JCI72171.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 3.Califano R, Landi L, Cappuzzo F. Prognostic and predictive value of K-RAS mutations in non-small cell lung cancer. Drugs. 2012;72(suppl 1):28–36. doi: 10.2165/1163012-S0-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Singh A, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6):489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28(1–2):151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 8.Gregory PA, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 9.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283(22):14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wellner U, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 11.Bracken CP, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68(19):7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 12.Zheng S, El-Naggar AK, Kim ES, Kurie JM, Lozano G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene. 2007;26(48):6896–6904. doi: 10.1038/sj.onc.1210493. [DOI] [PubMed] [Google Scholar]
- 13.Gibbons DL, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23(18):2140–2151. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bleau AM, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubrovska A, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A. 2009;106(1):268–273. doi: 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta S, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129(5):957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 17.Hambardzumyan D, et al. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22(4):436–448. doi: 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim CF, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 19.Muranen T, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21(2):227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, et al. Phosphatidylinositol 3-kinase mediates bronchioalveolar stem cell expansion in mouse models of oncogenic K-ras-induced lung cancer. PLoS One. 2008;3(5):e2220. doi: 10.1371/journal.pone.0002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creighton CJ, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010;12(3):R40. doi: 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chitale D, et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28(31):2773–2783. doi: 10.1038/onc.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ihle NT, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3(7):763–772. [PubMed] [Google Scholar]
- 24.Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11(9):670–677. doi: 10.1038/embor.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn YH, et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest. 2012;122(9):3170–3183. doi: 10.1172/JCI63608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17(1):1–16. doi: 10.1615/CritRevOncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu ZH, et al. Mammalian target of rapamycin activator RHEB is frequently overexpressed in human carcinomas and is critical and sufficient for skin epithelial carcinogenesis. Cancer Res. 2010;70(8):3287–3298. doi: 10.1158/0008-5472.CAN-09-3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okudela K, et al. PIK3CA mutation and amplification in human lung cancer. Pathol Int. 2007;57(10):664–671. doi: 10.1111/j.1440-1827.2007.02155.x. [DOI] [PubMed] [Google Scholar]
- 30.Rychahou PG, et al. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc Natl Acad Sci U S A. 2008;105(51):20315–20320. doi: 10.1073/pnas.0810715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bastian P, Lang K, Niggemann B, Zaenker KS, Entschladen F. Myosin regulation in the migration of tumor cells and leukocytes within a three-dimensional collagen matrix. Cell Mol Life Sci. 2005;62(1):65–76. doi: 10.1007/s00018-004-4391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark IB, Jarman AP, Finnegan DJ. Live imaging of Drosophila gonad formation reveals roles for Six4 in regulating germline and somatic cell migration. BMC Dev Biol. 2007;7:52. doi: 10.1186/1471-213X-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall EH, Daugherty AE, Choi CK, Horwitz AF, Brautigan DL. Tensin1 requires protein phosphatase-1alpha in addition to RhoGAP DLC-1 to control cell polarization, migration, and invasion. J Biol Chem. 2009;284(50):34713–34722. doi: 10.1074/jbc.M109.059592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo SH. Tensin. Int J Biochem Cell Biol. 2004;36(1):31–34. doi: 10.1016/S1357-2725(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 35.Zhao WJ, Schachner M. Neuregulin 1 enhances cell adhesion molecule l1 expression in human glioma cells and promotes their migration as a function of malignancy. J Neuropathol Exp Neurol. 2013;72(3):244–255. doi: 10.1097/NEN.0b013e3182863dc5. [DOI] [PubMed] [Google Scholar]
- 36.Roybal JD, et al. miR-200 Inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol Cancer Res. 2011;9(1):25–35. doi: 10.1158/1541-7786.MCR-10-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest. 2011;121(4):1373–1385. doi: 10.1172/JCI42579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Byers LA, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19(1):279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ebi H, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121(11):4311–4321. doi: 10.1172/JCI57909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Navas C, et al. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(3):318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong P, et al. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32(27):3286–3295. doi: 10.1038/onc.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iliopoulos D, et al. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39(5):761–772. doi: 10.1016/j.molcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chou J, Provot S, Werb Z. GATA3 in development and cancer differentiation: cells GATA have it! J Cell Physiol. 2010;222(1):42–49. doi: 10.1002/jcp.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kouros-Mehr H, et al. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13(2):141–152. doi: 10.1016/j.ccr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chlon TM, Crispino JD. Combinatorial regulation of tissue specification by GATA and FOG factors. Development. 2012;139(21):3905–3916. doi: 10.1242/dev.080440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Luca A, et al. New mutations in ZFPM2/FOG2 gene in tetralogy of Fallot and double outlet right ventricle. Clin Genet. 2011;80(2):184–190. doi: 10.1111/j.1399-0004.2010.01523.x. [DOI] [PubMed] [Google Scholar]
- 48.Zhou B, et al. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J Clin Invest. 2009;119(6):1462–1476. doi: 10.1172/JCI38723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar MS, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149(3):642–655. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 50.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCaffrey LM, Montalbano J, Mihai C, Macara IG. Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell. 2012;22(5):601–614. doi: 10.1016/j.ccr.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK. Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nat Cell Biol. 2013;15(2):189–200. doi: 10.1038/ncb2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ocana OH, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22(6):709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 54.Korpal M, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17(9):1101–1108. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iwanaga K, et al. Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res. 2008;68(4):1119–1127. doi: 10.1158/0008-5472.CAN-07-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Creighton CJ, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26(25):4078–4085. doi: 10.1200/JCO.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saldanha AJ. Java Treeview — extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 58.Creighton CJ, Nagaraja AK, Hanash SM, Matzuk MM, Gunaratne PH. A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. RNA. 2008;14(11):2290–2296. doi: 10.1261/rna.1188208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beer DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8(8):816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 60.Bhattacharjee A, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98(24):13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bild AH, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(7074):353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 62.Botling J, et al. Biomarker discovery in non-small cell lung cancer: integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clin Cancer Res. 2013;19(1):194–204. doi: 10.1158/1078-0432.CCR-12-1139. [DOI] [PubMed] [Google Scholar]
- 63.Hou J, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5(4):e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Okayama H, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72(1):100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 65.Shedden K, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14(8):822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang H, et al. A 12-Gene Set Predicts Survival Benefits from Adjuvant Chemotherapy in Non-Small Cell Lung Cancer Patients. Clin Cancer Res. 2013;19(6):1577–1586. doi: 10.1158/1078-0432.CCR-12-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomida S, et al. Relapse-related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. J Clin Oncol. 2009;27(17):2793–2799. doi: 10.1200/JCO.2008.19.7053. [DOI] [PubMed] [Google Scholar]
- 68.Zhu CQ, et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010;28(29):4417–4424. doi: 10.1200/JCO.2009.26.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gibbons DL, et al. Expression signatures of metastatic capacity in a genetic mouse model of lung adenocarcinoma. PLoS One. 2009;4(4):e5401. doi: 10.1371/journal.pone.0005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kessler JD, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335(6066):348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.