

Summary
Mutation or epigenetic silencing of the transcription factor C/EBPα is observed in ~10% of patients with acute myeloid leukemia (AML). In both cases, a common global gene expression profile is observed, but down-stream targets relevant for leukemogenesis are not known. Here we identify Sox4 as a direct target of C/EBPα whereby its expression is inversely correlated with C/EBPα activity. Downregulation of Sox4 abrogated increased self-renewal of leukemic cells and restored their differentiation. Gene expression profiles of leukemia initiating cells (LICs) from both Sox4 overexpression and murine C/EBPα mutant AML models clustered together, but differed from other types of AML. Our data demonstrate that Sox4 overexpression resulting from C/EBPα inactivation contributes to the development of leukemia with a distinct LIC phenotype.
Introduction
Acute myeloid leukemia (AML) is characterized by a differentiation block and aberrant clonal growth of hematopoietic blasts. It has been classified into individual subtypes with respect to morphology, immunophenotype and genetic abnormalities. In recent years, genome-wide gene-expression profiling has further identified distinct subsets (Valk et al., 2004), which may reflect the underlying biology of these subtypes and potentially reveal critical downstream targets for therapeutic intervention.
Transcription factor CEBPA is differentially translated into two isoforms of 42 kDa and 30 kDa (Lin et al., 1993). Two thirds of AML cases with acquired point mutations of CEBPA have one allele harboring N-terminal frame-shift mutations leading to increased 30 kDa isoform; and the other allele harboring C-terminal in-frame insertions or deletions resulting in deficient DNA-binding and/or homodimerization activities (Gombart et al., 2002; Pabst et al., 2001). CEBPA double mutant cases and cases where CEBP has been epigenetically silenced demonstrate similar gene expression signatures, suggesting a common mechanism of disease (Valk et al., 2004).
C/EBPα regulates the expression of myeloid lineage-specific genes and cell cycle regulators, and impacts on self-renewal and myeloid lineage commitment of hematopoietic stem cells (HSCs), as well as inducing growth arrest (Nerlov, 2004). However, the 30 kDa isoform fails to induce differentiation of granulocytes and to block cell proliferation (Nerlov, 2004). Cebpa knockout mice die at birth with a complete lack of mature granulocytes, while adult mice with induced loss of C/EBPα demonstrate a block from common myeloid progenitors (CMP) to granulocyte monocyte progenitors (GMP) and accumulation of myeloid blasts (Ye et al., 2013; Zhang et al., 2004). Knock-in mice carrying engineered bi-allelic CEBPA mutations as found in human AML developed leukemia (Bereshchenko et al., 2009), but the key molecular downstream events required to trigger leukemogenesis remain unclear.
Sox4 belongs to the Sox (SRY-related HMG-box) transcription factor family (Jafarnejad et al., 2012). T-cell development in Sox4-deficient embryos is severely impaired (Kuwahara et al., 2012), and mice receiving Sox4−/− fetal liver cells exhibit a block at the pro-B cell stage (Schilham et al., 1996). SOX4 is up-regulated in various types of human solid tumors and is a frequent target of retroviral insertional mutagenesis in many murine B-cell lymphoma and myeloid leukemia models (Jafarnejad et al., 2012). Its overexpression is associated with clonal dominance of HSC (Kustikova et al., 2007), stem/progenitor cells repopulation advantage (Deneault et al., 2009), a block in differentiation of myeloid progenitor 32DCl3 cells (Boyd et al., 2006), and can induce myeloid leukemia (Du et al., 2005)(Kvinlaug et al., 2011). However, the precise role of Sox4 gene in AML and how it is involved in specific AML subtypes is poorly understood.
Results
A shRNA screen identifies Sox4 as a mediator of enhanced replating ability and decreased differentiation of Cebpa-deficient cells in culture
Previous studies have revealed that disruption of C/EBPα in the hematopoietic system resulted in abnormal expansion and an altered transcription program of hematopoietic stem cells (Ye et al., 2013; Zhang et al., 2004). To identify the downstream effectors, we performed genome-wide gene expression profiling and verified expression changes of the top 30 candidates of up-regulated genes upon loss of C/EBPα in LSK cells (lin−Sca1+kit+) (Figure 1A; Table S1). We then functionally evaluated the effect of knocking-down these genes on Cebpa KO cells (Mx1-Cre+ Cebpa loxP/loxP–from here on referred to as Cebpa KO following Cre mediated deletion) after serially replating in methylcellulose cultures, a cell culture assay which has been correlated with the ability to induce leukemia in mice (Huntly et al., 2004; Lavau et al., 1997; Moran-Crusio et al., 2011). We transduced Cebpa KO LSK cells with lentiviruses carrying either a mix of scrambled shRNA (control) or a pool of five shRNAs all targeting one specific candidate and assessed their capability to undergo serial replating (Figure S1A). Among the 30 candidates, shRNA-mediated knock-down of Sox4 exhibited the strongest reduction of serial replating capability of Cebpa KO LSK cells, with only a few colonies formed after 2 rounds of replating and none at 4th round, while scrambled controls maintained colony formation even after 4 rounds of replating (Figure 1B, S1B). Moreover, Sox4 shRNA transduced Cebpa KO LSK cells differentiated into macrophages (Figure 1C); as compared to morphologically immature control colonies (aberrantly differentiated leukemic blast cells). Flow cytometry analysis also revealed significantly increased expression of the mature myeloid marker Mac1 (Figure 1D). Genotyping PCR results confirmed complete excision of Cebpa alleles in these cells (Figure S1C, S1D).
Figure 1. Sox4 is required for abnormal serial-replating ability and myeloid differentiation block of Cebpa KO stem/progenitor cells.

(A) Sox4 expression was analyzed by qPCR in LSK cells (Lin−c-kit+Sca1+) sorted from Mx1-Cre−CebpaloxP/loxP (WT) and Mx1-Cre+ CebpaloxP/loxP (KO) mice 14 days after polyI:C injection. Relative gene expression levels were determined as % Gapdh.
(B) Cebpa KO LSK cells were transduced with lentiviruses harboring either scrambled shRNAs or Sox4 shRNAs and replated in methylcellulose plus puromycin. The bar chart shows the colony number for four rounds.
(C) Wright-Giemsa staining of cells from the first plating round in Figure 1B. Data are representative of 3 independent experiments. Macrophages are indicated with arrows. Scale bar, 20 μm.
(D) Flow cytometry analysis of cells from the first plating round in Fig 1B. Shadow histogram indicates scrambled shRNA-infected cells, and black line indicates Sox4 shRNA-infected cells. Percent cells are shown for the indicated gates. Data is representative of 3 independent experiments.
(E) Wild-type LSK, CMP (Lin−c-kit+Sca1−CD34+FcRgII/IIIlo) and GMP (Lin−c-kit+Sca1−CD34+FcRgII/IIIhigh) were transduced with an empty retrovirus (Vector) or a retrovirus expressing Sox4 (Sox4) and replated as Figure 1B.
(F) Wild-type LSK cells were transduced as in Figure 1E and grown in liquid culture plus puromycin and cytokines. 7 days later, Mac1 (left) and Gr1 (right) expression was analyzed by flow cytometry. Shadow histograms indicate cells infected with empty virus, and black lines indicate cells overexpressing Sox4. Percent cells are shown for the indicated gates. Data are representative of 3 independent experiments. Error bars indicate the mean ± SEM of 3 independent experiments. **: p <0.001. See also Figure S1 and Table S1.
Since knocking-down of Sox4 impaired the outgrowth capability of Cebpa KO cells we asked if Sox4 overexpression was sufficient to confer replating ability to wild-type cells. We therefore overexpressed Sox4 in wild-type LSK and progenitor cells (CMP, GMP) and again performed serial replating assays. Aberrant Sox4 expression resulted in enhanced replating ability for more than 4 rounds in all tested cell types (Figure 1E, S1E). Moreover in cytokine-supplemented myeloid liquid culture of Sox4 transduced cells, myeloid differentiation was impaired (Figure 1F, S1F).
Collectively, our experiments identified the oncogene Sox4 as a factor mediating increased serial-replating ability and blocked differentiation of Cebpa-deficient progenitors.
Sox4 expression is directly regulated by C/EBPα
To understand the precise regulatory interaction between Cebpa and Sox4 during hematopoiesis, we examined their expression patterns during different stages of blood cell differentiation. qPCR analysis of adult wild-type mice revealed that expression of Cebpa increased from SLAM+-HSC (enriched for HSCs), LSK cells (enriched for both HSCs and multipotent progenitors), common myeloid progenitors (CMP) to granulocyte-macrophage progenitors (GMP), and finally dropped in mature myeloid cells (Mac1+Gr1+) (Figure 2A). In contrast, Sox4 demonstrated an inverse expression pattern during myeloid lineage commitment, with highest expression in LSK, and gradually decreasing towards myeloid maturation with barely detectable levels in terminally differentiated myeloid cells (Mac1+Gr1+) (Figure 2A).
Figure 2. Sox4 is a direct target of repression by C/EBPα.

(A) qPCR analysis of Cebpa (top) and Sox4 (bottom) level in SLAM+-LSK, LSK, CMP, GMP and myeloid cells (Mac1+Gr1+). Relative gene expression levels were determined as % Gapdh.
(B) (Top) Schematic representation of the Sox4 promoter extending from −500bp to +889bp relative to TSS of murine Sox4 gene. Triangles denote the ChIP-Seq C/EBPα binding peaks as shown in Figure S2A. (Bottom) HEK293 cells were transfected with the Sox4 promoter reporter and increasing amounts of pCDNA3-C/EBPα. Luciferase activities were normalized to renilla activities and presented as percentage of empty vector.
(C) (Left) Schematic representation of truncated constructs of the Sox4 promoter reporter. (Right) Luciferase values of reporter constructs with an empty vector (white bar) or C/EBPα expressing constructs (black bars).
(D) EMSA using a probe containing a potential C/EBPα binding site from the Sox4 promoter was performed with nuclear extracts from HEK293T cells overexpressing Flag tagged C/EBPα (HEK293T/C/EBPα-Flag) and Flag antibody (anti-Flag) or C/EBPa antibody (anti-C/EBPα). “C/EBPα”, C/EBPα complex; ss, supershifted band; x, migration of nonspecific protein complexes binding to the probes.
(E) (Left) Schematic representation of luciferase reporters carrying wild-type Sox4 promoter (WT) or Sox4 promoter with mutated C/EBPα binding sites (C/EBPα(m)) marked with a cross. The putative C/EBPα binding sites and the mutated version are underlined. (Right) Luciferase values of reporter constructs carrying wild-type or site-mutated Sox4 promoter with an empty vector (white bar) or C/EBPα expressing constructs (black bars).
(F) ChIP-qPCR analysis of specific binding of C/EBPα to the upstream proximal region of murine Sox4 gene in wild-type primary stem/progenitor cells (Lin−c-kit+) or myeloid cells (Mac1+Gr1+). (Top) Schematic of Sox4 gene with three qPCR primer sets (arrows below). Oval denotes the C/EBPα binding site identified in Figure 2E. Primer sets “mSox4-1” and “mSox4-2” were used to amplify regions −171 to +77 bp and −215 to −16 bp (relative to the TSS) of Sox4 gene and primer set “NC” were used as a negative control. (Bottom) Relative DNA enrichment was measured by qPCR and is presented as percentage of input chromatin. Black bars represent ChIP-qPCR signals from the C/EBPα pull-down (C/EBPα) while white bars are from IgG control (IgG).
Error bars indicate the mean ± SEM of 3 independent experiments. *: p<0.01; **: p<0.001. See also Figure S2.
We next examined whether the reciprocal expression of Cebpa and Sox4 during myeloid commitment was due to a direct transcriptional repression of Sox4 by C/EBPα. ChIP-seq analysis of murine bone marrow derived primary macrophages indeed revealed two adjacent C/EBPα-binding regions upstream of the murine Sox4 gene (Figure S2A). A major binding region was approximately 500bp upstream of the Sox4 transcriptional start site (TSS) (−500bp~+1bp relative to TSS) and a minor one was observed within the 5′-UTR region of Sox4 open reading frame (+1bp~+200bp relative to TSS) (Figure S2A, 2B). Luciferase assays using a reporter carrying a DNA fragment with the Sox4 promoter (−500bp to 889 bp relative to the TSS) revealed that C/EBPα repressed the transcription activity from the Sox4 promoter in a dose-dependent manner (Figure 2B). Such repression was not due to the cellular saturation or toxicity of C/EBPα protein, since a parallel experiment indicated that within a similar dose range, C/EBPα transactivated a luciferase reporter carrying six consensus C/EBPα binding sites (Figure S2B).
Previous studies from our laboratory demonstrated that C/EBPα inhibits Myc and Cebpg transcription through interfering with E2F1 transactivation on these promoters (Alberich-Jorda et al., 2012; Johansen et al., 2001). We therefore first examined whether C/EBPα mediated Sox4 repression through a similar mechanism. However, luciferase assays revealed that neither E2F1 nor other E2F family members were able to activate the Sox4 promoter (Figure S2C, S2D), although they were all fully capable of transactivating a reporter carrying consensus E2F binding sites. Increasing the amount of E2F1 did not antagonize the repression of Sox4 promoter by C/EBPα (Figure S2E). Furthermore, to determine whether C/EBPα-mediated repression of E2F1 activity suppressed Sox4 mRNA expression, we transduced Cebpa KO LSK cells with either wild-type Cebpa or a series of Cebpa mutants (Porse et al., 2005). These experiments showed that C/EBPα mutants BRM2 and BRM5, which were defective in inhibiting E2F1-dependent transcription but still binding efficiently to C/EBPα sites, still repressed aberrant Sox4 expression in Cebpa KO LSK cells at levels comparable to that of wild-type C/EBPα (Figure S2F). In these experiments, C/EBPα wild-type and mutant constructs were expressed at similar levels (Figure S2F). Taken together, these observations ruled out a role of E2F1 in the repression of Sox4 by C/EBPα.
The Sox4 promoter contains multiple C/EBPα binding sites. We therefore asked whether C/EBPα repressed Sox4 promoter through direct DNA binding. We generated a series of truncation mutants of the Sox4 promoter corresponding to the location of the putative C/EBPα binding sites (Figure 2C, S2G). Luciferase assays with these truncation mutants revealed that a fragment between −183bp and −83bp upstream of the Sox4 transcription start site was essential for C/EBPα-mediated repression (Figure 2C). EMSA combined with supershift assay and competitor assay further revealed specific association of C/EBPα protein with potential cis-elements at −174bp to −150bp upstream of the Sox4 transcription start site (Figure 2D, S2H). Mutation of a highly evolutionally conserved C/EBPα binding site in this region resulted in loss of repression of the Sox4 promoter by C/EBPα (Figure 2E, S2I). To further verify direct binding of C/EBPα to the identified region within the Sox4 promoter, we performed ChIP assays on primary cells isolated from adult wild-type mice. Using two sets of primers amplifying the −183~−83bp region (relative to the TSS), we confirmed binding of endogenous C/EBPα to this region in both a stem/progenitor cell enriched population (Lin−c-kit+) and mature myeloid cells (Mac1+Gr1+) (Figure 2F). Moreover, the binding of C/EBPα was specific to this region, since no enrichment of a distal region of the Sox4 gene was observed. In summary, our data demonstrate that C/EBPα negatively regulates Sox4 transcription via direct DNA-binding.
Sox4 mediates the abnormal expansion of LSK cells in the absence of C/EBPα in vivo
To functionally dissect the role in which dysregulated Sox4 expression is contributing to the Cebpa knockout phenotype in vivo, we designed an inducible Sox4/Cebpa double knockout model. We generated conditional Sox4 deficient mice alone (Mx1-Cre+ Sox4loxP/loxP– referred to as Sox4 KO following Cre mediated deletion) (Penzo-Mendez et al., 2007) and inducible Sox4/Cebpa double knockouts (Mx1-Cre+ Sox4loxP/loxPCebpa loxP/loxP– refer to as dKO after deletion) (Figure S3A). Flow cytometry analysis of bone marrow from mice 21 days after induction of Cre recombinase mediated deletion with polyI:C demonstrated that Cebpa KO mice exhibited a remarkable expansion of LSK cells, both in terms of percentage of total bone marrow (4-fold, Figure 3A) and in absolute number (18-fold, Figure 3B). LSK cells in Sox4 KO mice did not display any gross phenotypical differences compared to wild-type LSKs (Figure 3A, 3B). However, in concordance with the opposing roles of Sox4 and C/EBPα, LSK expansion of Cebpa KO mice was partially rescued in dKO mice with a 2-fold proportional (Figure 3A) and a 3-fold absolute reduction in LSK numbers (Figure 3B).
Figure 3. Sox4 mediates the aberrant progenitor expansion following loss of C/EBPα.
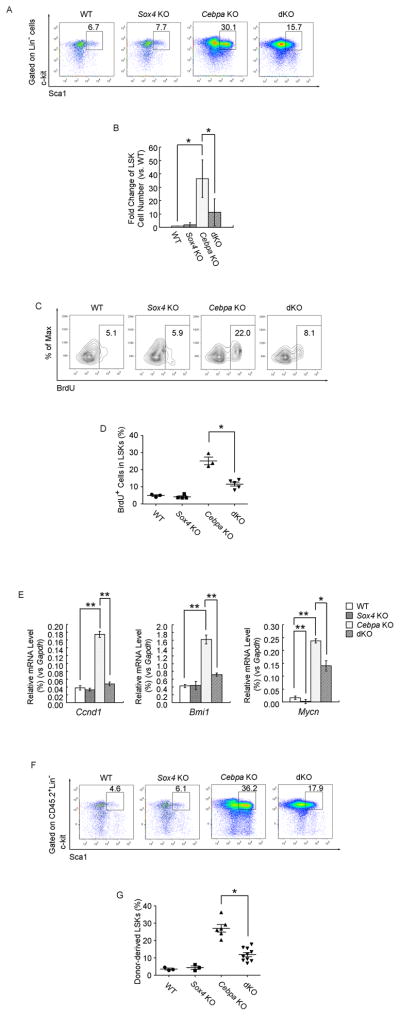
(A) Flow cytometry analysis of LSK frequency (Lin−c-kit+Sca1+) in bone marrow of Mx1-Cre−Sox4loxP/loxPCebpaloxP/loxP (WT), Mx1-Cre+Sox4loxP/loxPCebpa+/+ (Sox4 KO), Mx1-Cre−Sox4+/+CebpaloxP/loxP (Cebpa KO), and Mx1-Cre−Sox4loxP/loxPCebpaloxP/loxP (dKO) mice 21 days after polyI:C treatment. Representative FACS plots of 6 independent experiments are shown.
(B) Comparison of LSK cell number in the bone marrow of WT, Sox4 KO, Cebpa KO and dKO mice. Numbers are presented as fold change relative to the WT.
(C) Flow cytometry of BrdU incorporation in LSK cells of single and double knockouts. Percent BrdU+ cells are shown for the indicated gates. One representative experiment of three is shown.
(D) Quantitative analysis of BrdU incorporation in LSK cells of single and double knockouts.
(E) qPCR analysis of Ccnd1, Bmi1 and Mycn expression in LSK cells from single and double knockouts. Relative mRNA levels were determined as % Gapdh.
(F) Flow cytometry analysis of donor-derived LSK frequency (CD45.2+Lin−c-kit+Sca1+) in the bone marrow of recipient mice transplanted with LSK cells from mice with indicated phenotypes. Analysis was performed 4 months post transplantation. One representative of ten recipients is shown.
(G) Quantitative analysis of donor-derived LSK frequency in the bone marrow of recipient mice. Error bars indicate the mean ± SEM of 3 independent experiments. *: p<0.01; **: p<0.001. See also Figure S3.
We next assessed the impact of C/EBPα and Sox4 on LSK proliferation utilizing 5-bromodeoxyuridine (BrdU) incorporation in vivo. The proliferative fraction of LSKs was remarkably enhanced (~4-fold) in Cebpa KO mice, but was lower in the dKO mice, comparable to wild-type (Figure 3C, 3D). Gene expression analysis further identified several candidate genes potentially mediating the differences in proliferation between Cebpa KO and dKO LSK cells (Figure 3E, S3B, S3C). In Cebpa KO LSK cells, among the major cell cycle regulators, Cnnd1 was up-regulated whereas p21 and p57 (Pietras et al., 2011) were down-regulated compared to the wild-type and Sox4 KO counterparts. Increased expression was also observed for Bmi1 and Mycn, both of which are implicated in HSC proliferation and self-renewal in the absence of C/EBPα (Ye et al., 2013; Zhang et al., 2004). However, the additional disruption of Sox4 significantly impeded such deregulation in the dKO LSK cells, providing a putative molecular mechanism of how Sox4 induce LSK expansion in Cebpa KO mice.
To investigate if Sox4 modulates the Cebpa KO HSC phenotype in a cell-intrinsic manner, LSK cells from wild-type mice or conditional deficient mice (CD45.2+) 21 days post-polyI:C injection were transplanted into lethally irradiated recipients (CD45.1+). 4 months after transplantation, flow cytometry confirmed aberrant LSK expansion in recipients of Cebpa KO LSK cells and a partial rescue in recipients of dKO LSK cells (Figure 3F, 3G). Genotyping of LSK cells isolated from the conditional deficient mice or donor-derived LSK cells isolated from the recipients verified complete excision of the Cebpa and Sox4 alleles (Figure S3D, S3E, S3F). These data demonstrate an HSC cell intrinsic role for Sox4 on proliferation induced by loss of C/EBPα.
Targeting Sox4 restores myeloid differentiation of Cebpa KO cells in vivo
Knockdown of Sox4 enables Cebpa KO LSK cells to undergo myeloid differentiation in vitro (Figure 1C, 1D). To test the effect of Sox4 on the impaired myeloid differentiation of Cebpa KO LSKs in vivo, we examined whether the disruption of Sox4 could restore myeloid differentiation in Cebpa KO mice. Flow cytometry analysis of bone marrow cells from wild-type or Sox4 KO mice 21-day post-polyI:C injection revealed a similar proportion of Mac1+Gr1+ myeloid cells, whereas in Cebpa KO mice the myeloid population was barely detectable (Figure S4A, deletion efficiency shown in Figure S4B). Although a Mac1lo/+Gr1− population appeared in the dKO mice, they were morphologically immature blasts as the Mac1−Gr − population (data not shown).
In contrast, dKO LSK cells were able to give rise to a significant population of mature myeloid (Mac1+Gr1+) 4 months following transplantation (Figure 4A, 4B). Cytology of sorted donor-derived Mac1+Gr+ cells confirmed the presence of morphologically differentiated mature myeloid cells, similar to wild-type cells (Figure 4C). Genotyping analysis confirmed complete excision of Cebpa and Sox4 in the donor-derived Mac1+Gr1+ population (Figure S4C).
Figure 4. Loss of Sox4 restores myeloid differentiation of Cebpa-deficient LSK cells in vivo.

(A) Flow cytometry analysis of donor-derived myeloid cell (CD45.2+Mac1+Gr1+) in the bone marrow of recipient mice transplanted with LSK cells from mice with indicated phenotypes. Analysis was performed 4 months post transplantation. Representative FACS plots of 6 independent experiments are shown.
(B) Quantitative analysis of donor-derived myeloid cell frequency in the bone marrow of recipient mice. Error bars indicate the mean ± SEM of 6 independent experiments. *: p<0.01.
(C) Wright-Giemsa staining of donor-derived Mac1+Gr1+ cells in the bone marrow of recipient mice. Granulocytic cells are indicated with arrows. Scale bar, 10 μm.
(D) Flow cytometry analysis of donor-derived myeloid cells in the bone marrow of mice receiving Cebpa KO LSK cells transduced with scrambled shRNA or mSox4 shRNA. The percentages of Mac1+Gr1+ population within donor-derived (CD45.2+) GFP+ and GFP− cells are indicated. Representative FACS plots of 5 independent experiments are shown.
(E) Quantitative analysis of donor-derived shRNA-transduced myeloid cell frequency (CD45.2+GFP+ Mac1+Gr1+) in the bone marrow of recipient mice. Error bars indicate the mean ± SEM of 5 independent experiments. p=0.03.
See also Figure S4.
We also transplanted Cebpa KO LSK cells which had been transduced with GFP containing retroviruses expressing either Sox4 shRNAs (shRNA 672 and shRNA 928) or scrambled shRNA (Figure S4D, S4E). Mature myeloid cells (Mac1+Gr1+) were only present in the GFP+ donor population of mice receiving Sox4 shRNA-transduced Cebpa KO LSK cells (Figure 4D, 4E). Genotyping of donor-derived GFP+Mac1+Gr1+ cells confirmed a complete excision of Cebpa allele (Figure S4F). Taken together, these data demonstrate that the disruption of Sox4 restores myeloid programming of Cebpa KO LSKs in vivo, further substantiating the notion that Sox4 is epistatic to Cebpa.
Sox4 is crucial for the oncogenic activity of mutated C/EBPα
In a murine model of human AML expressing bi-allelic CEBPA mutations (K/L mice), only a distinct phenotypically defined group of cells (leukemia initiating cells, or LIC) gives rise to leukemia (Bereshchenko et al., 2009). To examine the role of Sox4 in those cells we examined its expression in cellular subpopulations of leukemic K/L mice, and observed that Sox4 mRNA was exclusively increased in the LIC fraction (fraction II: Lin*loSca1−Mac1loc-kit+), whereas it appeared to be decreased or absent in the other fractions (fraction I: Lin*loSca1+; fraction III: Lin*loSca1−Mac1+c-kit lo/−) (Figure 5A, S5A). It had previously been shown that mice transplanted with Sox4-transduced 5-FU treated bone marrow cells develop myeloid leukemia (Du et al., 2005). We confirmed this result by transplanting Sox4-transduced LSK cells and showed that mice died with a median latency of 4 months (Figure S5B, S5C). Flow cytometry revealed that Sox4-induced leukemia cells shared key phenotypical features with the model for bi-allelic Cebpa mutants (K/L leukemia; Figure 5B). Most importantly, as described for K/L leukemia, in Sox4-induced leukemia, only cells from fraction II gave rise to leukemia in recipient mice (Figure 5C, 5D).
Figure 5. The LIC in Sox4-induced AML resembles the LIC in murine C/EBPα mutant AML model.

(A) qPCR analysis of Sox4 expression in donor-derived fractions I (Lin*loSca1+), II (Lin*loSca1−Mac1loc-kit+) and III (Lin*loSca1−Mac1+c-kitlo/−) from wild-type control (WT) or leukemic K/L mice. Relative expression levels were determined as % Gapdh and presented as the mean ± SEM of 3 independent experiments. *: p<0.01
(B) Flow cytometry analysis of mice transplanted with bone marrow from wild-type mice, K/L mice or with LSK cells overexpressing Sox4. Total bone marrow cells were subjected to lineage depletion excluding Mac1 and Gr1 (Lin*) followed by FACS analysis. Representative FACS plots of 3 independent experiments are shown.
(C) Kaplan-Meier survival analysis of mice that received fractions I, II, and III from Sox4 leukemic mice. Number of recipients for each fraction (n) is indicated.
(D) Flow cytometry analysis of mice that received fractions I, II, and III from Sox4 leukemic mice. Donor-derived cells (CD45.2+) in the bone marrow were analyzed at indicated time points. Representative FACS plots of 3 independent experiments are shown.
(E) Donor-derived fractions II from leukemic K/L mice were transduced with either scrambled shRNAs or Sox4 shRNAs and serially replated. The bar chart shows the mean ± SEM of colony number of 3 independent experiments.
See also Figure S5.
To determine if Sox4 contributes to leukemogenesis not only in Cebpa deficient AML but also AML with mutated Cebpa, we examined the effect of Sox4 ablation on the self-renewal ability of K/L LIC. As Cebpa KO cells, LIC from leukemic K/L mice exhibited enhanced replating ability (Figure S5D). In addition, sorted K/L LIC cells were lentivirally transduced with either scrambled shRNAs (ϕ-1, ϕ-2) or Sox4 shRNAs (#80, #81 and #82) (Figure S5E). Cells transduced with Sox4 shRNA exhibited decreased ability to undergo serial-replating and barely gave rise to colonies in the second round of plating, while the control cells transduced with scrambled shRNA still gave rise to colonies even after the third round of plating (Figure 5E), demonstrating that increased Sox4 levels are crucial for the self-renewal properties of Cebpa mutant LICs.
A common gene expression signature defines Cebpa KO, Sox4 AML and K/L AML
Hierarchical clustering analysis of gene expression profiles further revealed that the LIC populations from K/L and Sox4 murine leukemia models clustered together, and were distinct from their immunophenotypic counterparts from wild-type mice (WT FrII) as well as the leukemic stem cell enriched L-GMP from MLL-AF9 and MOZ-TIF2 murine AML models (MF9 L-GMP and MT2 L-GMP) (Krivtsov et al., 2006; Kvinlaug et al., 2011) (Figure 6A). This suggested the existence of a common leukemogenic gene expression signature characterizing Sox4 LIC and mutated C/EBPα LIC. Intriguingly, Cebpa deficient HSCs also clustered together with LICs from K/L and Sox4 AML by sharing similar expression pattern of 686 genes, indicating underlying similarities among these three populations. We next addressed the question whether our results from the mouse models were of relevance to human AML. Previous unsupervised hierarchical gene-expression clustering of leukemic blasts from 562 AML patients identified a homogenous group (Cluster 4) in which both CEBPA silenced and bi-allelic mutated cases clustered together (Valk et al., 2004). Intriguingly, SOX4 expression was significantly increased within the very same group (Figure 6B), further confirming that SOX4 and mutated/silenced CEBPA are within one oncogenic pathway.
Figure 6. Gene expression signatures of Sox4 and K/L LICs are distinct from MLL-AF9 and MOZ-TIF2 L-GMPs.

(A) Hierarchical clustering of fraction II population from leukemic K/L and Sox4 recipients and their wild-type counterparts; L-GMP population from leukemic MLL-AF9 and MOZ-TIF2 recipients; and HSCs from Cebpa KO mice (KO 21D).
(B) Pair wise correlations between CEBPA and SOX4 in gene expression profiles of human AML patients. The bar and histograms next to each sample indicate: CEBPA allele status (mutations are shown in red and wild-type are shown in green), CEBPA and SOX4 mRNA expression levels. The black lines designate cluster 4 AML patients that have either mutated or silent CEBPA allele(s).
(C) Heatmap shows the 80-gene self-renewal associated LIC signature that are specifically up-regulated in the wild-type HSCs (vs. committed progenitors) as well as Sox4 and K/L LICs (vs. WT FrII) (fold change cutoff=1.5; p value cutoff=0.05).
(D) The Venn diagram shows the overlap between the “self-renewal associated LIC signature” (80 genes) and the “self-renewal associated L-GMP signature” (420 genes) in a single 2-class comparison. Shadow area indicates the “unique C/EBPα-Sox4 LIC signature” (56 genes).
(E) Supervised analysis of gene expression profiles from wild-type HSCs, CMPs, GMPs, MEPs as well as those from MLL-AF9 L-GMP, MOZ-TIF2 L-GMP, Sox4 LICs, K/L LICs and their control counterparts identified a set of self-renewal associated genes that show increased expression only in the L-GMPs but not in the LICs (fold change cutoff=1.5; p value cutoff=0.05).
(F) GSEA demonstrated significant enrichment of the murine “unique C/EBPα-Sox4 LIC signature” in human AML with mutated CEBP (left panel, p=0.005) or with silent CEBP (right panel, p<0.001) as compared to other human AML with wild-type CEBP.
See also Figure S6 and Table S2~S5.
Supervised clustering analysis identified a set of genes that were highly expressed in the wild-type HSCs and LICs populations from Sox4 and K/L leukemias, but exhibited decreased expression in committed progenitors (referred to as “self-renewal associated LIC signature”) (Figure 6C; Table S2). We next compared this signature with a previously reported L-GMP self-renewal associated signature (Krivtsov et al., 2006). This revealed that only a very limited set of genes crucial for acquisition of self-renewal properties in L-GMP (24 out of 420) are present in the LICs from Sox4 and K/L leukemias (24 out of 80) and the majority of this self-renewal associated LIC signature (56 genes) are unique to the Sox4 and K/L leukemias (refer to as “unique C/EBPα-Sox4 LIC signature”) (Figure 6D, S6A; Table S3~S5). Within the unique C/EBPα-Sox4 LIC signature, two key elements of TGFβ-FOXO signaling which have been implicated in maintaining leukemia-initiating cell in chronic myeloid leukemia (Naka et al., 2010), Foxo3a and type II TGFβ receptor are specifically upregulated. In addition, a set of genes whose upregulation are functionally important for L-GMPs transformation by MLL-AF9 and MOZ-TIF2 (Krivtsov et al., 2006; Kvinlaug et al., 2011) were either downregulated or unaltered in LICs, including TCF4, p57, HoxA5, HoxA10 and Lmo2 (Figure 6E). These observations suggested a distinct transformation programming resulting from the underlying initiating mutation of L-GMPs and the LICs from Sox4 and K/L leukemias. On the other hand, Mef2C (a cooperating oncogene in leukemogenesis) (Du et al., 2005; Krivtsov et al., 2006) and prostaglandinendoperoxide synthase 1 (Ptgs1) (a key enzyme in Wnt/β-catenin pathway-related prostaglandin synthesis) (Castellone et al., 2005; Goessling et al., 2009) are upregulated in the self-renewal associated signature of both LICs and L-GMPs (Wang et al., 2010). These observations suggested that certain molecular pathways were commonly required for leukemic transformation regardless of the underlying initiating mutation of L-GMPs and LICs.
Gene set enrichment analysis (GSEA) further demonstrated that the unique C/EBPα-Sox4 LIC signature was highly enriched in gene expression profiles from human AML samples with bi-allelic CEBPA mutation or silent CEBPA but not in those of human AML samples with wild-type CEBPA (Figure 6F, S6B), supporting the relevance of the LIC signature identified from murine AML model in human AML.
Targeting SOX4 rescues myeloid differentiation in human CEBP mutant AML
To functional evaluate the relevance of our findings in murine models to the human leukemia, we first determined the ability of leukemogenic human CEBPA mutations to repress Sox4 mRNA expression. We analyzed Sox4 mRNA levels after transducing Cebpa KO LSK cells with viruses encoding either wild-type human CEBPA or a series of mutant proteins identified from human AML patients (Pabst et al., 2001). Interestingly, CEBPA mutant proteins 22N, 22C and 10, which exhibit an increased ratio of 30 kDa to 42 kDa CEBPA and are defective in DNA-binding, failed to repress Sox4, in contrast to wild-type human CEBPA (Figure 7A). Of note, CEBPA mutant 128, which exhibited the wild-type p42 to p30 ratio and maintained the ability to bind efficiently to C/EBPα binding sites, was still capable of repressing Sox4 expression at levels comparable to those of wild-type CEBPA (Figure 7A). In all of these experiments, CEBPA wild-type and mutant constructs were expressed at similar levels (Figure S7A). We next analyzed the effects of individual CEBPA mutant proteins on Sox4 promoter activity using reporter assays. Consistent with the findings of Sox4 mRNA expression analysis (Figure 7A), DNA-binding defective CEBPA mutants (22N, 22C, and 10) failed to repress Sox4 promoter activity (Figure 7B) when expressed at comparable protein levels (Figure S7B). Taken together, these data indicate that dysfunctional CEBPA proteins known to underlay the development of human AML fail to suppress SOX4.
Figure 7. Abnormal expression of SOX4 is correlated with mutations or silencing of CEBPA in human AML.

(A) Expression of Sox4 levels in Cebpa KO LSK cells transduced with empty virus (MIGR1) or viruses expressing wild-type human CEBPA or its mutants identified from AML patients (22N, 22C, 10 and 128). Relative gene expression levels were determined by qPCR as % Gapdh.
(B) HEK293 cells were transfected with murine Sox4 promoter reporter and either empty vector (pCDNA3) or constructs expressing wild-type CEBP (WT) or CEBPA mutants. Luciferase activities were normalized to renilla activities and are presented as percentage of the empty vector.
(C) Primary human AML patient cells #20073 and #15251 were transduced with lentiviruses carrying scrambled shRNA or human SOX4 shRNA and transplanted into NSG mice. 5 weeks later, flow cytometry analysis was performed on the human GFP+ population in the bone marrow of transplanted mice. Differentiated myeloid cells were identified by CD15+.
(D) Morphologically distinct cells were identified by Wright-Giemsa staining of donor-derived GFP+ cells from the bone marrow of recipient mice. Scale bar, 5 μm.
Error bars indicate the mean ± SEM of 3 independent experiments. *: p<0.01; **: p<0.001. See also Figure S7.
To determine if targeting SOX4 could revert human CEBPA bi-allelic mutant AML, we next studied the effect of SOX4 shRNA knockdown in such cells in vivo. Two CEBPA mutant AML patient samples were transduced with a lentivirus carrying a GFP marker and either SOX4 shRNA or a scrambled shRNA (Figure S7C). Transduced cells were then transplanted into sublethally irradiated NSG mice. Flow cytometry of bone marrow cells 5 weeks after transplantation revealed pronounced neutrophilic differentiation as evidenced by CD15 surface marker expression in the GFP+ population of SOX4 shRNA-transduced human patient cells (Figure 7C). Furthermore, cytology of sorted GFP+ human cells (CD45+GFP+) showed the presence of mature neutrophils in SOX4 shRNA-transduced cells only (Figure 7D). Taken together; down-regulation of SOX4 in human CEBP mutant AML samples partially restores granulocytic differentiation in vivo, thus providing strong experimental evidence that enhanced SOX4 is a major contributor to the leukemic phenotype in human leukemia with dysfunctional CEBPA.
Discussion
Sox4 is a central mediator of AML with defective C/EBPα
In this study, we demonstrate that Sox4 contributes to the leukemic phenotype of C/EBPα mutant AML in murine models as well as in human AML. C/EBPα is both a transcription activator and repressor, depending on the target genes and cell context. As a tumor suppressor, C/EBPα modulates cell growth through repression of oncogenes Myc, Mycn, and Bmi1 (Johansen et al., 2001; Ye et al., 2013; Zhang et al., 2004). Here we identified and validated Sox4 as a C/EBPα downstream target.
Previous studies reported that Sox4 is upregulated in L-GMPs from murine MOZ-TIF2, AML1-ETO, and NUP98-HOXA9 AML models in an immediate leukemia initiation signature (Kvinlaug et al., 2011). However, among 16 groups of AML patients classified on the basis of molecular signatures (Valk et al., 2004), SOX4 is only upregulated in AML subtypes carrying bi-allelic mutated and silent CEBPA, or internal tandem duplication mutation of FLT3 (FLT3-ITD). Previous observations that FLT3-ITD blocks C/EBPα through transcriptional repression or posttranslational modification (Radomska et al., 2006; Zheng et al., 2004), and that CEBP bi-allelic mutant AML patients have a lower coincidence of FLT3-ITD mutations (Green et al., 2010), suggest that aberrant levels of SOX4 in FLT3-ITD subtypes is likely due to CEBP deficiency.
Previous studies based on knock-in mice demonstrated that bi-allelic Cebpa mutations induce leukemia by expanding stem/progenitor cells with development of a transformed progenitor pool blocked in myeloid differentiation (Bereshchenko et al., 2009). Here we demonstrate that dysregulated Sox4 contributes to these leukemia phenotypes. Furthermore, the expansion of a highly proliferative premalignant LSK compartment caused by DNA-binding defective Cebpa mutations is alleviated in dKO mice, suggesting that enhanced Sox4 levels are crucial to maintain this transformable pool. The exclusive increase of Sox4 level in C/EBPα mutant LICs also suggests its potential role in LIC function. Unfortunately, complete loss of LIC leukemogenic potential after even a very short period of liquid culture precludes addressing this issue by viral-mediated RNAi knock-down followed by transplantation (Claus Nerlov and Jorg Cammenga, personal communication, and confirmed by us). However, serial-replating assays, a surrogate for self-renewal potential which has been validated with transplantation assays in a number of studies (Huntly et al., 2004; Lavau et al., 1997; Moran-Crusio et al., 2011), suggest a causative role for Sox4 in the acquisition of LIC self-renewal.
Although myelopoiesis in Sox4 KO mice appears normal, Sox4 can inhibit myeloid differentiation in 32DCl3 cells (Boyd et al., 2006) and primary stem/progenitor cells treated with cytokines, consistent with the progressive decrease in Sox4 expression during normal myeloid lineage commitment and terminal differentiation. Our finding that suppression of Sox4 partially restored myeloid differentiation of dKO LSK and human CEBPA mutant AML samples further demonstrates that enhanced Sox4 expression blocks myeloid differentiation in this AML subtype. Collectively, we demonstrate a central role of Sox4 in C/EBPα mutant induced leukemogenesis.
Our findings in murine C/EBPα mutant AML suggest that abrogation of SOX4 may also affect the function of LICs from human CEBPA mutated or silent AML. However, a lack of phenotypic definition of human CEBPA mutated/silent LICs omitted further investigations, since LICs cannot be universally defined for all AML samples on the basis of the CD34+CD38− phenotype (Eppert et al., 2011; Sarry et al., 2011; Taussig et al., 2008; Taussig et al., 2010).
Sox4 and C/EBPα mutant leukemias share a unique gene expression signature
The overlap between gene expression patterns of Sox4 LICs, K/L LICs and Cebpa KO HSCs suggests the existence of a transformation signature specific to Sox4-mediated C/EBPα defective LICs which is distinct from that of L-GMPs from MLL-AF9 and MOZ-TIF2 AML (Krivtsov et al., 2006; Kvinlaug et al., 2011). This suggests that Sox4 and K/L LICs use different transformation pathways from MLL-AF9 and MOZ-TIF2 L-GMPs, a notion further strengthened by very limited overlap between their self-renewal signatures. Intriguingly, gene expression profile comparisons demonstrated increased Foxo3a and type II TGFβ receptor levels only in the unique C/EBPα-Sox4 LIC signature and in human AML samples with mutated and silent CEBPA. Although the exact role of this signaling cascade in AML remains unknown, its relevance to LICs in CML has been demonstrated (Naka et al., 2010). In a BCR-ABL induced CML model, pharmaceutical inhibition of TGFβ-FOXO signaling in combination with a tyrosine kinase inhibitor decreased the colony-forming capacity of LICs and depleted CML in vivo (Naka et al., 2010). It will be of interest to investigate whether the TGFβ-FOXO signaling pathway is specifically activated and required for maintaining Sox4-mediated C/EBPα defective LICs.
Self-renewal associated signatures of both Sox4/C/EBPα LICs and MOZ-TIF2/MLL-F9 L-GMPs share a central enzyme of the prostaglandin E (PGE) biosynthetic pathway, Ptgs1. In addition, our gene expression profiling analysis revealed that not only the synthase for PGE (Ptgs1), but the receptors for PGE (PTGERs) also exhibit increased expression in both LICs and L-GMPs compared to their normal counterparts. This suggests that this pathway or its related regulatory network might serve as a crucial node for leukemic transformation in general. Previous studies have suggested that PGE activated the Wnt signaling pathway by relieving the inhibitory phosphorylation of β-catenin (Castellone et al., 2005; Goessling et al., 2009). Investigating whether the Wnt/β-cateinin pathway is commonly required for leukemogenic transformation regardless of their underlying initiating mutation will be of future interest.
The findings of our mouse model are directly translatable to human AML. SOX4 levels are strongly enhanced in human AML patients carrying mutated or silent CEBPA and a set of self-renewal associated genes that are specifically up-regulated in Sox4 and C/EBPα LICs are also highly enriched in such human AML samples. Furthermore, human AML cells with CEBPA deficiency recapitulate the response of murine cells to Sox4 knock-down and are driven into differentiation. Considering the selective toxicity of Sox4 ablation to leukemic cells but not to normal hematopoietic stem cells (Schilham et al., 1996), it may therefore be a promising target for effective and leukemia subtype-specific therapies.
Experimental Procedures
Serial replating assay
Cells were plated in Methocult M4343 media (StemCell Technologies) and every 10 days, the colony number was counted and subjected to replating.
Mouse strains
Cebpa conditional knockout mice, Sox4loxP/+ heterozygous mice, and K/L mice have been described previously (Bereshchenko et al., 2009; Penzo-Mendez et al., 2007; Zhang et al., 2004). Sox4 conditional knockout mice were generated by breeding Sox4loxP/+ mice to Sox4loxP/loxP homozygous mice and intercross of Sox4loxP/loxP mice with Mx1-Cre mice. Sox4/Cebpa conditional double knockout mice were generated by intercross of the CebpaloxP/loxP mice carrying the Mx1-Cre transgene with Sox4loxP/loxP mice.
Chromatin immunoprecipitation
ChIP assays were performed on stem/progenitor cells (Lin−c-kit+) or myeloid cells (Mac1+Gr1+) as described previously (Alberich-Jorda et al., 2012). Immunoprecipitation was performed with rabbit antibodies against C/EBPα (sc-61X, Santa Cruz) or normal rabbit IgG (12–370, Millipore). Specific regions were quantified by qPCR with SYBR reagent (Bio-Rad).
Bone Marrow Transplantation
1×104 LSKs or 2×105 viral-ransduced cells were transplanted into lethally irradiated congenic B6.SJL-Ptprca mice by retro-orbital injection with 2×105 supporting bone marrow cells from unirradiated B6.SJL-Ptprca mice. For transplantation of cells from leukemic mice, 2×103 cells of fraction I, II, or III were transplanted into sublethally irradiated mice. Mice were monitored by peripheral blood analysis between 2~6 months post-transplantation and were considered moribund when they were severely anemic.
Human AML samples were transduced with lentiviruses for 6 hours and immediately transplanted into sublethally irradiated (150rads) NSG (NOD.Cg-Prkdcscid IL2rgtm1Wjl/szJ) mice. Donor reconstitution of recipient hematopoiesis was analyzed 5 weeks post-transplantation.
Microarray Analysis
Total RNA was purified using Qiagen RNeasy Plus Micro kit and cDNA was amplified by Nugen Ovation® Pico WTA System. cDNA library was labeled and hybridized to Affymetrix Mouse430 2.0 chips. RMA was run for Cel files generated from our lab (GSE45430) or public databases (GSE3725, GSE24797). Integrated data was normalized using the Cross-Correlation method and further log2 transformed. Subtraction of the mean of the means was carried out for each gene for each sample set. Hierarchical clustering was executed to generate the clustering tree. Human data of AML with mutated or silent CEBPA and normal CEBPA were taken from GSE14468 and normalized as above. Gene Set Enrichment Analysis (GSEA) was performed as previously described (Ye et al., 2013).
Statistical Analysis
Survival analysis was performed using the Kaplan-Meier method. Differences between survival distributions were analyzed using the log-rank test. All other statistical analyses were performed using the unpaired Student’s t test. Statistical computations were performed using GraphPad Prism.
Study approval
The animal study was approved by the Institutional Animal Care and Use Committee (Protocol # 201–2011) and the study involving human subjects was approved by the Committee on Clinical Investigations of Beth Israel Deaconess Medical Center. All experiments conform to the relevant regulatory standards. Patients’ informed consent was obtained in accordance with the Declaration of Helsinki.
Accession Numbers
Gene expression data for LICs from Sox4 and C/EBPα mutant leukemic mice and their normal counterparts and ChIP-seq data of genome binding/occupancy profiling of C/EBPα in primary macrophage cells are deposited in the Gene Expression Omnibus (GSE45430 and GSE50565, respectively).
Supplementary Material
Highlights.
C/EBPα represses Sox4 transcription in a DNA-binding dependent manner;
SOX4 is overexpressed in human AML samples with mutated or silent CEBP;
Sox4 mediates leukemic outgrowth due to defective C/EBPα in murine and human models;
LICs in Sox4- or mutated C/EBPα-driven AML models share a gene expression signature.
Significance.
The alteration of the activity of transcription factors crucial for blood cell development with a concomitant deregulation of their target genes is a recurrent pattern in leukemia. However, the precise nature and the role of their downstream targets are still poorly understood. This also holds true for AML with loss of C/EBPα activity. Here, we identified Sox4 as a direct target of transcriptional repression by C/EBPα and functionally validate it as a central mediator of C/EBPα deficiency. Our data provide an explanation for the high levels of SOX4 expression in a human AML subtype carrying mutated or silent CEBPA and highlight SOX4 as a potential therapeutic target for a subtype of AML with currently limited therapeutic options.
Acknowledgments
This study was supported by the National Institutes of Health grant HL 56745 and CA118316 the Harvard Stem Cell Institute grant DP-0086-10-00. GA was supported by Collegio Ghislieri Fellowship. PS was supported by Austrian Research Foundation and European Union. ADR was supported by the Societa’ Italiana di Ematologia Sperimentale borsa di studio “Dr.Tito Bastianello”. EL was supported by FAMRI CIA. VL was supported by National Institutes of Health grant NIH/NIAMS R01 AR54153. We thank all members of the Tenen laboratory for helpful discussions; the Harvard Stem Cell Institute/Beth Israel Deaconess Medical Center Flow Cytometry Core for their expertise; Angie Tan Lay Keng and Lee Ming Hui from the NUS-Duke genomic facility in Singapore for their expertise in microarray analysis; Archibald Perkins, Joseph R. Nevins and Kristian Reckzeh for providing us experiment reagents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberich-Jorda M, Wouters B, Balastik M, Shapiro-Koss C, Zhang H, Di Ruscio A, Radomska HS, Ebralidze AK, Amabile G, Ye M, et al. C/EBPgamma deregulation results in differentiation arrest in acute myeloid leukemia. J Clin Invest. 2012;122:4490–4504. doi: 10.1172/JCI65102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereshchenko O, Mancini E, Moore S, Bilbao D, Mansson R, Luc S, Grover A, Jacobsen SE, Bryder D, Nerlov C. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell. 2009;16:390–400. doi: 10.1016/j.ccr.2009.09.036. [DOI] [PubMed] [Google Scholar]
- Black EP, Hallstrom T, Dressman HK, West M, Nevins JR. Distinctions in the specificity of E2F function revealed by gene expression signatures. Proc Natl Acad Sci U S A. 2005;102:15948–15953. doi: 10.1073/pnas.0504300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd KE, Xiao YY, Fan K, Poholek A, Copeland NG, Jenkins NA, Perkins AS. Sox4 cooperates with Evi1 in AKXD-23 myeloid tumors via transactivation of proviral LTR. Blood. 2006;107:733–741. doi: 10.1182/blood-2003-05-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- Deneault E, Cellot S, Faubert A, Laverdure JP, Frechette M, Chagraoui J, Mayotte N, Sauvageau M, Ting SB, Sauvageau G. A functional screen to identify novel effectors of hematopoietic stem cell activity. Cell. 2009;137:369–379. doi: 10.1016/j.cell.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Spence SE, Jenkins NA, Copeland NG. Cooperating cancer-gene identification through oncogenic-retrovirus-induced insertional mutagenesis. Blood. 2005;106:2498–2505. doi: 10.1182/blood-2004-12-4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombart AF, Hofmann WK, Kawano S, Takeuchi S, Krug U, Kwok SH, Larsen RJ, Asou H, Miller CW, Hoelzer D, Koeffler HP. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood. 2002;99:1332–1340. doi: 10.1182/blood.v99.4.1332. [DOI] [PubMed] [Google Scholar]
- Green CL, Koo KK, Hills RK, Burnett AK, Linch DC, Gale RE. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J Clin Oncol. 2010;28:2739–2747. doi: 10.1200/JCO.2009.26.2501. [DOI] [PubMed] [Google Scholar]
- Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Jafarnejad SM, Ardekani GS, Ghaffari M, Li G. Pleiotropic function of SRY-related HMG box transcription factor 4 in regulation of tumorigenesis. Cell Mol Life Sci. 2012 doi: 10.1007/s00018-012-1187-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR, Tenen DG. c-Myc is a critical target for c/EBPalpha in granulopoiesis. Mol Cell Biol. 2001;21:3789–3806. doi: 10.1128/MCB.21.11.3789-3806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E, Theilgaard-Monch K, Mansson R, Pedersen TA, Pabst T, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13:299–310. doi: 10.1016/j.ccr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Kustikova OS, Geiger H, Li Z, Brugman MH, Chambers SM, Shaw CA, Pike-Overzet K, de Ridder D, Staal FJ, von Keudell G, et al. Retroviral vector insertion sites associated with dominant hematopoietic clones mark “stemness” pathways. Blood. 2007;109:1897–1907. doi: 10.1182/blood-2006-08-044156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara M, Yamashita M, Shinoda K, Tofukuji S, Onodera A, Shinnakasu R, Motohashi S, Hosokawa H, Tumes D, Iwamura C, et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-beta and suppresses T(H)2 differentiation. Nat Immunol. 2012;13:778–786. doi: 10.1038/ni.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvinlaug BT, Chan WI, Bullinger L, Ramaswami M, Sears C, Foster D, Lazic SE, Okabe R, Benner A, Lee BH, et al. Common and overlapping oncogenic pathways contribute to the evolution of acute myeloid leukemias. Cancer Res. 2011;71:4117–4129. doi: 10.1158/0008-5472.CAN-11-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997;16:4226–4237. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FT, MacDougald OA, Diehl AM, Lane MD. A 30-kDa alternative translation product of the CCAAT/enhancer binding protein alpha message: transcriptional activator lacking antimitotic activity. Proc Natl Acad Sci U S A. 1993;90:9606–9610. doi: 10.1073/pnas.90.20.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, Hirao A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676–680. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4:394–400. doi: 10.1038/nrc1363. [DOI] [PubMed] [Google Scholar]
- Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–270. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- Penzo-Mendez A, Dy P, Pallavi B, Lefebvre V. Generation of mice harboring a Sox4 conditional null allele. Genesis. 2007;45:776–780. doi: 10.1002/dvg.20358. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195:709–720. doi: 10.1083/jcb.201102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porse BT, Bryder D, Theilgaard-Monch K, Hasemann MS, Anderson K, Damgaard I, Jacobsen SE, Nerlov C. Loss of C/EBP alpha cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J Exp Med. 2005;202:85–96. doi: 10.1084/jem.20050067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porse BT, Pedersen TA, Xu X, Lindberg B, Wewer UM, Friis-Hansen L, Nerlov C. E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell. 2001;107:247–258. doi: 10.1016/s0092-8674(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Radomska HS, Basseres DS, Zheng R, Zhang P, Dayaram T, Yamamoto Y, Sternberg DW, Lokker N, Giese NA, Bohlander SK, et al. Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med. 2006;203:371–381. doi: 10.1084/jem.20052242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, Swider CR, Strzelecki AC, Cavelier C, Recher C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J Clin Invest. 2011;121:384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilham MW, Oosterwegel MA, Moerer P, Ya J, de Boer PA, van de Wetering M, Verbeek S, Lamers WH, Kruisbeek AM, Cumano A, Clevers H. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature. 1996;380:711–714. doi: 10.1038/380711a0. [DOI] [PubMed] [Google Scholar]
- Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115:1976–1984. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Zhang H, Amabile G, Yang H, Staber PB, Zhang P, Levantini E, Alberich-Jordà M, Zhang J, Kawasaki A, Tenen DG. C/EBPa controls acquisition and maintenance of adult hematopoietic stem cell quiescence. Nature Cell Biology. 2013 doi: 10.1038/ncb2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, Shigematsu H, Levantini E, Huettner CS, Lekstrom-Himes JA, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21:853–863. doi: 10.1016/j.immuni.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Zheng R, Friedman AD, Levis M, Li L, Weir EG, Small D. Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPalpha expression. Blood. 2004;103:1883–1890. doi: 10.1182/blood-2003-06-1978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.