

Abstract
Purpose
Current cancer chemotherapy is gradually shifting to the application of drug combinations that prevent development of drug resistance. Many anticancer drugs have poor solubility and limited oral bioavailability. Using an innovative approach, we developed dual-drug nanoformulations of a polymeric nanogel conjugate with anticancer 5-FU nucleoside analog, floxuridine (FLOX), and the second anticancer drugs, paclitaxel (PCL), or a geldanamycin analog,17-AAG, for combination therapy.
Methods
PCL or17-AAG had been encapsulated in the cholesteryl-polyvinyl alcohol-floxuridine nanogel (CPVA-FLOX) by simple solution mixing and sonication. Dual nanodrugs formed particles with diameter 180 nm and either drug content (5–20%) that were stable and could be administered orally. Their cytotoxicity in human and mouse cancer cells was determined by MTT assay, and cellular target inhibition – by Western blot analysis. Tumor growth inhibition was evaluated using an orthotopic mouse mammary 4T1 cancer model.
Results
CPVA-FLOX was more potent than free drug in cancer models including drug-resistant ones; while dual nanodrugs demonstrated a significant synergy(CPVA-FLOX/PCL), or showed no significant synergy (CPVA-FLOX/17-AAG) compared to free drugs (PCL or 17-AAG). Dual nanodrug CPVA-FLOX/17-AAG effect on its cellular target (HSP70) was similar to 17-AAG alone. In animal model, however, both dual nanodrugs effectively inhibited tumor growth compared to CPVA-FLOX after oral administration.
Conclusion
Oral dual-drug nanoformulations of poorly-soluble drugs proved to be a highly efficient combination anticancer therapy in preclinical studies.
Keywords: dual-drug nanoformulations, oral delivery, 17-AAG, paclitaxel, floxuridine
INTRODUCTION
Cancer cells develop drug resistance after prolonged and repeated exposure to many anticancer drugs (1). This is a major clinical setback of chemotherapy resulting in tumor relapse. As a drug resistance-preventive strategy, combinations of drugs that include different mechanisms of action have been evaluated, such as gemcitabine and cisplatin for treatment of non-small cell lung carcinoma (2), or capecitabine and docetaxel for treatment of breast cancer (3). Paclitaxel (PCL) is a widely prescribed drug with activity against various cellular pathways (4). Previous studies have shown that PCL can be combined with other drugs such as gemcitabine, capecitabine, and fludarabine due to its non-overlapping toxicity with nucleoside analogs as DNA and microtubule damaging agents (5, 6). Considerable interest has been recently shown to the development of new anticancer drug 17-AAG, a promising heat shock protein 90 (HSP90) inhibitor (7). Prior research of 17-AAG demonstrated that it can induce functional loss of client proteins and sensitize tumor cells to nucleoside analogs that are active in the S-phase during replication (8, 9). Both PCL and 17-AAG alone are poorly soluble in water and usually ineffective as oral drugs. Nanoencapsulation can increase drug bioavailability and targeted delivery into tumors. Rational design of drug delivery systems is currently one of the most rapidly developing areas of cancer therapy. Drug nanoformulations which are able to deliver and release drugs in a controlled manner in tumor microenvironment can significantly increase the efficacy of chemotherapy while reducing nonspecific toxicities and side effects (10).
We previously demonstrated that nanogel-drug conjugates (nanodrugs) of floxuridine and gemcitabine are efficient against regular and drug-resistant tumors (11, 12). The nanodrugs containing nucleoside analogs attached via the tetraphosphate linker to CPVA demonstrated strong tumor growth inhibition activity even against drug-resistant cancers. The free drugs can be released in an active phosphorylated form from nanodrugs inside cancer cells over extended time period, by passing the critical nucleoside activation step. The aim of the current study was to apply these nanodrugs for encapsulation and delivery of poorly soluble anticancer drugs in order to obtain more efficient dual-drug nanoformulations, even for oral administration. Drugs such as PCL and 17-AAG have never been used for oral administration due to their low bioavailability and instability in gastrointestinal (GI) tract. Here, we demonstrated the preparation and superior activity of oral dual nanodrugs over the mixture of two drugs in preclinical cancer models.
MATERIALS AND METHODS
Materials
Chemical reagents, solvents, and polymers were purchased from Sigma-Aldrich (St. Louis, MO) and Alfa Aesar (Wardhill, MA) with the highest available purity and used without purification unless otherwisestated.5-Fluro-2'-deoxyuridine (Floxuridine, FLOX) was obtained from SynQuest Laboratories (Alachua, FL).17-(allylamino)-17-demethoxygeldanamycin (17-AAG) was obtained from ChemieTek (Indianapolis, IN). Centrifuge filter devices (MWCO 3500 Da) were purchased from Millipore (Bedford, MA). CPVA-FLOX conjugate was synthesizedas earlier described(11).
Cells
Human breast carcinoma,MDA-MB-231 and BT-474,were maintained in alpha-modified Minimum Essential Medium (α-MEM) supplemented with 5% fetal bovine serum (FBS),1% L-glutamine, and 1% penicillin-streptomycin. MIAPaCa-2 and Capan-1cells were a kind gift from Surinder Batra, UNMC; the cells were maintained in DMEM high glucose medium supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin-streptomycin. 4T1 mouse mammary carcinoma cells were a kind gift from Joseph Vetro (UNMC). The cells were cultured in RPMI 1640 supplemented with 2 mML-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 1% penicillin-streptomycin, and 10% FBS. All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Nanoformulation
The CPVA-FLOX conjugate used for nanoformulation contained PVA with M.w. 31,000 and the FLOX content 0.45 μmol/mg(11). CPVA-FLOX(5.0 mg) was mixed with 0.5 mg 17-AAG (10% by weight) or 0.25 mg paclitaxel (5% by weight)in 0.5 mL DMSO. This solution was added drop wise to 50 mLPBS and sonicated for 5 min (Branson Sonifier 200). Drug loading in CPVA-FLOX/17-AAG was determined by UV absorbance at 320 nm (ε 19,300) for 17-AAG and at 260 nm (ε 7,000) for FLOX. Paclitaxel (PCL) content in CPVA-FLOX/PCL was determined by reverse-phase HPLC analysis using an Ascentis-C18 column (10 μm, 15cm x 4.6mm) at flow rate of 1 mL/min. The elution was performed with buffer A: 5% acetonitrile/water; and buffer B: 95% acetonitrile/water in a linear gradient mode (100% B in 20 min), using detection at 223 nm.
The hydrodynamic diameter and polydispersity of nanogels and dual nanodrugs was measured by dynamic light scattering using a Zetasizer Nano-ZS90 (Malvern, Worcestershire, UK). All samples were sonicated to prepare uniform dispersions and centrifuged for 4 min at 10,000 rpm before measurements. Size distribution of the samples was characterized by the polydispersity index. Zeta-potential was calculated on the base of electrophoretic mobility at 25°C using the Zetasizer software. The data reported in Table 1 represents an average of three measurements.
Table I.
Physicochemical characteristics of dual nanodrugs
| Nanodrugs* | dh, nm | PDI | ζ, mV |
|---|---|---|---|
| CPVA | 35 ± 1.3 | 0.36 ± 0.01 | 0 ± 3.7 |
| CPVA-FLOX | 42 ± 6.4 | 0.40 ± 0.04 | −8.5 ± 3.6 |
| CPVA-FLOX/17-AAG | 169±2.1 | 0.32 ± 0.04 | −17.9 ± 4.3 |
| CPVA-FLOX/PCL | 189± 6.3 | 0.45 ± 0.02 | −18 ± 3.4 |
Particle size (dh), polydispersity index (PDI), and zeta-potential (ζ) were measured in 1% aqueous solutions after sonication (30 min). The results are means ± SD of three measurements.
In vitro drug release
In vitro drug release was studied in simulated gastric fluid (SGF) and in physiological conditions (PBS). Briefly, SGF was prepared by dissolving 3.2 g/L of pepsin (800–2500 U/mg) from porcine stomach mucosa in 34 mM NaCl and 84 mM HCl adjusted to final pH 1.2. CPVA-FLOX/17-AAG and PCL formulations (10 mg) were placed in dialysis tubes (MWCO 3500) contained 0.5 mL of SGF or PBS and incubated against the same solutions at 37°C. Samples (50 μL) withdrawn from the tubes at selected time points have been analyzed by UV absorbance for FLOX (260 nm, ε 7,000) and 17-AAG (320 nm, ε 19,300) for in triplicate (Spectramax M2 spectrophotometer, Molecular devices, Sunnyvale, CA). PCL release was analyzed by reverse-phase HPLC using and Ascentis-C18 column (10 μm, 15cm x 4.6mm) at flow rate of 1 mL/min. The elution was performed with buffer A: 5% acetonitrile/water; and buffer B: 95% acetonitrile/water in a linear gradient mode (100% B in 20 min), using detection at 223 nm.
Cell viability assay
Cytotoxicity of dual-drug nanoformulations was analyzed in various breast and pancreatic cancer cell lines by MTT assay. Briefly, BT-474,MDA-MB-231, 4T1, Capan-1 and MIA PaCa-2 cells were seeded in 96-well plates at a density of 3,000 cells/200μL growth medium per well. Cells were allowed to attach overnight and serial dilutions of drugs were added. Solutions of PCL and 17-AAG were prepared with Cremaphor®EL as solubilizer. PCL-containing samples were incubated in full medium for 3 days (MIA PaCa-2 and Capan-1 cells), and 17-AAG samples for 7 days (BT-474, MDA-MB-231 and 4T1 cells) at 37°C. Metabolic mitochondrial activity was determined by adding 20 μL of a 5 mg/mL MTT solution in 100 μL of Phenol red-free DMEM medium. The samples were then incubated for 4 h at 37°C, and 100 μL of extraction buffer (20% SDS in DMF/water, 1:1, pH 4.7) was added to each well. Samples were incubated for 24 h at 37°C. Optical absorbance was measured at 560 nm using a Model 680 microplate reader (BioRad, Hercules, CA), and cytotoxicity was expressed as a percentage of survived cells compared to a non-treated control. All samples were analyzed by an average of eight measurements (means ± SEM). Percentage of viable cells was plotted against the log of the drug concentrations, and drug concentrations resulting in 50% cellular viability (IC50 values) have been calculated using a trapezoid rule as averages of two independent cellular experiments.
Western blot
Treated cells or tumor tissues were lysed with ice-cold lysis buffer containing 50 mM Tris-HCl (pH7.5), 150mM sodium chloride, 10% SDS, 1% Triton X-100, and 1mM phenylmethanesulfonylfluoride. BCA protein assay kit (Pierce, Rockford, IL) was used to measure protein concentration. Samples with equal amounts of total protein were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred onto poly(vinylidenedifluoride) membrane. The membrane was blocked with 1% bovine serum albumin in TBS-T buffer (10mM Tris-HCl pH 8.0, 150mM sodium chloride, 0.05% Tween-20, 0.02% sodium azide) for 2 h at 25°C. The membrane was then incubated with an optimal concentration of antibody in TBS-T buffer overnight at 4°C. The next day, the membrane was washed 3 times with TBS-T buffer and incubated with horse radish peroxidase-conjugated protein (1:20,000 dilution in TBS-T buffer) for 2 hours at 25°C. Protein signal was measured using Pierce ECL Western Blotting Substrate.
Tumor growth inhibition
Female BALB/c mice (6-8 weeks old, 20–25 g) were obtained from the Charles River Laboratories (Wilmington, MA). The mice were housed five per cage in a sterile and temperature-controlled facility, receiving food and water ad libitum. This research was conducted under a protocol approved by the University of Nebraska Medical Center Animal Care and Use Committee. All manipulations with animals were performed using sterile solutions. To induce tumors, mice were injected subcutaneously in the right flank mammary fat pads with 1 × 106 early-passage 4T1 cells re-suspended in 400 μL of serum-free medium containing 20% Matrigel (Becton-Dickinson, San Diego, CA). Approximately one week after injection, when tumors could be palpitated, mice were randomly divided into treatment and control groups (n = 5-6). Solutions of CPVA-FLOX, CPVA-FLOX/PCL and CPVA-FLOX/17-AAG in 2% sucrose were administered via oral gavage (50 μL) twice a week at doses of 16 mg/kg (FLOX), 8mg/kg (PCL), and 16mg/kg (17-AAG), respectively. Tumor size was measured using electronic calipers, and the volumes were calculated according using the formula: , where L and W are length and width of tumor in millimeters. Experiments were terminated when TV reached 15% of mouse weight, or when mice became moribund.
Statistical analysis
Tumor growth inhibition results between control and treatment animal groups were compared on selected days by a two-tailed Student’s t-test. Differences in tumor volumes are considered statistically significant when p < 0.05.
RESULTS
Characterization of dual nanodrugs
Dual-drug formulations could be easily prepared by mixing DMSO solutions of hydrophobic drugs (17-AAG or PCL) with aqueous solution of CPVA-FLOX at ultrasonication. Small nanogel particles with a hydrophobic core consisting of cholesterol and the second drug molecules form during this process (Fig. 1). Anionic chains of CPVA-FLOX in the hydrophilic outer layer of dual nanodrugs prevent aggregation and precipitation of encapsulated hydrophobic drugs. We found that dual nano-drugs formed stable dispersions with a hydrodynamic diameter of 179±10 nm in aqueous media. This method was capable of attaining dual nanodrugs with FLOX content of 0.4 μmol/mg, 17-AAG content of 0.16 μmol/mg, and PCL content of 0.08 μmol/mg. The CPVA-FLOX/17-AAG and CPVA-FLOX/PCL demonstrated at least 400 times higher solubility of hydrophobic drugs in aqueous media and better solubility compared to Cremaphor®EL.
Figure 1.

Formation of nanogels (CPVA-FLOX) and dual nanodrugs (CPVA-FLOX/17-AAG or PCL), and chemical structures of all dual nanodrug components: CPVA-FLOX, 17-AAG and PCL.
The average size of CPVA-FLOX was 42 nm with narrow polydispersity index (PDI), while dual nanodrugs formed particles with 4 times higher diameter and the PDI in the range of 0.25-0.4 (Table 1). The size and morphology of dual nanodrugs was confirmed by the transmission electron microscopy (TEM) (Fig. 2). In the TEM results, formation of a compact hydrophobic core inside nanoformulations following the loading of the second drug can be observed after negative staining.
Figure 2.
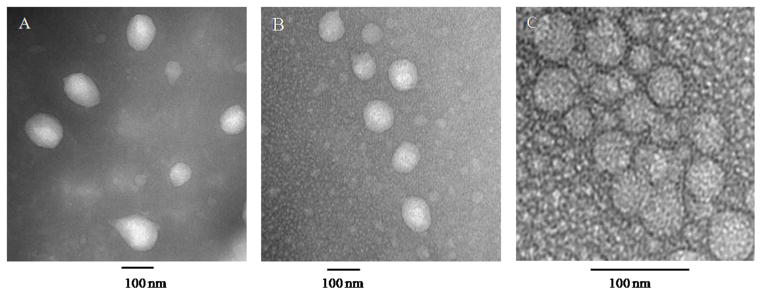
Transmission electron microscopy (TEM) images of (A) CPVA-FLOX/PCL, (B) CPVA-FLOX/17-AAG dual nanodrugs, and (C) CPVA-FLOX nanogel-drug conjugate. Samples were sonicated for 30 min before vanadate staining. The bar is 100 nm.
Overall charge (zeta-potential) of all nanoformulations was negative, and the inclusion of hydrophobic drugs resulted only in slight increase of zeta-potential, evidently due to the higher surface area of dual nanodrugs. Storage stability of dual nanodrugs was determined by monitoring size and zeta potential of dispersions. When a sufficient amount of charged polymer chains was located on the surface, hydrophobic drug molecules in the core were protected from aggregation. Stability of dual nanodrugs evidently depended on the second drug structure. For example, 17-AAG content in stable nanoformulation can be as high as 15-20%, while the PCL content usually was 2-3 times lower. Heavy loaded CPVA-FLOX/17-AAG formulations remained stable for 6 weeks loosing less than 10% of the second drug. CPVA-FLOX/PCL nanoformulations remained stable for 3-7 days with the loss of the second drug less than 10%. At 37°C, the stability remained high; for example, CPVA-FLOX/17-AAG was stable for 3 weeks. Stable dual-drug formulations with optimized drug contents (10% 17-AAG and 5% PCL) have been used in all subsequent experiments.
In vitro drug release
Stability of the dual nanodrug CPVA-FLOX/17-AAG and CPVA-FLOX/PCL in gastric conditions was monitored at 37°C in simulated gastric fluid. The maximum transit time in stomach is generally 5 h. We observed a very slow drug release (< 1 % after 6 h) for all three drugs in these conditions (Fig. 3A, B, C). FLOX release at pH 7.4 was also slow (Fig. 3D). We do not expect any significant drug loss during the dual nanodrug delivery to the tumor site. Previously, we demonstrated sustained release of nanogel-FLOX conjugate at both pHs (11). Both dual nanoformulations were stable enough in gastric conditions to be used for oral administration. The second drug (FLOX) release is slower than the first drug release. Thus, when nanodrugs interact with cellular membrane, they are incorporated and allowed the second drug penetrate into cytoplasm by previously suggested mechanism (27).
Figure 3.

In vitro drug release from the dual nanodrug CPVA-FLOX/17-AAG and CPVA-FLOX/PCL. (A) 17-AAG, and (B) PCL (C) FLOX release in simulated gastric fluid (pH 1.2). (D) FLOX release from CPVA-FLOX nanogel-drug conjugate at physiological condition (pH 7.4). Shown data are means ± SD (n = 3).
Cytotoxicity studies
Cytotoxicity of CPVA-FLOX, dual nanodrugs CPVA-FLOX/PCL and CPVA-FLOX/17-AAG, dual-drug mixtures FLOX+PCL and FLOX+17-AAG, and individual drugs was compared in human breast carcinoma MDA-MB-231 and BT-474 cells, human pancreatic adenocarcinoma MIA PaCa-2 and Capan-1 cells, and murine mammary carcinoma 4T1cells using a MTT cell proliferation assay. The cell viability was assessed in MTT assay based on the activity of mitochondrial dehydrogenases, converting the MTT dye into a blue-colored formazan precipitatedin living cells. Dead cells or cells with damaged mitochondria do not develop the blue staining. Both CPVA-FLOX/17-AAG and 17-AAG demonstrated an equal activity with IC50 0.03±0.01 μM in BT-474 cells and 0.35±0.1μM in MDA-MB-231 cells (Fig. 4A, B). CPVA-FLOX/PCL exhibited a 3-10-fold enhanced cytotoxicity in MIA PaCa-2 and Capan-1 cells compared to PCL alone, e.g. IC50 0.35±0.07nM versus 1.2±0.18nM in MIA PaCa-2 cells and 0.45±0.04nM versus 4.5±0.14nM in Capan-1 cells (Fig. 4E, F). Cytotoxicity of CPVA-FLOX/17-AAG and CPVA-FLOX/PCL after incubation with aggressive metastatic 4T1 cells was practically the same as for Cremaphor®EL-stabilized drug solutions. CPVA-FLOX nanogel-drug conjugate alone showed lower activity than dual nanodrugs in all investigated cell lines.
Figure 4.

Cytotoxicity of dual nanodrugs, CPVA-FLOX/17AAG (10% 17-AAG) and CPVA-FLOX/PCL (5% PCL). Cancer cell lines: (A) BT-474, (B)MDA-MB-231, (C-D) 4T1, (E) MIA PaCa-2, and (F) Capan-1. Nanoformulations were compared with dual-drug mixtures at the same molar ratio (17-AAG:FLOX,1: 2.3; PCL:FLOX,1: 6.8). Cell viability was measured after 7-daysincubation (17-AAG) or 3-days incubation (PCL). Data are means ± SD(n = 8).
Effect on the HSP70 expression
Biological effect of 17-AAG was studied in high level ErbB2-expressing BT-474 cells and low level ErbB2-expressing MDA-MB-231 and 4T1 cells. ErbB2 is a member of an epidermal growth factor receptor (EGFR) family that is expressed in most human breast cancers. Stability of ErbB2 that is important for tumor development is supported by the HSP90 molecular chaperone complex that plays an important role in the maturation and re-folding of certain signaling proteins in cells exposed to environmental stress (13, 14). 17-AAG can interfere with its function and inhibit tumor growth through its strong binding to an ATP/ADP binding pocket of HSP90 and induction of the HSP70, which leads to degradation of ErbB2 (15). The effect of dual nanodrugs on the HSP70 expression in BT-474 and 4T1 cell lines was measured using a Western blot technique. CPVA-FLOX/17-AAG induced the same level of HSP70 protein in BT-474 cells as the treatment with 17-AAG (Fig. 5A, C). 4T1 cells treated with CPVA-FLOX/17-AAG revealed the 1.75-2.50-fold higher HSP70 levels compared to 17-AAG-treated cells (Fig. 5A,C). Complete loss of ErbB2 in ErbB2-overexpressing BT-474 cells was observed within only 8-24 hours of the treatment by CPVA-FLOX/17-AAG.
Figure 5.
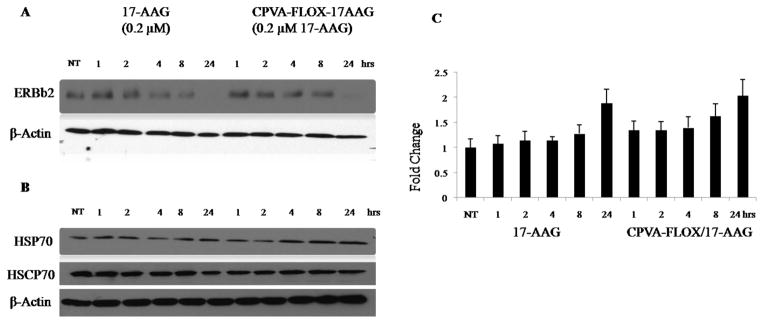
Effect of the CPVA-FLOX/17-AAG nanoformulation on: (A) down-regulation of ErbB2 protein, and (B) HSP70 expression in BT-474 cells compared to free 17-AAG dissolved in 2% DMSO. Cell lysates were analyzed by Western blot. HSC70 protein and β-actin served as internal controls. (C) Increase in HSP70 levels compared to non-treated BT-474 cells calculated by Western blot densitometry. Shown data are means ± SEM (n = 3), NT: non-treated cells.
In vivo tumor growth inhibition
The therapeutic effect of dual nanodrugs was evaluated in an orthotopic 4T1 mammary carcinoma model that produced fast growing and aggressive tumors. Theses specific tumors spontaneously metastasize from the primary tumor site to multiple distant sites (including lymph nodes, blood, liver, lung, brain, and bone) in approximately 6 weeks and represent a good breast cancer model (16). Tumor cells were transplanted subcutaneously into mammary fat pads of female BALB/c mice to initiate tumors in anatomically correct locations. The animals were randomly separated into treatment groups (n=6), and aqueous solutions of saline (control group), CPVA-FLOX, CPVA-FLOX/17-AAG, and CPVA-FLOX/PCL were administered by oral gavage using doses of 2 and 4 mg nanodrug per mouse (Fig. 7). At the dose 4 mg per mouse, the individual drug doses were equivalent to 16 mg/kg (FLOX and 17-AAG), and 8 mg/kg (PCL). The oral gavage was performed twice a week for four consecutive weeks. These doses are well-tolerated at oral administration as we previously determined (12). 17-AAG and PCL alone were not administered as controls because these drugs are not compatible with oral administration. In this experiment, we compared the anticancer effect of single and dual nanodrugs including exclusively intravenous and low soluble drugs (17-AAG and PCL) in order to evaluate the advantages of combination therapy.
Figure 7.
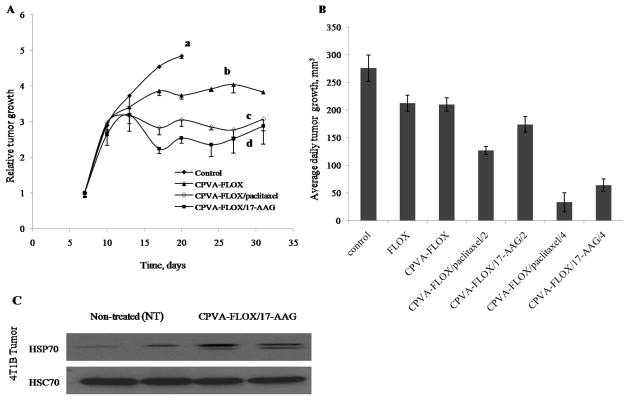
Tumor growth inhibition in mouse mammary carcinoma 4T1 model following the oral administration (gavage) of CPVA-FLOX nanogel or dual nanodrugs. (A) Relative tumor growth in BALB/c mice inoculated with 4T1 cells expressed as a ratio of tumor volume at any given time (V) to the tumor volume at the beginning of treatment (Vo). The treatment started on Day 7 after inoculation. (a) Control group: 2% sucrose, (b) CPVA-FLOX (dose: 16 mg/kg FLOX), (c) CPVA-FLOX/PCL (dose: 8mg/kg PCL, 16 mg/kg FLOX), and (d) CPVA-FLOX/17-AAG (dose: 16mg/kg 17-AAG, 16 mg/kg FLOX). (B) Average daily tumor growth calculated for the period of treatment between Days 10 and 17. The differences between CPVA-FLOX/PCL or CPVA-FLOX/17AAG-treated mice and control group were found significant (P<0.05). (C) HSP70 expression in 4T1 tumors after the oral treatment by CPVA-FLOX/17-AAG in the end of experiment on Day 32. Tumor cell lysates were analyzed by western blot. HSC70 protein served as internal control.
Relative tumor volume (V/Vo) was used in this study to measure the therapeutic effect of nanodrugs, because the initial tumor volumes (Vo) showed significant variability between mice. Tumor growth inhibition curves were obtained by plotting mean relative tumor volumes as a function of time for each experimental group. We observed a complete tumor growth inhibition at an early stage after the treatment by dual nanodrugs, while only a moderate inhibition was observed for CPVA-FLOX at later stage (Fig. 7A). Compared to control group, the inhibitory effect of dual nano drugs was lower at the dose of 2 mg per mouse. The differences in tumor growth between the control and CPVA-FLOX/17-AAG and CPVA-FLOX/PCL groups were statistically significant (P<0.05) from Day 17 (Fig. 7A). 4T1 tumors reached volumes corresponding to 15% of animal weight in the control group within 2-3 weeks. Mice treated with CPVA-FLOX showed tumor growth up to 17 days, and thereafter, the effect was cytostatic and tumor volume remained unchanged for more than 30 days. A similar effect was observed for both dual nanodrugs, but the tumor volume remained lower during the first 10-12 days of treatment and was at least 25-30% smaller in the end of the experiment compared to the group treated by the CPVA-FLOX alone. These data suggest that PCL or 17-AAG release from nanogels is faster than the release of FLOX. Our results well correlate with in vitro drug release data we previously observed (11). The effect of 17-AAG or PCL was predominant during the first several days, when the drug concentration was sufficient enough to inhibit the tumor growth. Thereafter, the cytostatic effect on tumor growth was evidently maintained due to the continuous release of FLOX over a longer period of time.
To analyze the dosage effect on tumor growth rate, we plotted an average daily increase in tumor volume between Days 10 and 17 for each group (Fig. 7B). Animals treated with the doses of 4 mg/mouse for CPVA-FLOX/PCL (8 mg/kg PCL) or CPVA/17-AAG (16 mg/kg 17-AAG) showed significantly slower tumor growth, respectively 12% and 24% compared to control group. At lower doses of 2 mg/mouse, the growth inhibition by dual nanodrugs expressed the same trend, respectively 46% and 63% compared to control group. However, the effect at the dose of 2 mg/mouse was not statistically significant compared to CPVA-FLOX or free FLOX, while it was statistically significant at the dose of 4 mg/mouse. The FLOX alone reduced the tumor growth rate only by 24%, indicating that the drug is not effective enough to stop the aggressive 4T1 tumors. Previous observations confirm this conclusion, e.g. FLOX failed to inhibit 4T1tumor growth at the dose of 31mg/kg but helped to significantly reduce secondary bone metastases (17). As shown in Fig.7A, during the first 5 days after the beginning of treatment, the tumor volumes in all groups grew at the same rate, evidently until the release of PCL or 17-AAG strongly affected it, even decreasing tumor volumes. Later, the tumor volumes showed no significant changes during the treatment (cytostatic effect). In the tumor sections analyzed by Western blot, we observed a significant induction of HSP70 as compared to the control group (Fig.7C). As described above, the HSP70 induction is a measure of therapeutic activity of 17-AAG. This test confirms the in vivo targeted activity of dual nanodrugs following the oral administration. Earlier, we demonstrated an efficient GI transport of CPVA-FLOX nano-drugs using an in vitro Caco-2 model (12). Evidently, CPVA-FLOX-based dual nanodrugs are transported from the GI tract into the blood and, consequently, accumulated in tumor cells due to the enhanced permeability and retention effect. Our animal experiments also demonstrated that the PCL is 50-100% more efficient than 17-AAG in nanodrugs (Fig. 7B).
DISCUSSION
Several drug delivery systems such as liposomes and polymeric micelles have been recently explored for simultaneous delivery of two drugs, including 17-AAG and PCL (18, 19). However, many of them could not achieve therapeutic drug concentrations inside tumors because of the rapid drug loss in blood circulation (20, 21). Treatment of solid tumors with nanocarriers requires efficient retention and penetration in tumor tissues, which may be problematic for relatively large particles such as liposomes (22). To bypass some of these problems, we recently reported synthesis and application of novel nanogel-drug conjugates (11, 12, 23). These nanodrugs could deliver activated phosphorylated nucleoside analogs, releasing drugs in a controlled manner over an extended time period (11, 12). Nanogel-drug conjugates showed high efficacy against drug-resistant tumors. Another advantage of nanodrugs was their compatibility with oral route of administration. As nanogel-drug conjugates form small hydrophilic particles with a hydrophobic cholesterol core at sonication, we proposed them for encapsulation of poorly soluble hydrophobic drugs. This approach can potentially be a safer and more effective alternative to many combination treatments by excluding toxic solvents or solubilizers used currently to solubilize low-soluble drugs and extending the drug release period during cancer chemotherapy.
Cholesteryl-PVA drug conjugates form small nanogels after sonication due to the stacking of cholesterol moieties in the core of a coiled PVA molecule. When a solution of hydrophobic drug in DMSO is mixed with aqueous solution of CPVA-FLOX at sonication, the formation of larger dual-drug nanoformulations that encapsulate the poorly soluble drug in the core can be observed. The obtained dual nanodrugs fit to the critical size range between 50 and 300 nm that is optimal for longer circulation and slower clearance from the blood(24). PVA-based nanocarriers are highly hydrophilic, which makes them similar to PEGylated nanocarriers by biodistribution after intravenous administration (25). Long-term circulation in the blood is one of the most important factors that enhance drug accumulation in the tumor site. Narrow size distribution is another positive factor leading to better distribution of nano drugs in vivo and in tumor volume.
Dual-drug formulations can carry high content of the incorporated insoluble drug (10-20% for 17-AAG and 5-10% for PCL) and release therapeutic levels of these drugs after accumulation in cancer cells. As we discovered, the CPVA-FLOX/17-AAG is more stable than CPVA-FLOX/PCL in aqueous media. It might be the cause of the observed higher efficacy of PCL vs. 17-AAG nanodrugs in bioassays. Faster PCL release dramatically increased cytotoxicity during the first three days period as seen in our in vitro cytotoxicity data. In contrast, an equal or less pronounced effect was observed for CPVA-FLOX/17-AAG compared to free 17-AAG, even at the extended 7 day period of the assay. PCL is an antitumor drug that binds to microtubules and prevents their normal functions, resulting in the suppression of cell division and induction of apoptosis. It was confirmed in several clinical studies that anticancer nucleoside analogs such as gemcitabine and 5-flurouracil can be used with PCL for the treatment of non-small cell lung, breast, and ovarian cancers due to their potency as single agents and non-overlapping activities (26). No synergistic effect of dual-drug mixtures other than an additive effect was observed in these studies (5,6). On the contrary, we observed a clearly synergistic effect resulting in 8-10 times higher cytotoxicity of CPVA-FLOX/PCL formulation compared to the identical mixture of FLOX and PCL in pancreatic cancer cell lines. Until now, only few studies have been performed on the combination effect of 17-AAG and nucleoside analogs (8, 9). They illustrated only a limited combination activity because the therapeutic levels of both drugs existed only for a short period of time in the body (8). We detected no synergistic effect in cytotoxicity studies after the treatment with CPVA-FLOX/17-AAG. However, in this research, a remarkable tumor growth inhibition of very aggressive breast cancer was achieved after oral administration of CPVA-FLOX/17-AAG. The stability in gastric conditions and drug release kinetics of nanodrugs is critically important factor in oral administration. The CPVA-FLOX/17-AAG nanoformulation was very stable in gastric conditions releasing only 0.4-0.6% of free drug after 6 h-incubation. This time is generally considered as a maximum transit time in stomach. Once a nanodrug is released from stomach and absorbed in the small intestine, it will circulate in the blood stream at physiological conditions. The second drug release is evidently faster compared to the conjugated FLOX over an extended period of time. Our results demonstrate the importance of the second drug release kinetics on the anticancer effect of dual nanodrugs after accumulation in tumors. Tailoring dual nanodrugs would allow controlled and sustained maintaining of therapeutic drug levels in the body at minimal frequency of drug administration. Our observations extended to other tumor models and different drug combinations can serve as a basis for optimal design of novel types of combination therapies.
CONCLUSIONS
We have made evident here that poorly soluble anticancer drugs can be encapsulated in polymeric nanogel-drug conjugates forming novel dual nanodrugs that significantly enhanced therapeutic outcome of cancer treatment. CPVA-FLOX/PCL and CPVA-FLOX/17-AAG dual nanodrugs revealed a higher activity against pancreatic cancer cells than dual drug mixtures. They allowed oral administration of PCL and 17-AAG, even though these drugs have been never used as oral drugs, and demonstrated a significant tumor growth inhibition in aggressive tumor animal model. Thus, the development of dual nanodrugs can be a promising nanomedicine strategy for increasing therapeutic activity and reducing undesirable side effects of combination chemotherapy.
Figure 6.
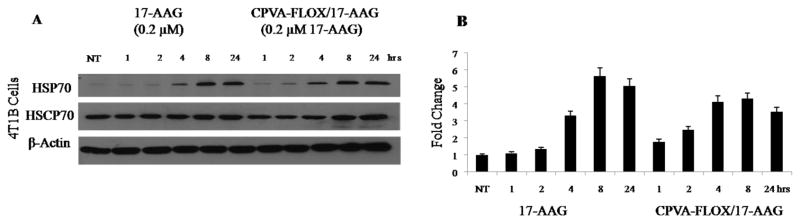
Comparative effect of the CPVA-FLOX/17-AAG nanoformulation and free 17-AAG (2% DMSO solution) on the HSP70 expression in 4T1 cells.(A) Cell lysates analyzed by Western blot using HSC70 protein and β-actin as internal controls. (B) Normalized HSP70 levels obtained by Western blot densitometry. Shown data are means ± SEM (n = 3). NT: non-treated cells.
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under R01 award numbersCA136921 (for S.V.V.),CA116552, CA99163, CA87986 and CA105489 (for H.B.) and DODW81XWH-11-1-0167 (for H.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.M.R. acknowledges support from Nebraska Department of Health and Human Services and Nebraska Center for Nanomedicine-Center for Biomedical Research Excellence (NCN-COBRE seed grant).The authors thank Tom Bargar for assistance with transmission electron microscopy (UNMC Core), and Xin Wei for valuable help with some cell cultures.
References
- 1.Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002;3(7):415–24. doi: 10.1016/s1470-2045(02)00788-x. [DOI] [PubMed] [Google Scholar]
- 2.Abratt RP, Sandler A, Crino L, Steward WP, Shepherd FA, Green MR, et al. Combined cisplatin and gemcitabine for non-small cell lung cancer: influence of scheduling on toxicity and drug delivery. Semin Oncol. 1998;25(4 Suppl 9):35–43. [PubMed] [Google Scholar]
- 3.Fujimoto-Ouchi K, Tanaka Y, Tominaga T. Schedule dependency of antitumor activity in combination therapy with capecitabine/5'-deoxy-5-fluorouridine and docetaxel in breast cancer models. Clin Cancer Res. 2001;7(4):1079–86. [PubMed] [Google Scholar]
- 4.Rowinsky EK, Donehower RC. Paclitaxel (taxol) The New England journal of medicine. 1995;332(15):1004–14. doi: 10.1056/NEJM199504133321507. [DOI] [PubMed] [Google Scholar]
- 5.Shord SS, Faucette SR, Gillenwater HH, Pescatore SL, Hawke RL, Socinski MA, et al. Gemcitabine pharmacokinetics and interaction with paclitaxel in patients with advanced non-small-cell lung cancer. Cancer Chemother Pharmacol. 2003;51(4):328–36. doi: 10.1007/s00280-002-0560-1. [DOI] [PubMed] [Google Scholar]
- 6.Sawada N, Ishikawa T, Fukase Y, Nishida M, Yoshikubo T, Ishitsuka H. Induction of thymidine phosphorylase activity and enhancement of capecitabine efficacy by taxol/taxotere in human cancer xenografts. Clin Cancer Res. 1998;4(4):1013–9. [PubMed] [Google Scholar]
- 7.Neckers L, Neckers K. Heat-shock protein 90 inhibitors as novel cancer chemotherapeutic agents. Expert Opin Emerg Drugs. 2002;7(2):277–88. doi: 10.1517/14728214.7.2.277. [DOI] [PubMed] [Google Scholar]
- 8.Kaufmann SH, Karp JE, Litzow MR, Mesa RA, Hogan W, Steensma DP, et al. Phase I and pharmacological study of cytarabine and tanespimycin in relapsed and refractory acute leukemia. Haematologica. 2011;96(11):1619–26. doi: 10.3324/haematol.2011.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mesa RA, Loegering D, Powell HL, Flatten K, Arlander SJ, Dai NT, et al. Heat shock protein 90 inhibition sensitizes acute myelogenous leukemia cells to cytarabine. Blood. 2005;106(1):318–27. doi: 10.1182/blood-2004-09-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61(13):1203–13. doi: 10.1016/j.addr.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Senanayake TH, Warren G, Vinogradov SV. Novel anticancer polymeric conjugates of activated nucleoside analogues. Bioconjug Chem. 2011;22(10):1983–93. doi: 10.1021/bc200173e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senanayake TH, Warren G, Wei X, Vinogradov SV. Application of activated nucleoside analogs for the treatment of drug-resistant tumors by oral delivery of nanogel-drug conjugates. J Control Release. 2013;167(2):200–9. doi: 10.1016/j.jconrel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yonehara M, Minami Y, Kawata Y, Nagai J, Yahara I. Heat-induced chaperone activity of HSP90. J Biol Chem. 1996;271(5):2641–5. doi: 10.1074/jbc.271.5.2641. [DOI] [PubMed] [Google Scholar]
- 14.Schneider C, Sepp-Lorenzino L, Nimmesgern E, Ouerfelli O, Danishefsky S, Rosen N, et al. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc Natl Acad Sci U S A. 1996;93(25):14536–41. doi: 10.1073/pnas.93.25.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raja SM, Clubb RJ, Bhattacharyya M, Dimri M, Cheng H, Pan W, et al. A combination of Trastuzumab and 17-AAG induces enhanced ubiquitinylation and lysosomal pathway-dependent ErbB2 degradation and cytotoxicity in ErbB2-overexpressing breast cancer cells. Cancer Biol Ther. 2008;7(10):1630–40. doi: 10.4161/cbt.7.10.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast cancer research : BCR. 2000;2(5):331–4. doi: 10.1186/bcr77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiraga T, Hata K, Ikeda F, Kitagaki J, Fujimoto-Ouchi K, Tanaka Y, Yoneda T. Preferential inhibition of bone metastases by 5'-deoxy-5-fluorouridine and capecitabine in the 4T1/luc mouse breast cancer model. Oncol Rep. 2005;14(3):695–699. [PubMed] [Google Scholar]
- 18.Xiong MP, Yanez JA, Kwon GS, Davies NM, Forrest ML. A cremophor-free formulation for tanespimycin (17-AAG) using PEO-b-PDLLA micelles: characterization and pharmacokinetics in rats. J Pharm Sci. 2009;98(4):1577–86. doi: 10.1002/jps.21509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasenstein JR, Shin HC, Kasmerchak K, Buehler D, Kwon GS, Kozak KR. Antitumor activity of Triolimus: a novel multidrug-loaded micelle containing Paclitaxel, Rapamycin, and 17-AAG. Mol Cancer Ther. 2012;11(10):2233–42. doi: 10.1158/1535-7163.MCT-11-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H, Kim S, He W, Wang H, Low PS, Park K, et al. Fast release of lipophilic agents from circulating PEG-PDLLA micelles revealed by in vivo forster resonance energy transfer imaging. Langmuir. 2008;24(10):5213–7. doi: 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]
- 21.Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, et al. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436(7050):568–72. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 22.Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci U S A. 2001;98(8):4628–33. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei X, Senanayake TH, Warren G, Vinogradov SV. Hyaluronic acid-based nanogel-drug conjugates with enhanced anticancer activity designed for the targeting of CD44-positive and drug-resistant tumors. Bioconjug Chem. 2013;24(4):658–68. doi: 10.1021/bc300632w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318. [PubMed] [Google Scholar]
- 25.Yamaoka T, Tabata Y, Ikada Y. Comparison of body distribution of poly(vinyl alcohol) with other water-soluble polymers after intravenous administration. J Pharm Pharmacol. 1995;47(6):479–86. doi: 10.1111/j.2042-7158.1995.tb05835.x. [DOI] [PubMed] [Google Scholar]
- 26.Abratt RP, Bezwoda WR, Goedhals L, Hacking DJ. Weekly gemcitabine with monthly cisplatin: effective chemotherapy for advanced non-small-cell lung cancer. J Clin Oncol. 1997;15(2):744–9. doi: 10.1200/JCO.1997.15.2.744. [DOI] [PubMed] [Google Scholar]
- 27.Vinogradov SV, Kohli E, Zeman AD. Cross-linked polymeric Nanogel formulations of 5’-triphosphates of nucleoside analogs: role of the cellular membrane in drug release. Mol Pharm. 2005;2:449–61. doi: 10.1021/mp0500364. [DOI] [PMC free article] [PubMed] [Google Scholar]