

Abstract
Objective
To assess cerebrospinal fluid (CSF) β-site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1) activity in relation to Alzheimer’s disease (AD) and to correlate the enzyme activity with protein markers of APP metabolism and axonal degeneration.
Methods
BACE1 activity and protein concentrations were measured and analysed in 342 participants of the AD Neuroimaging Initiative, including 99 normal controls, 75 stable mild cognitive impairment (MCI), 87 progressive MCI and 79 AD dementia cases. All statistical analyses were Bonferroni corrected for multiple comparisons.
Results
No significant differences between controls and any of the three patient groups were detected for BACE1 activity and soluble APP (sAPP)β concentrations in CSF. Significant correlations with BACE1 activity were found for CSF APPβ and total tau in all four groups; and for CSF phosphorylated tau181 in all groups but the progressive MCI group. There were no correlations for CSF amyloid β (Aβ)1-42 nor for plasma Aβ1-42 and Aβ1-40.
Conclusions
The consistent correlation between BACE1 activity and sAPPβ supports their role as biomarkers of target engagement in clinical trials on BACE1 inhibition.
Keywords: Biomarker, dementia, mild cognitive impairment, diagnosis, prognosis, amyloid precursor protein, amyloid beta, tau, correlation
1. Introduction
Alzheimer’s disease (AD) in neuropsychiatric tradition is a clinical diagnosis characterised by an amnestic type of progressive dementia and the exclusion of alternative causes. These simple clinical criteria are neither sufficiently sensitive for early changes nor specific enough for AD; major efforts of academia and the pharmaceutical industry to identify prodromal AD, i.e. the stage before full-blown dementia develops, and to treat pathophysiological processes rather than their end products are driving forward the search for potential biomarkers capable of identifying neurodegeneration independently of its clinical manifestations. Individuals with asymptomatic early AD would probably benefit most from interventions aiming to prevent further neural damage in order to maintain their independence, ability to work and fulfilment of social roles. Furthermore, pathophysiological markers may also offer the added benefit of directly assessing response to treatment options that target core processes of AD pathogenesis. The application of novel therapeutics with potentially significant side effects could thereby be restricted to patients with biological evidence of treatment response in line with the notion of personalised medicine.
A principal problem with current biomarkers is their insensitivity to initial, or upstream, pathophysiological events, which limits their value in identifying pre-clinical or early clinical AD and to monitor treatment response to novel compounds targeting the cerebral accumulation of amyloid β (Aβ). One feasible approach to improve the diagnostic and prognostic performance is to measure upstream events of amyloid precursor protein (APP) processing, which are at the core of AD pathogenesis according to the prevalent school of thought (1). The β-site APP-cleaving enzyme 1 (BACE1) is responsible for the first and rate-limiting APP processing step (2) and is therefore a major target in biomarker research. Previous studies indicate the suitability of BACE1 activity as diagnostic and prognostic marker in AD (3, 4). However, the evidence basis is still inconclusive, and partly contradictory, which warrants replication and validation of previous findings in a multi-centric setting involving sufficient numbers of patients in different disease stages and matched healthy controls. The aim of the present study was therefore to explore BACE1 activity differences between patients and controls and to characterise the stage-dependent correlations between BACE1 activity and protein biomarker concentrations in the AD Neuroimaging Initiative (ADNI). Multi-centre studies are an important part of the biomarker validation process since they provide important advantages over single-centre studies, such as larger sample size and the recruitment of participants from a wider population assuring a more representative sample of the target population making it easier to generalise the findings of the study (5). In addition to the general benefits of a large multi-centre study, the ADNI offers the added benefit of uniform laboratory assessments, which help to overcome issues with differences across different immunoassay platforms and measurements in different laboratories (6).
2. Materials and Methods
2.1 Study design and sample
The data used in this study were obtained from the ADNI database at www.loni.ucla.edu/ADNI on 29 April 2013. Information from 402 samples with BACE1 activity measurements and CSF concentrations of its main cleavage product soluble APP (sAPP)β were available, including 106 elderly normal controls (NL), 92 patients with stable mild cognitive impairment (sMCI), 92 patients who had progressed to AD dementia during the follow-up period (pMCI), 92 patients with AD dementia and 20 technical replications (repeated measurements for quality control purposes, not included in the analyses). Fourty-two participants were excluded because of missing biomarker data, resulting in a final dataset of 342 individuals including 99 NL, 75 sMCI, 87 pMCI and 79 AD. The study was approved by the institutional review boards of all participating centres and written informed consent was obtained from all participants or authorised representatives after extensive description of the ADNI according to the 1975 Declaration of Helsinki. The study is registered at www.ClinicalTrials.gov (identifier, NCT00106899). BACE1 activity and sAPPβ concentration in CSF were measured simultaneously, using aliquots obtained from the same vial at the same thaw using analytically validated assays and according to published protocols (7, 8). More information on the ADNI including CSF sampling and analysis is provided in the supplementary material.
2.2 Statistical analysis
Data were analysed in IBM SPSS, v21. Normal distribution was checked using the Kolmogorov–Smirnov test; non-parametric comparisons between groups were performed using the Kruskal-Wallis test, followed by the Mann-Whitney test since some of the biomarker data were skewed. The correlations between CSF BACE1 activity and other variables of interest including biomarker concentrations, age, gender and APOE (binarised as carriers vs non-carriers) were assessed using Spearman rank correlation coefficients. The correlations were assessed separately for each of the four diagnostic groups. Bonferroni correction (separately for the group comparisons and for each of the group-wise correlation analyses) was applied with α = 0.05 in order to minimise the likelihood of false positive findings due to multiple testing. All tests were two-sided.
3. Results
All reported p values are after Bonferroni correction. As expected, in contrast to the NL group, CSF Aβ1-42 concentrations were decreased in all three patient groups (sMCI, p=0.05; pMCI, p<0.001; AD, p<0.001), CSF tTau was increased in all three patient groups (sMCI, p<0.001; pMCI, p<0.001; AD, p<0.001) and pTau181 was increased in the pMCI and AD groups (sMCI, p=0.07; pMCI, p<0.001; AD, p<0.001); all three patient groups showed lower MMSE scores than the NL group (sMCI, p<0.001; pMCI, p<0.001; AD, p<0.001). The pMCI group had lower CSF Aβ1-42 and higher pTau181 concentrations than the sMCI group (both p<0.01). No other significant biomarker differences were detected between the NL group and the four patient groups as well as between the sMCI and the pMCI groups. The distribution of the APOE ε4 allele followed the previously reported pattern, with 70 % carriers in the AD group and only 25 % carriers in the NL group (Table 1).
Table 1.
Characteristics of the study population
| Variable | NL group | sMCI group | pMCI group | AD group |
|---|---|---|---|---|
| N | 99 | 75 | 87 | 79 |
| Age, years | 76 (28) | 74 (33) | 74 (34) | 77 (32) |
| MMSE, points | 29 (5) | 28 (6)* | 27 (6)* | 24 (7)* |
| Men : women | 50 : 49 | 52 : 23 | 56 : 33 | 42 : 38 |
| APOE ε4, %carriers | 24.24 | 45.33 | 59.77 | 69.62 |
| CSF BACE1, pM | 42 (80) | 48 (85) | 45 (64) | 43 (71) |
| CSF Aβ1-42, ng/L | 222 (225) | 178 (211)* | 141 (272)* | 138 (213)* |
| CSF tTau, ng/L | 61 (152) | 72 (226)* | 93 (301)* | 115 (328)* |
| CSF pTau181, ng/L | 21 (71) | 25 (62) | 37 (70)* | 36 (105)* |
| CSF sAPPβ, pM | 3964 (6439) | 3510 (7384) | 4260 (7781) | 3695 (5608) |
| Plasma Aβ1-42, pg/mL | 150 (228) | 161 (323) | 155.80 (238.30) | 158 (250) |
| Plasma Aβ1-40, pg/mL | 37 (74) | 37 (56) | 35 (52) | 39 (55) |
Data presented as median (range) where appropriate.
significant difference compared to the NL group at α = 5% (Bonferroni corrected).
NL: normal controls; sMCI: stable mild cognitive impairment; pMCI: progressive mild cognitive impairment; AD: Alzheimer’s disease; MMSE: Mini-Mental-State Examination; APOE: Apolipoprotein E; CSF: cerebrospinal fluid; BACE1: beta-site amyloid precursor protein cleaving enzyme 1; tTau: total-Tau; pTau181: phosphorylated Tau181; sAPPβ: soluble amyloid precursor protein β; Aβ1-40: amyloid β1-40; Aβ1-42: amyloid β1-42.
Significant correlations with BACE1 activity in all four study groups were found for APPβ (NL, r=0.30, p=0.02; sMCI, r=0.37, p=0.01; pMCI, r=0.33, p=0.02; AD, r=0.33, p=0.02) and tTau (NL, r=0.57, p<0.001; sMCI, r=0.56, p<0.001; pMCI, r=0.31, p=0.04; AD, r=0.44, p<0.001). BACE1 activity was also significantly correlated with pTau181 in all groups with the exception of the pMCI group (NL, r=0.32, p=0.02; sMCI, r=0.40, p<0.01; pMCI, r=0.11, p=0.31; AD, r=0.40, p<0.01) (Figure 1). There were no correlations with BACE1 activity in any of the four study groups for CSF Aβ1-42, plasma Aβ1-40 and Aβ1-42, age, gender or APOE (r range, -0.10 to 0.24; p>0.17).
Figure 1.
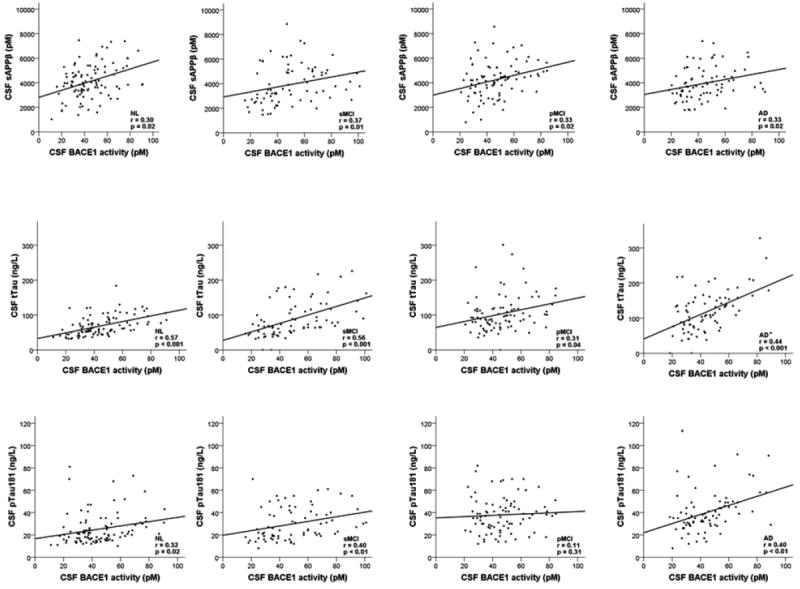
Scatterplots showing the correlations between BACE1 activity and the concentrations of cerebrospinal fluid proteins (rows) in the different study groups (columns)
NL: normal controls; sMCI: stable mild cognitive impairment; pMCI: progressive mild cognitive impairment; AD: Alzheimer’s disease; BACE1: beta-site amyloid precursor protein cleaving enzyme 1; sAPPβ: soluble amyloid precursor protein β; tTau: total-Tau; pTau181: phosphorylated Tau181.
4. Discussion
The findings of this multicentre study confirm and extend some earlier results, while they contradict others. We did not find any CSF BACE1 activity differences between the control group and any of the patient groups. This aspect of our research is in line with one study (6), but in contrast to other previous studies with partly contradictory findings, showing increased BACE1 activity in MCI but not AD (7, 8); increased activity in both MCI and AD (9); or even decreased activity in AD (5). Part of the discrepancy may be explained by the different properties of the applied laboratory assays, the characteristics of the study samples and the definitions of patient groups, but the wide range of BACE1 activity measurements and the large overlap between the groups may also have a significant impact.
Some earlier studies found increased sAPPβ CSF levels in AD vs controls (11, 12) and stable vs progressive MCI (13). Other published reports do not support these results (3, 9, 14, 15), which is in line with the findings of the present study. Our negative findings in relation to the influence of demographic and genetic factors on BACE1 activity confirm previous reports on age (3), gender (3, 4, 10) and APOE (3). However, increased BACE1 activity has also been shown in relation to older age (8) and the APOE ε4 allele (10) before.
We show that BACE1 activity positively correlates with sAPPβ, tTau and pTau181 in CSF across the spectrum from physiological ageing to clinically diagnosable AD (the lacking correlation with pTau181 in pMCI is probably a spurious finding). On the other hand, we also show that BACE1 activity is not associated with Aβ1-42 in CSF nor with Aβ1-42 and Aβ1-40 in blood. Our findings confirm the consistent correlation of BACE1 activity with markers of upstream events of APP metabolism and markers of neurodegeneration (3, 16, 17). The absence of an association between BACE1 activity and CSF Aβ1-42 underlines the notion that CSF levels of Aβ1-42 most likely reflect its deposition in senile plaques, which is determined by decreased clearance from brain rather than increased production in sporadic AD (18); this may also explain the missing correlation between central BACE1 and peripheral Aβ levels.
The large sample size, the recruitment at multiple sites and the availability of plasma markers are the advantages of our study compared to previous efforts in this field. Testing the relationship of the Aβ burden measured by biomarkers in different biological compartments is needed in order to characterise the complex dynamic balance between blood and CSF biomarkers (19). The usual limitations of clinical cohorts recruited at specialised centres apply, including the lack of histopathological verification of the clinical diagnoses and the limited generalisability of the findings to the population of interest. To sum up, two key conclusions emerge from our study and the literature review. Firstly, BACE1 activity and sAPPβ concentration changes in CSF due to AD do not seem to follow a consistent pattern, which limits their utility as diagnostic markers. Encouraging results in blood (11, 20) need replication and validation before further conclusions can be drawn. Secondly, correlations between BACE1 activity and upstream markers of APP cleavage and axonal degeneration are highly consistent. Even though correlations are moderate in most studies, including the present report, these markers may be candidates for target engagement measures in on-going and future trials of BACE1 inhibitors (21).
Supplementary Material
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organisation is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514. The sponsors did not have any role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript. The corresponding author had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors wish to thank Dr Dorottya Ruisz for proofreading.
Footnotes
Author contributions: Study concept and design: Perneczky, Alexopoulos. Analysis and interpretation of data: Perneczky, Alexopoulos. Drafting of the manuscript: Perneczky. Critical revision of the manuscript for important intellectual content: Perneczky, Alexopoulos. Statistical analysis: Perneczky. Administrative, technical, and material support: Perneczky. Study supervision: Perneczky.
There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Vassar R. BACE1: the beta-secretase enzyme in Alzheimer’s disease. J Mol Neurosci. 2004;23(1-2):105–14. doi: 10.1385/JMN:23:1-2:105. [DOI] [PubMed] [Google Scholar]
- 3.Zetterberg H, Andreasson U, Hansson O, Wu G, Sankaranarayanan S, Andersson ME, et al. Elevated cerebrospinal fluid BACE1 activity in incipient Alzheimer disease. Arch Neurol. 2008;65(8):1102–7. doi: 10.1001/archneur.65.8.1102. [DOI] [PubMed] [Google Scholar]
- 4.Zhong Z, Ewers M, Teipel S, Burger K, Wallin A, Blennow K, et al. Levels of beta-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch gen psychiatry. 2007;64(6):718–26. doi: 10.1001/archpsyc.64.6.718. [DOI] [PubMed] [Google Scholar]
- 5.U S Food and Drug Administration draft guidance for industry, clinical investigators, and Food and Drug Administration staff – design considerations for pivotal clinical investigations for medical devices. Rockwell, MD: Food and Drug Administration; 2011. [Google Scholar]
- 6.Kang JH, Korecka M, Toledo JB, Trojanowski JQ, Shaw LM. Clinical utility and analytical challenges in measurement of cerebrospinal fluid amyloid-beta(1-42) and tau proteins as Alzheimer disease biomarkers. Clin chem. 2013;59(6):903–16. doi: 10.1373/clinchem.2013.202937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu G, Sankaranarayanan S, Hsieh SH, Simon AJ, Savage MJ. Decrease in brain soluble amyloid precursor protein beta (sAPPbeta) in Alzheimer’s disease cortex. J neurosci res. 2011;89(6):822–32. doi: 10.1002/jnr.22618. [DOI] [PubMed] [Google Scholar]
- 8.Wu G, Sankaranarayanan S, Tugusheva K, Kahana J, Seabrook G, Shi XP, et al. Decrease in age-adjusted cerebrospinal fluid beta-secretase activity in Alzheimer’s subjects. Clin biochem. 2008;41(12):986–96. doi: 10.1016/j.clinbiochem.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 9.Rosen C, Andreasson U, Mattsson N, Marcusson J, Minthon L, Andreasen N, et al. Cerebrospinal fluid profiles of amyloid beta-related biomarkers in Alzheimer’s disease. Neuromolecular med. 2012;14(1):65–73. doi: 10.1007/s12017-012-8171-4. [DOI] [PubMed] [Google Scholar]
- 10.Ewers M, Zhong Z, Burger K, Wallin A, Blennow K, Teipel SJ, et al. Increased CSF-BACE 1 activity is associated with ApoE-epsilon 4 genotype in subjects with mild cognitive impairment and Alzheimer’s disease. Brain. 2008;131(Pt 5):1252–8. doi: 10.1093/brain/awn034. [DOI] [PubMed] [Google Scholar]
- 11.Wu G, Sankaranarayanan S, Wong J, Tugusheva K, Michener MS, Shi X, et al. Characterization of plasma beta-secretase (BACE1) activity and soluble amyloid precursor proteins as potential biomarkers for Alzheimer’s disease. J neurosci res. 2012;90(12):2247–58. doi: 10.1002/jnr.23122. [DOI] [PubMed] [Google Scholar]
- 12.Lewczuk P, Popp J, Lelental N, Kolsch H, Maier W, Kornhuber J, et al. Cerebrospinal fluid soluble amyloid-beta protein precursor as a potential novel biomarkers of Alzheimer’s disease. J Alzheimers dis. 2012;28(1):119–25. doi: 10.3233/JAD-2011-110857. [DOI] [PubMed] [Google Scholar]
- 13.Perneczky R, Tsolakidou A, Arnold A, Diehl-Schmid J, Grimmer T, Forstl H, et al. CSF soluble amyloid precursor proteins in the diagnosis of incipient Alzheimer disease. Neurology. 2011;77(1):35–8. doi: 10.1212/WNL.0b013e318221ad47. [DOI] [PubMed] [Google Scholar]
- 14.Brinkmalm G, Brinkmalm A, Bourgeois P, Persson R, Hansson O, Portelius E, et al. Soluble amyloid precursor protein alpha and beta in CSF in Alzheimer’s disease. Brain res. 2013;1513:117–26. doi: 10.1016/j.brainres.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 15.Olsson A, Hoglund K, Sjogren M, Andreasen N, Minthon L, Lannfelt L, et al. Measurement of alpha- and beta-secretase cleaved amyloid precursor protein in cerebrospinal fluid from Alzheimer patients. Exp neurol. 2003;183(1):74–80. doi: 10.1016/s0014-4886(03)00027-x. [DOI] [PubMed] [Google Scholar]
- 16.Tsolakidou A, Alexopoulos P, Guo LH, Grimmer T, Westerteicher C, Kratzer M, et al. Beta-Site amyloid precursor protein-cleaving enzyme 1 activity is related to cerebrospinal fluid concentrations of sortilin-related receptor with A-type repeats, soluble amyloid precursor protein, and tau. Alzheimers dement. 2013;9(4):386–91. doi: 10.1016/j.jalz.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 17.Mulder SD, van der Flier WM, Verheijen JH, Mulder C, Scheltens P, Blankenstein MA, et al. BACE1 activity in cerebrospinal fluid and its relation to markers of AD pathology. J Alzheimers dis. 2010;20(1):253–60. doi: 10.3233/JAD-2010-1367. [DOI] [PubMed] [Google Scholar]
- 18.Grimmer T, Faust M, Auer F, Alexopoulos P, Forstl H, Henriksen G, et al. White matter hyperintensities predict amyloid increase in Alzheimer’s disease. Neurobiol aging. 2012;33(12):2766–73. doi: 10.1016/j.neurobiolaging.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 19.Figurski MJ, Waligorska T, Toledo J, Vanderstichele H, Korecka M, Lee VM, et al. Improved protocol for measurement of plasma beta-amyloid in longitudinal evaluation of Alzheimer’s Disease Neuroimaging Initiative study patients. Alzheimers dement. 2012;8(4):250–60. doi: 10.1016/j.jalz.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perneczky R, Guo LH, Kagerbauer SM, Werle L, Kurz A, Martin J, et al. Soluble amyloid precursor protein beta as blood-based biomarker of Alzheimer’s disease. Transl psychiatry. 2013;3:e227. doi: 10.1038/tp.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullard A. Sting of Alzheimer’s failures offset by upcoming prevention trials. Nat rev drug discov. 2012;11(9):657–60. doi: 10.1038/nrd3842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.