

Abstract
C-, N-, P-, and S-nucleophiles reacted with symmetrical tris(2,3,5,6-tetrathiaaryl)methyl cations, generated from the corresponding triarylmethanols by strong acids, to give a variety of asymmetrical monosubstituted persistent triaryl-methyl (TAM) radicals as the major products. The only byproducts were symmetrical TAMs.
Keywords: Carbocations, Radicals, Electron transfer, Nucleophilic addition, Reaction mechanisms
Introduction
Persistent triarylmethyl radicals[1] (trityls, TAMs, see Figure 1) have recently been proposed as ideal spin probes for numerous applications in biology,[2–5] analytical chemistry,[6] and materials science.[7] Trityls are stable towards the majority of biological oxidants and reductants. In addition, they have long relaxation times in liquid solutions and a particularly narrow EPR singlet line, which make them especially useful for in vivo 3D EPR oxygen imaging,[8] studies of dynamic nuclear polarization,[9] site-directed spin labeling of proteins, and the measurement of nanometer distances by means of pulsed EPR dipolar spectroscopy.[10]
Figure 1.

Molecular structures of typical TAMs.
The growing demand for trityls has sparked numerous searches for the efficient synthesis of these intricate substances, resulting, inter alia, in practical and convenient strategies for the large-scale preparation of the most extensively employed and simplest representative of persistent TAMs, that is, tris(8-carboxy-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl)methyl (also known as Finland trityl, see Figure 1).[11,12]
A different situation arises with asymmetrical monofunctionalized derivatives of TAMs. Typically, these compounds can be obtained in low yields from highly challenging multistep procedures requiring, as a rule, the laborious purification of products by preparative HPLC.[6,13] These methods are not sufficiently versatile to fit the variety of chemical scenarios and would prove difficult to adapt to the large-scale synthesis of functional derivatives of TAMs. Therefore it is not surprising that most of the trityls synthesized so far have a symmetrical framework composed of three identical aryl groups.
Thus, it would seem that a direct entry to diversely monofunctionalized TAMs through a simple and unified synthetic strategy could notably facilitate their wider availability and broaden the scope of trityl applications. Recently, we observed new reactions of nucleophiles with tris(2,3,5,6-tetrathiaaryl)methyl cations, generated from the corresponding triarylmethanols in the presence of strong acids.[12] Nucleophilic quenching afforded the trityl radicals 4a,b as the products of formal one-electron reduction of carbocations 2a,b (see Scheme 1 and Scheme 2). With water, the only byproducts were diamagnetic quinone methides 6a,b. In the presence of diethylamine, the asymmetrical monosubstituted trityl radical 8 was obtained as the major product, which could be isolated in 42–82% yield (see Scheme 2) depending on the reaction conditions.
Scheme 1.

Quenching of carbocations 2a,b with water. Proposed rationale for the formation of symmetrical TAMs 4a,b and quinone methides 6a,b through a two-step ET reaction.[12]
Scheme 2.

Reaction of carbocation 2a with diethylamine. Concurrent generation of trityl 4a and asymmetrical monofunctionalized trityl 8.[12]
These encouraging results prompted us to extend this approach to a broader series of nucleophiles. Thus, in this paper we report that cations 2a,b readily react with C-, P-, S-, and N-centered nucleophiles to yield new asymmetrical TAM derivatives in which the nucleophile is inserted into the para position of one of three aryl rings. This new formal aromatic nucleophilic substitution reaction clearly has potential for the preparative synthesis of a wide variety of new, diversely substituted, persistent triarylmethyl radicals.
Results and Discussion
Schemes 1 and 2[14] show the reactions of cations 2a,b with nucleophiles to give the 4-methylenecyclohexa-2,5-diene intermediates 3a,b (see Scheme 1) or 7 (see Scheme 2). When water was used as nucleophile, the one-electron oxidation of intermediates 3a,b with cations 2a,b afforded the trityls 4a,b (with a concomitant decarboxylation proceeding in the particular case of cyclohexadiene 3b) and hypothetical transient trityls 5a,b. These intermediate species were oxidized again with the eventual formation of diamagnetic quinoids 6a,b and symmetrical trityls 4a,b. The trityls 4a,b and quinone methides 6a,b were obtained in a ratio close to 2:1, in good agreement with the prediction in Scheme 1.
However, with diethylamine as the nucleophile, the reaction gave two paramagnetic products, that is, trityl 4a and monosubstituted trityl 8 (see Scheme 2). The proportion of these TAMs strongly depends on both the trityl cation 2a/ diethylamine ratio and the order of reagent mixing. If a small excess of diethylamine was added to a homogeneous solution of 2a in dichloromethane (DCM), trityls 4a and 8 were isolated in yields of 47 and 42%, respectively, close to the 1:1 ratio expected for the fast oxidation of hypothetical intermediate 7 (k1[HNEt2] ≪ k2[2a]).[12]
When a solution of cation 2a was added slowly to a large excess of diethylamine, trityl 8 was obtained in 82% isolated yield. Most likely the large excess of the nucleophile meets the condition for the preferential formation of the intermediate cyclohexadiene 7 (k1[HNEt2] ≫ k2[2a]) and, hence, strong suppression of the reaction pathway giving the symmetrical TAM 4a.[12] Further work-up, in which the resulting reaction mixture was exposed to air, afforded trityl 8 as the major product, apparently derived from the oxidation of intermediate 7 by atmospheric oxygen.
These observations led to the assumption that an efficient protocol for the synthesis of monosubstituted TAMs must satisfy the following conditions: 1) A large excess of nucleophile in the reaction with the trityl cation,[15] 2) the use of anhydrous solvents and carefully dried reagents to minimize the concentration of water and thus avoid the unwanted reaction pathway leading to quinoids 6a,b,[16] 3) exposure of the crude product to air in the final work-up to oxidize the intermediate cyclohexadienes to monosubstituted TAMs, and 4) the prevention of adverse reaction channels, such as the addition of the counter ion Z− to the trityl cation (see Scheme 2) with further oxidative transformations of the corresponding cyclohexadiene.
Of the four conditions listed above, little is known of the potential participation of counter ions in the nucleophilic quenching of their positively charged “native” cations. To study the reactivity of these anions, we turned to a model reaction of diethylamine with carbocation 2a, generated by strong acids under various conditions.
When trifluoromethanesulfonic acid or an ethereal solution of HBF4 was used in small excess (1.2–1.4 equiv. per alcohol 1a in DCM solution), the resulting cation gave two paramagnetic products derived from the reaction of 2a with diethylamine (4 equiv.): TAMs 4a and 8.[17] They were obtained in yields of 48 and 44%, respectively, close to a 1:1 ratio (see the Exp. Sect.). No side-products were observed. This suggests that CF3SO3− and BF4− anions do not act as nucleophiles in the cation generation step and do not compete with diethylamine in the nucleophilic quenching of the carbocation.
A different situation arose when neat TFA or DCM/TFA solutions were used as acidic reagents for cation generation. We found that the products of the reaction of cation 2a with diethylamine depended upon the method of preparation of the cation solution. Thus, a neutral solution of cation 2a in DCM, prepared by the complete removal of excess TFA, slowly reacted over 96 h with diethylamine (4 equiv.) to give the trityl 4a and quinone methide 6a after standard work-up. These products were obtained in a ratio of 1:1.02, with yields of 45 and 46%, respectively (see the Exp. Sect.).[18] The monofunctionalized trityl 8 was not observed at all.
This may be explained by the reaction of cation 2a with the trifluoroacetate anion to yield the intermediate cyclohexadiene 9 (see Scheme 3). The oxidation of 9 by cation 2a followed by acid-catalyzed hydrolysis of the resulting trityl 10 affords the transient trityl 5a with parallel formation of unsubstituted symmetrical trityl 4a. The trityl 5a is oxidized further by atmospheric oxygen to quinone methide 6a during work-up when the reaction mixture was exposed to air for the first time. This proposed mechanism predicts the trityl/quinoid ratio to be 1:1, in good agreement with the experimental result.
Scheme 3.

Proposed mechanism for the reaction of carbocation 2a with the CF3CO2− counter anion in the absence of acids: The formation of trityl 4a and quinone methide 6a.
On the other hand, cation 2a, prepared by the dissolution of 1 molequiv. of CF3SO3H and alcohol 1a in neat TFA with subsequent evaporation of the highly volatile TFA, smoothly reacted with diethylamine to afford trityls 4a (isolated yield 47%) and 8 (isolated yield 42%) as the only products (see the Exp. Sect.). The quinone methide 6a was not detected.
To rationalize the observed effect, one could assume that evaporation of TFA in the presence of poorly volatile trifluoromethanesulfonic acid should result in a very efficient substitution of the trifluoroacetate counter ion by the less nucleophilic CF3SO3− anion. In addition, one may expect substantial inhibition of the nucleophilicity of trifluoroacetate anions by residual superacid.
These data suggest the potential ability of the CF3CO2− anion to act as an “internal” nucleophile in acid-free solutions of trityl carbocations. When neat TFA is used both as a solvent and strongly acidic reagent for the generation of trityl cations, this special feature must be taken into account. The precaution is especially important in the design of syntheses of monofunctionalized derivatives of Finland trityl 4b by the reaction of nucleophiles with the carbocation 2b. Seemingly, the latter could be generated by the treatment of a DCM solution of triarylmethanol 1b with CF3SO3H or HBF4, similarly to the generation of cation 2a. Unfortunately, the insolubility of 1b in low-polar solvents (e.g., DCM, chloroform) obstructs the application of this easy and attractive method. In contrast, homogeneous solutions of both carbocations 2a and 2b can be generated by using highly polar, neat TFA or a 1:1 mixture of DCM and TFA.[12] This means that TFA may be used, when the occasion requires, with the understanding that the involvement of CF3CO2− anions in nucleophilic quenching must be minimized.
With these observations in mind we examined a broader set of nucleophiles. A series of C-centered anions was studied first (see Scheme 4). To avoid the formation of undesirable 3,6-dimethylidenecyclohexa-1,4-dienes 15 (the carbon analogues of quinone methide 6a) and to simplify the course of the reaction, we first turned to the easily obtainable α-monosubstituted derivative of malonic ester 11. The slow addition of carbocation 2a to a vigorously stirred suspension of the sodium salt of 11 (13 equiv.) in anhydrous toluene readily gave trityl 12, which was isolated in 54% yield. Trityl 4a was obtained as a less abundant but very notable byproduct (isolated yield 27%). The parallel formation of both forms of TAMs suggests that the reaction conditions do not favor the preferential formation of the intermediate cyclohexadiene (k1[Nu] ≫ k2[2a], in which [Nu] is the concentration of the nucleophile, see, for example, Scheme 2), even though the nucleophile was in large excess.
Scheme 4.

C-Nucleophiles in the synthesis of monofunctionalized TAMs 12–14.
This result may be explained by the low solubility of the sodium salt of 11, which does not provide the required high concentration of nucleophile. To increase the concentration of anionic C-nucleophiles, one could modify the reaction conditions, for example, by replacement of toluene with the more polar aprotic solvent. Unfortunately, our preliminary experiments performed in acetonitrile and tetrahydrofuran (THF) did not result in an improvement of this problem. We proposed to continue these studies by using phase-transfer catalysts and dimethylformamide (DMF) as a polar solvent.
Analogously, the reaction of cation 2a with the sodium salt of the malonic ester afforded trityl 13 in 54% isolated yield. The only byproduct was TAM 4a (18%). With acetyl-acetone as a precursor of the anionic nucleophile, we obtained TAMs 14 and 4a in yields of 56 and 22%, respectively. TAMs 13 and 14 demonstrate the properties of weak C–H acids. Although both compounds are insoluble in water and water/ethanol solutions, their deprotonated forms are sparingly soluble in these polar solvents. Thus, a homogeneous solution of 14 (0.5 mM) may be readily obtained by dissolving the trityl in a 1:1 (v/v) mixture of ethanol and 5 mM aqueous KOH. This deprotonation is easily detectable in the EPR spectrum of 13 (see the Supporting Information), which shows a three-fold decrease in the line-widths of each component of the 1:2:1 triplet signal, from 1269 to 415 mG, after the addition of KOH to a solution of TAM 13 in methanol. The g factor and hyperfine constant aH did not change notably on addition of a strong base.
In the case of radical 14, the linewidth is small, even in neutral methanol solution (496 mG), and does not depend on the concentration of KOH. This implies the absence of nuclear spins in the immediate vicinity of the aryl ring, that is, close enough to the central carbon atom to cause broadening of the EPR signal. Thus, in deciding between the two tautomeric forms of trityl 14, we prefer the enol structure. This assumption is in good agreement with its IR spectrum: Trityl 14 shows a broad absorbance at 1616 cm−1, which is common to the superposition of C=O and C=C stretches and is typical of the enol form of acetylacetones.[21]
Taking into account the easy homolysis of the C–H bond of the side methine groups, we could imagine a facile oxidation of trityls 13 and 14 to diamagnetic compounds 15 (see Scheme 4), which are the direct analogues of the p-quinone dimethanes obtained in the analogous reactions of C-carbanions with bulky triarylmethyl cations.[22] Contrary to expectations, TAMs 13 and 14 showed good stability for weeks in the crystalline state in contact with atmospheric oxygen. This is also the case for solutions of trityl 14 in methanol and chloroform. However, trityl 13 is less stable in solution. On exposure to air, a methanolic solution of 13 transforms slowly over 10 d into a multicomponent mixture of diamagnetic products. Their identities are currently being investigated.
Recently, a series of phosphonated TAMs was reported. They were synthesized based on the reaction of the corresponding aryllithium derivatives with diethyl chlorophosphate followed by conversion of the resulting arylphosphonates, ArPO(OC2H5)2, into arylphosphonic acids, Ar-PO(OH)2.[6a,6b] The newly found ability of trityl cations to produce monosubstituted TAMs by reaction with nucleophiles suggests a new route to phosphonated TAMs.
We reasoned that these highly valuable compounds could be obtained by the reaction of trityl cations with the anionic forms of dialkyl phosphites, [PO(OR)2]−. The latter are known to behave as P-centered nucleophiles, for example, in the reactions with various alkyl halides.[23] To validate this suggestion, we studied the reaction of cation 2a, which was slowly added to a stirred suspension of sodium diethyl phosphite, NaPO(OR)2, in acetonitrile. As anticipated, we obtained two products: The required phosphonated trityl 16 (56%; see Figure 2) and symmetrical TAM 4a (31%). The undesired quinoid 6a was detected by TLC as a negligible byproduct. The EPR spectrum of trityl 16 shows the expected 1:2:1 triplet (aH = 2.263 G), split further by the phosphorus nucleus. The observed phosphorus doublet splitting constant (aP = 3.717 G) is in good agreement with the literature data recently reported for phosphonated TAMs (3.70–3.80 G).[6a,6b]
Figure 2.
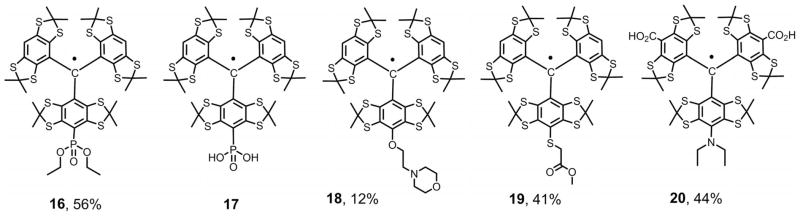
P-, O-, S-, and N-nucleophiles in the synthesis of monofunctionalized TAMs 16–20.
The ester functions present in the diethyl phosphonic group of trityl 16 were smoothly cleaved by trimethylsilyl bromide (TMSBr). The phosphonic acid derivative 17 was obtained in a high isolated yield of 94%. Its EPR spectrum shows a 1:2:1 triplet pattern (aH = 2.248 G), each component of which is further split by the phosphorus (aP = 3.720 G) to give the expected six lines (see the Supporting Information).
Although similar patterns were seen in the EPR spectra of these two trityls, the conversion of diethyl phosphonate 16 into phosphonic acid 17 results in a more than two-fold decrease of the linewidth, from 758 to 312 mG, with negligible changes in the g factor and hyperfine constants aH and aP. Trityl 17 is soluble in organic solvents (e.g., DCM, DMF, methanol) and water/ethanol mixtures. It is sparingly soluble in water in the presence of strong bases. For example, a homogeneous solution of 17 (0.1–0.2 mM) may readily be obtained with the use of 5 mM aqueous KOH. Potentially, this trityl may be used to measure acid and base concentrations in various media, including non-aqueous solutions. A detailed study of the EPR spectral sensitivity of 17 to pH is currently in progress.
To further evaluate the generality of the proposed synthetic approach we studied the reactions of a series of O-and S-nucleophiles. When methanol and 2-propanol were subjected to an experimental procedure similar to those described above for C-centered anions, the only products detected by TLC and MS (ESI) were trityl 4a and quinoid 6a.[24] The only promising result was obtained for the reaction of cation 2a with the sodium derivative of N-(2-hydroxyethyl)morpholine, which afforded the trityl 18, but in a very low yield of 12%. In addition to TAM 18, this reaction produced abundant byproducts: Trityl 4a (41%) and quinone methide 6a (29%). These data suggest that the monoalkoxy TAMs are not sufficiently stable to survive in the presence of water and atmospheric oxygen. In the course of the final work-up they may be hydrolyzed to the hypothetical monohydroxy TAM 5a (see Scheme 1 and Scheme 3) and then oxidized to diamagnetic quinoid 6a.
A better result was obtained with methyl thioglycolate as a typical aliphatic thiol. Its reaction with the cation 2a smoothly produced trityl 19 (41%) along with the less abundant trityl 4a (34%) and quinone methide 6a (11%). The EPR spectra of trityls 18 and 19 exhibit the expected 1:2:1 triplet signals, but with notably different linewidths: 372 and 212 mG, respectively. Clearly, in the case of TAM 18, the line broadening results from the unresolved hyperfine splitting caused by the pair of adjacent methylene groups in the side-substituent, whereas only one methylene group is present in 19. In addition, replacement of the S-linkage with an O bond may affect the transmission of electronic effects induced by remote side-substituents.[25]
Water-soluble monofunctionalized trityls are our particular interest. First, they may act as spin reagents for site-specific labeling of oligonucleotides and peptides.[10] Therefore it was very tempting to apply the above approach to the more polar trityl cation 2b (see Scheme 1). Presumably its reaction with nucleophiles could provide a variety of asymmetrical trityls, which, in addition to a properly chosen specific functional group, must contain two residual carboxy functions.
To verify this hypothesis, we studied the reaction of cation 2b with methyl thioglycolate. To prepare a persistent form of 2b, we treated triarylmethanol 1b with a mixture of neat TFA and CF3SO3H (1.2 molar equiv.) with the subsequent complete evaporation of volatile trifluoroacetic acid (see above and the Exp. Sect.). Contrary to expectation, we obtained the symmetrical TAM 4b (82%) as the major product with quinoid 6b also isolated as a minor byproduct (ca. 2%). This result implies that thiol participates as a sufficiently strong reducing agent to enable the one-electron reduction of 2b with the eventual formation of trityl 4b.
To prevent this unwanted reduction, we turned to diethylamine. Being a typical aliphatic amine, it should possess a much stronger resistance towards oxidation. Indeed, reaction of cation 2b with diethylamine smoothly gave the desired trityl 20 (isolated yield 44%) along with symmetrical trityl 4b (31%). The monofunctionalized trityl 20 is soluble in polar solvents (ethanol, methanol) and in aqueous solutions of strong bases. The EPR spectrum shows the expected 1:1:1 triplet pattern (aN = 191 mG), each component of which is further split by the hydrogen nuclei of the methylene groups N(CH2) (aH = 150 mG) into a 1:4:6:4:1 pattern to give a poorly resolved multiplet (see the Supporting Information).
Conclusions
The reactions between C-, P-, S-, and N-nucleophiles with bulky tris(2,3,5,6-tetrathiaaryl)methyl cations have been reported herein for the first time. Persistent triaryl-methyl radicals are obtained in which the nucleophiles are inserted into the para position of one of the three aryl rings. Di- and trisubstitution were never observed. This new, formal, aromatic nucleophilic substitution reaction suggests an easy route to a variety of new, diversely substituted, persistent triarylmethyl radicals that can be obtained on a preparative scale of 0.050–0.500 g with good or acceptable yields of 41–56%. The synthesis of trityl 20 shows that this new approach may be successfully applied to the preparation of functional derivatives of the Finland trityl. However, the method requires further detailed investigation and optimization of the reaction conditions with special emphasis on the reactions of cation 2b in order to obtain a diverse series of highly polar monofunctionalized TAMs.
Experimental Section
General
1H and 13C NMR spectra were recorded with a Bruker AV-400 spectrometer (1H NMR: 400.134 MHz; 13C NMR: 100.624 MHz) for solutions in CDCl3. Chemical shifts (δ) are given in ppm with reference to residual signals of [1H]chloroform (1H NMR: 7.26 ppm; 13C NMR: 77.16 ppm). IR spectra were recorded with Bruker Tensor 27 and Bruker Vector 22 FTIR spectrometers with the use of KBr pellets. UV/Vis absorption spectra were recorded by using a Varian Cary 5000 UV/Vis/NIR spectrophotometer; data were collected from dilute solutions (0.1 mM) in quartz spectroscopy cells. EPR spectra were recorded with a Bruker ELEXSYS E540 spectrometer (microwave power of 2 mW, modulation frequency of 100 kHz and modulation amplitude of 0.003 mT). MALDI-TOF mass spectra were recorded with an Ultraflex III MALDI-TOF mass spectrometer (Bruker Daltonics, Germany) equipped with a pulsed smart-beam laser (325 nm) in positive reflectron mode. Ions formed by a laser beam were accelerated to 25 keV. The final spectra were obtained by the accumulation of 200 single-laser-shot spectra. A solution (50 mg/mL) of 2,5-dihydroxybenzoic acid (DHB) in acetonitrile was used as matrix. A sample solution in chloroform was mixed with the same volume of matrix solution. Approximately 1 μL of the resulting solution was deposited on the 384 ground-steel target plate and allowed to dry before being introduced into the mass spectrometer. External calibration in positive mode was achieved by using a Peptide Calibration Standard II (Part No. 217498, Bruker Daltonics, Germany). Mass accuracy of about 0.1% was usually achieved. Mass spectra were processed by using flexAnalysis 2.4 software (Bruker Daltonik GmbH, Germany). Electrospray ionization mass spectra (ESI-MS) were recorded by using a hybrid quadrupole/ time-of-flight Bruker micrOTOF-Q spectrometer with methanol or dichloromethane (DCM) as solvent and scanning the spectra in the m/z range 100–3000 in positive and negative ionization modes. Nitrogen was used as the drying gas at 220 °C and a flow rate of 4 L/min. The nebulizer pressure was set to 1.0 bar. The capillary voltage was set to 4.0 kV. Sample solutions were infused into the ESI source by using an LC Agilent 1200 apparatus in FIA mode (Flow Injection Analysis, 2–3 μL at a solvent flow rate of 0.1 mL/ min). Preparative column chromatography was performed by using 60–200 μm silica gel purchased from Acros. Chemicals were purchased from Aldrich and Acros and used without further purification unless otherwise stated.
Tris(2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl)methyl (4a) and Trityl 8 from an Acid-Containing Solution of Cation 2a Generated with the Use of CF3SO3H
A solution of 1a (0.133 g, 0.150 mmol) in anhydrous dichloromethane (3 mL) and CF3SO3H (0.027 g, 0.18 mmol, 1.2 equiv.) was stirred at room temp. for 4 h[17] under argon. The resulting deep-green solution was added through a syringe to a stirred solution of diethylamine (0.044 g, 0.603 mmol, 4 equiv.) in DCM (0.5 mL). The green homogeneous solution was stirred overnight at room temp., after which water (6 mL) was added. The mixture was stirred in air for 30 min. The organic phase was separated and the aqueous phase extracted with DCM (3× 3 mL). The combined organic extracts were filtered through a short cotton plug and concentrated in vacuo. Trityls 4a and 8 were isolated by column chromatography on silica gel (TFA/DCM/hexane, 1:500:500 v/v/v, and then DCM saturated with aqueous ammonia) to give pure trityls 4a (0.063 g, 48%) and 8 (0.062 g, 44%) as the only products (see the Supporting Information for TLC analytical data). Similar results were obtained for experiments in which HBF4 was used as the acid in the cation generation step. The spectroscopic characteristics of these products (ESI-MS, EPR, and IR spectra) are identical to those of recently obtained authentic trityls 4a and 8.[12]
Tris(2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl)methyl (4a) and Quinone Methide 6a from a Neutral Solution of Cation 2a Generated with the Use of TFA
A solution of 1a (0.199 g, 0.225 mmol) in anhydrous dichloromethane (2 mL) and freshly distilled TFA (1.28 g, 0.84 mL, 50 equiv.) was stirred at room temp. overnight under argon. To remove DCM and the excess TFA, the resulting deep-green solution was concentrated and the residue was dried overnight in vacuo (0.2 Torr). The black crystalline TFA-free material (0.195 g) was dissolved in anhydrous DCM (4 mL) and the flask was flushed with argon. After standing at room temp. for 96 h,[18] the resulting brownish-green solution was added through a syringe to a solution of diethylamine (0.066 g, 0.90 mmol, 4 equiv.) in anhydrous DCM (2 mL). The mixture was stirred overnight under argon and then concentrated in vacuo. Column chromatography on silica gel (DCM/hexane, 1:1, v/v, then DCM, see the Supporting Information) afforded trityl 4a (0.088 g, 45%) and quinoid 6a (0.092 g, 46%). The spectroscopic characteristics of the products were identical to those of authentic trityl 4a and quinone methide 6a.[12]
Tris(2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl)methyl (4a) and Trityl 8 from Cation 2a Generated with the Use of TFA in the Presence of CF3SO3H
A solution of 1a (0.182 g, 0.205 mmol) and CF3SO3H (0.031 g, 0.207 mmol) in anhydrous dichloromethane (1.5 mL) and freshly distilled TFA (1.17 g, 0.76 mL, 50 equiv.) was stirred at room temp. overnight under argon. To remove DCM and the excess TFA, the resulting solution was concentrated and the residue was dried overnight in vacuo (0.16 Torr). The black crystalline TFA-free material (0.184 g) was dissolved in anhydrous DCM (4 mL) and the flask was flushed with argon. After standing at room temp. for 24 h, the resulting deep-green solution was added through a syringe to a solution of diethylamine (0.060 g, 0.82 mmol, 4 equiv.) in anhydrous DCM (2 mL). The mixture was stirred overnight under argon and then concentrated in vacuo. Column chromatography on silica gel (DCM/hexane, 1:1, v/v, then DCM) gave trityls 4a (0.084 g, 47%) and 8 (0.081 g, 42%). The spectroscopic characteristics of the products were identical to those of authentic trityls 4a and 8.[12]
1-Ethyl 4-Methyl 2-Ethoxycarbonylsuccinic Diester (11)
Compound 11 was prepared by analogy with a known literature procedure.[19] 1H NMR (400 MHz, CDCl3): δ = 1.27 (t, J = 7.16 Hz, 6 H, OCH2CH3), 2.92 (d, J = 7.40 Hz, 2 H, COCH2CH), 3.70 (s, 3 H, OCH3), 3.82 (t, J = 7.40 Hz, 1 H, COCHCH2), 4.21 (m, 4 H, OCH2) ppm. 13C NMR (100 MHz, CDCl3): δ = 14.10 (OCH2CH3), 33.02 (COCH2CH), 47.96 (COCHCH2), 52.17 (OCH3), 62.26 (OCH2), 168.49 (COOEt), 171.44 (COOMe) ppm. IR (KBr): ν̃ = 2986 (m), 1738 (s), 1441 (m), 1369 (m), 1335 (m), 1271 (s), 1213 (s), 1165 (s), 1097 (m), 1032 (m), 860 (m) cm−1. MS: calcd. for C10H15O6− [M – H]− 231.0863; found 231.0866.
Trityl 12
Sodium hydride (60 wt.-% paste in mineral oil, 0.151 g, 13 equiv.) was added portion wise over 10 min to a stirred solution of triester 11 (0.912 g, 3.93 mmol, 13.6 equiv.) in anhydrous toluene (5 mL). The thick slurry of the sodium derivative was flushed with argon, treated with ultrasound, and then vigorously stirred for 1 h at room temp. A solution of 1a (0.257 g, 0.290 mmol) in anhydrous dichloromethane (3 mL) and CF3SO3H (0.052 g, 0.348 mmol, 1.2 equiv.) was stirred at room temp. for 1.5 h under argon. Slowly, over 30 min, the resulting solution of cation 2a was added through a syringe to a stirred suspension of the sodium salt of 11. The mixture was stirred for 3 h at room temp. Water (5 mL) was added and the mixture was stirred for 5 h at room temp. in air. The organic phase was separated and the aqueous phase extracted with DCM (3× 3 mL). The combined organic extracts were filtered through a short cotton plug and concentrated in vacuo. Column chromatography on silica gel (hexane, then DCM/hexane, 1:1, v/v, then DCM) afforded trityl 4a (0.068 g, 27 %) and trityl 12 (0.172 g, 54%).[20] Data for 12: black powder, m.p. >280 °C (decomp.). IR (KBr): ν̃ = 2976 (m), 2957 (m), 2922 (m), 2912 (m), 1744 (vs), 1452 (m), 1433 (m), 1385 (m), 1365 (m), 1252 (s), 1232 (m), 1194 (s), 1169 (s), 1150 (s), 1022 (m), 862 (m) cm−1. MS (MALDI-TOF): calcd. for C47H53O6S12 [M]+ 1097.0491; found 1097.063. EPR spectrum for 0.50 mM solution in DCM: 1:2:1 triplet, aH = 2.321 G, linewidth = 651 mG, g = 2.0056.
Trityl 13
Sodium hydride (60 wt.-% paste in mineral oil, 0.144 g, 3.6 mmol, 18 equiv.) was added portionwise over 20 min to a stirred solution of malonic acid diethyl ester (0.640 g, 4.00 mmol, 20 equiv.) in anhydrous acetonitrile (15 mL). A very thick slurry was flushed with argon, treated with ultrasound, and then vigorously stirred overnight at room temp. A solution of 1a (0.177 g, 0.200 mmol) in anhydrous dichloromethane (2.5 mL) and CF3SO3H (0.042 g, 0.280 mmol, 1.4 equiv.) was stirred at room temp. for 2.5 h under argon. Slowly, over 1.5 h, the resulting solution of cation 2a was added through a syringe to a stirred suspension of the sodium derivative of malonic ester. The mixture was stirred for 2 h at room temp. Water (5 mL) was added and the mixture was stirred for 5 h at room temp. in air. The organic phase was separated and the aqueous phase extracted with DCM (3× 3 mL). The combined deep-green organic extracts were filtered through a short cotton plug and concentrated in vacuo. The resulting solid material was dissolved in toluene (20 mL), which then was evaporated to remove the residual malonic ester. Column chromatography on silica gel (hexane, then DCM/hexane, 1:1, v/v, then DCM) afforded trityl 4a (0.031 g, 18%) and trityl 13 (0.111 g, 54%). Data for 13: greenish-black powder, m.p. >260 °C (de-comp.). IR (KBr): ν̃ = 2972 (m), 2957 (m), 2922 (m), 2912 (m), 1736 (vs), 1452 (m), 1435 (m), 1383 (m), 1366 (s), 1300 (m), 1250 (vs), 1194 (m), 1167 (s), 1148 (vs), 1111 (m), 1101 (m), 1036 (m) cm−1. MS (ESI): calcd. for C44H49O4S12 [M]+ 1025.0279; found 1025.021. EPR for 0.5 mM solution in methanol: 1:2:1 triplet, aH(Ar) = 2.264 G, aH(CH) = 410 mG, linewidth = 1.269 G, g = 2.0055. EPR for 0.50 mM solution in methanol in the presence of KOH (20 mM): 1:2:1 triplet, aH(Ar) = 2.243 G, linewidth = 415 mG, g = 2.0054.
Trityl 14
TAMs 14 (0.542 g, isolated yield 56%) and 4a (0.192 g, isolated yield 22%) were obtained from triarylmethanol 1a (0.886 g, 1.000 mmol) and acetylacetone (3.301 g, 33.0 mmol, 33 equiv.) by the method used above for the synthesis of trityl 13. Data for 14: black powder, m.p. >280 °C (decomp.). IR (KBr): ν̃ = 2961 (s), 2922 (m), 2855 (m), 1616 (m), 1520 (m), 1452 (s), 1433 (s), 1383 (s), 1366 (vs), 1321 (m), 1300 (s), 1292 (m), 1246 (vs), 1167 (s), 1150 (vs), 1003 (m) cm−1. MS (MALDI-TOF): calcd. for C42H45O2S12 [M]+ 965.0068; found 965.090. EPR for 0.50 mM solution in methanol: 1:2:1 triplet, aH(Ar) = 2.299 G, aH(CH) = 382 mG, linewidth = 496 mG, g = 2.0056.
Trityl 16
TAMs 16 (0.140 g, isolated yield 56%) and 4a (0.067 g, isolated yield 31%) were obtained from triarylmethanol 1a (0.221 g, 0.250 mmol) and diethyl phosphite (0.413 g, 2.99 mmol, 12 equiv.) by the method used above for the synthesis of trityl 13. The analogous reaction with a suspension of sodium diethyl phosphite in anhydrous toluene afforded the title trityl 16 in a lower yield of 33%. Data for 16: fine black powder, m.p. >260 °C (de-comp.). IR (KBr): ν̃ = 2965 (s), 2924 (s), 2856 (m), 1452 (s), 1383 (m), 1364 (s), 1288 (m), 1251 (vs), 1221 (s), 1167 (s), 1150 (s), 1099 (m), 1051 (s), 1016 (vs), 970 (s), 941 (m), 881 (m), 793 (m), 681 (m), 548 (m) cm−1. MS (MALDI-TOF): calcd. for C41H48O3PS12 [M]+ 1002.999; found 1003.000. EPR spectrum for 0.50 mM solution in methanol: doublet of a 1:2:1 triplet, aH = 2.270 G, aP = 3.714 G, linewidth = 758 mG, g = 2.0057.
Trityl 17
A solution of trityl 16 (0.225 g, 0.224 mmol) in dry DCM (30 mL) and TMSBr (1.40 g, 9.15 mmol, 41 equiv.) was stirred at room temp. for 36 h under argon. The mixture was concentrated in vacuo, after which methanol (40 mL) was added. The resulting green solution was stirred at room temp. overnight under argon and then concentrated in vacuo. The title trityl 17 (0.200 g, 94%) was isolated by column chromatography on silica gel (DCM, then DCM/methanol 10:1, v/v) as the only product. Data for 17: fine greenish-black powder, m.p. >280 °C (decomp.). IR (KBr): ν̃ = 2957 (s), 2922 (s), 2860 (m), 1626 (br m), 1452 (s), 1433 (m), 1383 (m), 1366 (s), 1285 (m), 1250 (s), 1229 (s), 1167 (s), 1150 (vs), 1099 (br s), 1084 (s), 1036 (m), 978 (m), 681 (m), 565 (m), 554 (m) cm−1. MS (ESI): calcd. for C37H39O3PS12− [M – H]− 945.9243; found 945.93. EPR spectrum for 0.50 mM solution in methanol: doublet of a 1:2:1 triplet, aH = 2.248 G, aP = 3.720 G, linewidth = 312 mG, g = 2.0056.
Trityl 18
Sodium hydride (60 wt.-% paste in mineral oil, 0.067 g, 12 equiv.) was added portionwise over 10 min to a stirred solution of N-(2-hydroxyethyl)morpholine (0.227 g, 1.73 mmol, 12.4 equiv.) in anhydrous toluene (2.5 mL). A gel of sodium alkoxide was flushed with argon, treated with ultrasound, and then vigorously stirred for 1 h at room temp. A solution of 1a (0.124 g, 0.140 mmol) in anhydrous dichloromethane (2 mL) and CF3SO3H (0.031 g, 0.21 mmol, 1.5 equiv.) was stirred at room temp. for 1.5 h under argon. Slowly, over 30 min, the resulting solution of cation 2a was added through a syringe to a stirred suspension of sodium alcoholate. The mixture was stirred overnight at room temp. Water (5 mL) and DCM (5 mL) was added, and the mixture was stirred for 5 h at room temp. in air. The organic phase was separated and the water phase was extracted with DCM (4× 3 mL). The combined organic extracts were filtered through a short cotton plug and concentrated in vacuo. Column chromatography on silica gel (hexane, then DCM/hexane, 1:1, v/v, then DCM/methanol, 95:5, v/v) afforded trityl 4a (0.050 g, 41%), quinoid 6a (0.036 g, 29 %), and trityl 18 (0.017 g, 12%). Data for 18: greenish-black powder, m.p. >260 °C (decomp.). IR (KBr): ν̃ = 2955 (s), 2920 (s), 2912 (s), 2851 (m), 1452 (s), 1433 (m), 1383 (s), 1364 (vs), 1279 (s), 1252 (s), 1167 (s), 1148 (vs), 1117 (vs), 1032 (m), 854 (m) cm−1. MS (ESI): calcd. for C43H51NO2S12 [M + H]+ 997.056; found 997.051. EPR spectrum for 0.15 mM solution in methanol: 1:2:1 triplet, aH = 2.245 G, linewidth = 372 mG, g = 2.0055.
Trityl 19
A solution of 1a (0.140 g, 0.158 mmol) in anhydrous dichloromethane (2.5 mL) and CF3SO3H (0.033 g, 0.220 mmol, 1.4 equiv.) was stirred at room temp. for 2.5 h under argon. A solution of methyl thioglycolate (0.218 g, 2.05 mmol, 13 equiv.) in DCM (1 mL) was added through a syringe, all at once by fast driving the syringe plunger, to the resulting vigorously stirred solution of cation 2a. The mixture was stirred overnight at room temp under argon. Water (5 mL) was added and the mixture was stirred for 5 h at room temp. in air. The organic phase was separated and the aqueous phase was extracted with DCM (3× 3 mL). The combined deep-green organic extract was filtered through a short cotton plug and concentrated in vacuo. The resulting solid material was dissolved in toluene (20 mL), which then was evaporated to remove residual thioglycolate. Column chromatography on silica gel (hexane, then DCM/hexane, 1:1, v/v, then DCM) afforded trityl 4a (0.047 g, 34%), quinone methide 6a (0.015 g, 11%), and the title trityl 19 (0.063 g, 41%). Data for 19: fine black powder, m.p. >280 °C (decomp.). IR (KBr): ν̃ = 2955 (s), 2920 (s), 2912 (s), 2851 (m), 1736 (s), 1450 (s), 1433 (s), 1383 (m), 1364 (s), 1304 (m), 1244 (vs), 1167 (s), 1148 (vs), 1126 (s), 1105 (m), 847 (m) cm−1. MS (ESI): calcd. for C40H43O2S13 [M]+ 970.9632; found 970.955. EPR spectrum for 0.50 mM solution in methanol: 1:2:1 triplet, aH = 2.263 G, linewidth = 212 mG, g = 2.0056.
Trityl 4b from the Reaction of Cation 2b with Methyl Thioglycolate
A solution of 1b (0.105 g, 0.103 mmol) and CF3SO3H (0.021 g, 0.136 mmol, 1.40 equiv.) in freshly distilled TFA (2.0 mL) was stirred at room temp. overnight under argon. To remove the excess TFA, the resulting solution was concentrated and the residue was dried overnight in vacuo (0.2 Torr). The black crystalline TFA-free material (0.114 g) was dissolved in anhydrous DCM (4 mL) with the use of ultrasound, the flask was flushed with argon, and then a solution of methyl thioglycolate (0.162 g, 1.53 mmol, 14.9 equiv.) in anhydrous DCM (2 mL) was added by syringe. The resulting brownish-green solution was stirred overnight at room temp. under argon. The final work-up, analogous to that of trityl 20 (see below), gave symmetrical trityl 4b as the predominant product (0.084 g, 82%) and quinoid 6b as a minor byproduct (0.003 g, 2%). The spectroscopic characteristics of the products were identical to those of authentic trityl 4b and quinone methide 6b.[12]
Trityl 20
A solution of 1b (0.210 g, 0.206 mmol) and CF3SO3H (0.037 g, 0.249 mmol, 1.2 equiv.) in freshly distilled TFA (3.5 mL) was stirred at room temp. overnight under argon. To remove the excess TFA, the resulting brownish-green solution was concentrated and the residue was dried overnight in vacuo (0.2 Torr). The black crystalline TFA-free material (0.212 g) was suspended in anhydrous toluene (4 mL) with the use of ultrasound, the flask was flushed with argon, and then a solution of diethylamine (0.722 g, 9.89 mmol, 48 equiv.) in anhydrous DCM (6 mL) was added by syringe. The resulting brownish-green gel was stirred for 3.5 h at room temp. under argon. Water (10 mL) was added and the bi-phasic system was vigorously stirred in air. After the addition of HCl (2 N aqueous solution) and adjusting the pH of the aqueous phase to 6.5, the deep-brown organic phase was separated. The aqueous layer was extracted with DCM (3× 5 mL) and the combined organic extracts were filtered through a short cotton plug and then concentrated in vacuo to give a black cake. The latter was dissolved in NaOH (2 N solution, 2 mL, 5 mmol) and water (40 mL) to afford a deep-green solution. The addition of brine (40 mL) resulted in formation of an abundant amount of a fine precipitate. The mixture was left standing under argon for 2 h and filtered through slow filtering paper. The deep-green clear filtrate was acidified to pH 3 with HCl (2 M solution) to give the trityl 4b (0.062 g, 31%), which was isolated according to a known literature method.[12] The solid material collected by filtration was washed with a mixture of water and brine (1:1, v/v, 3×1 mL) and then dissolved in acidified methanol (25 μL of conc. HCl in 50 mL of methanol). The resulting solution was concentrated in vacuo and the crude product was purified by column chromatography on silica gel (DCM/methanol, from 20:1 to 3:1, v/v) to afford trityl 20 (0.094 g, 44 %) as a fine black powder. Data for 20: m.p. >280 °C (decomp.). IR (KBr): ν̃ = 2964 (s), 2922 (s), 2853 (m), 1574 (s), 1491 (m), 1452 (s), 1433 (m), 1385 (vs), 1366 (s), 1313 (m), 1236 (s), 1167 (m), 1150 (m), 1113 (m) cm−1. MS (ESI): calcd. for C43H47NO4S13− [M – H]− 1025.0159; found 1025.003. EPR spectrum for 0.20 mM solution in deoxygenated water/methanol (10:1, v/v) in the presence of KOH (2 mM): aN = 191 mG, aH = 150 mG, linewidth = 78 mG, g = 2.0056.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Alexander M. Genaev (Novosibirsk Institute of Organic Chemistry, Russian Academy of Sciences) for helpful discussions and suggestions, and Dr. Evgeny A. Demidov (The Institute of Cytology and Genetics, Russian Academy of Sciences) for registration of the MALDI-TOF mass spectra. This study was supported by The Ministry of Education and Sciences of the Russian Federation (project 8466), the Russian Foundation for Basic Research (grant numbers 13-04-00680A and 5P41EB002034) from the National Institute of Biomedical Imaging and Bioengineering (National Institutes of Health, NIH, USA). The NMR, IR, HRMS (ESI), and EPR experiments were performed in the Chemical Service Centre of the Siberian Branch of the RAS.
Footnotes
Supporting Information (see footnote on the first page of this article): TLC analyses of crude reaction mixtures, MS, IR and EPR spectroscopic data for trityls 12–14 and 16–20.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.201301161.
Contributor Information
Victor M. Tormyshev, Email: torm@nioch.nsc.ru.
Howard J. Halpern, Email: h-halpern@uchicago.edu.
References
- 1.a) Andersson S, Radner F, Rydbeck A, Servin R, Wistrand L-G. 5530140. U S Pat. 1996; Chem Abstr. 1996;125:142745. [Google Scholar]; b) Ardenkjaer-Larsen JH, Leunbach I. WO/9709633. PCT Int Appl. 1997; Chem Abstr. 1997;126:1274245. [Google Scholar]; c) Andersson S, Radner F, Rydbeck A, Servin R, Wistrand L-G. 5728370. U S Pat. 1998; Chem Abstr. 1998;128:244054. [Google Scholar]; d) Thaning M. WO/9839277. PCT Int Appl. 1998; Chem Abstr. 1998;129:245140. [Google Scholar]
- 2.a) Khan N, Swartz H. Mol Cell Biochem. 2002;234:341–357. [PubMed] [Google Scholar]; b) Kuppusamy P, Wang P, Chzhan M, Zweier JL. Magn Reson Med. 1997;37:479–483. doi: 10.1002/mrm.1910370402. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Villamena FA, Song Y, Sun J, Rockenbauer A, Zweier JL. J Org Chem. 2010;75:7796–7802. doi: 10.1021/jo1016844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Liu Y, Villamena FA, Sun J, Wang TY, Zweier JL. Free Radical Biol Med. 2009;46:876–883. doi: 10.1016/j.freeradbiomed.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Y, Villamena FA, Sun J, Xu Y, Dhimitruka I, Zweier JL. J Org Chem. 2008;73:1490–1497. doi: 10.1021/jo7022747. [DOI] [PubMed] [Google Scholar]
- 5.Rizzi C, Samouilov A, Kutala VK, Parinandi NL, Zweier JL, Kuppusamy P. Free Radical Biol Med. 2003;35:1608–1618. doi: 10.1016/j.freeradbiomed.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 6.a) Driesschaert B, Marchand V, Levêque P, Gallez B, Marchand-Brynaert J. Chem Commun. 2012;48:4049–4051. doi: 10.1039/c2cc00025c. [DOI] [PubMed] [Google Scholar]; b) Bobko AA, Dhimitruka I, Komarov DA, Khramtsov VV. Anal Chem. 2012;84:6054–6060. doi: 10.1021/ac3008994. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dhimitruka I, Bobko AA, Hadad CM, Zweier JL, Khramtsov VV. J Am Chem Soc. 2008;130:10780–10787. doi: 10.1021/ja803083z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Talmon Y, Shtirberg L, Harneit W, Yu O, Rogozhnikova, Tormyshev V, Blank A. Phys Chem Chem Phys. 2010;12:5998–6007. doi: 10.1039/b922060g. [DOI] [PubMed] [Google Scholar]
- 8.a) Elas M, Hleihel D, Barth ED, Haney CR, Ahn KH, Pelizzari CA, Epel B, Kocherginsky M, Weichselbaum RR, Halpern HJ. Mol Imaging Biol. 2011;13:1107–1113. doi: 10.1007/s11307-010-0436-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Halevy R, Tormyshev V, Blank A. Biophys J. 2010;99:971–978. doi: 10.1016/j.bpj.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Elas M, Ahn KH, Parasca A, Barth ED, Lee D, Haney C, Halpern HJ. Clin Cancer Res. 2006;12:4209–4217. doi: 10.1158/1078-0432.CCR-05-0446. [DOI] [PubMed] [Google Scholar]; d) Elas M, Williams DD, Parasca A, Mailer C, Pelizzari CA, Lewis MA, River JN, Karczmar GS, Barth ED, Halpern HJ. Magn Reson Med. 2002;49:682–691. doi: 10.1002/mrm.10408. [DOI] [PubMed] [Google Scholar]
- 9.a) Leggett J, Hunter R, Granwehr J, Panek R, Perez-Linde AJ, Horsewill AJ, McMaster J, Smith G, Köckenberger W. Phys Chem Chem Phys. 2010;12:5883–5892. doi: 10.1039/c002566f. [DOI] [PubMed] [Google Scholar]; b) Ardenkjær-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, Servin R, Thaning M, Golman K. Proc Natl Acad Sci USA. 2003;100:10158–10163. doi: 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Yang Z, Liu Y, Borbat P, Zweier JL, Freed JH, Hubbell WL. J Am Chem Soc. 2012;134:9950–9952. doi: 10.1021/ja303791p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Y, Villamena FA, Rockenbauer A, Song Y, Zweier JL. J Am Chem Soc. 2013;135:2350–2356. doi: 10.1021/ja311571v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Reginsson GW, Kunjir NC, Sigurdsson STh, Schiemann O. Chem Eur J. 2012;18:13580–13584. doi: 10.1002/chem.201203014. [DOI] [PubMed] [Google Scholar]
- 11.a) Tormyshev VM, Genaev AM, Sal’nikov GE, Yu O, Rogozhnikova, Troitskaya TI, Trukhin DV, Mamatyuk VI, Fadeev DS, Halpern HJ. Eur J Org Chem. 2012:623–629. doi: 10.1002/ejoc.201101243. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dhimitruka I, Velayutham M, Bobko AA, Khramtsov VV, Villamena FA, Hadad CM, Zweier JL. Bioorg Med Chem Lett. 2007;17:6801–6805. doi: 10.1016/j.bmcl.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Reddy TJ, Iwama T, Halpern HJ, Rawal VH. J Org Chem. 2002;67:4635–4639. doi: 10.1021/jo011068f. [DOI] [PubMed] [Google Scholar]
- 12.Yu O, Rogozhnikova, Vasiliev VG, Troitskaya TI, Trukhin DV, Mikhalina TV, Halpern HJ, Tormyshev VM. Eur J Org Chem. 2013:3447–3455. doi: 10.1002/ejoc.201300176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhimitruka I, Bobko AA, Eubank TD, Komarov DA, Khramtsov VV. J Am Chem Soc. 2013;135:5904–5910. doi: 10.1021/ja401572r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The hypothetical mechanisms shown in Schemes 1 and 2 are very similar to those proposed recently by C. Decroos and coworkers to interpret the unprecedented effect of the generation of TAMs through the reactions of nucleophiles with trityl cation 2b: Decroos C, Prange T, Mansuy D, Boucher JL, Li Y. Chem Commun. 2011;47:4805–4807. doi: 10.1039/c1cc10426h.Decroos C, Li Y, Soltani A, Frapart Y, Mansuy D, Boucher JL. Arch Biochem Biophys. 2010;502:74–80. doi: 10.1016/j.abb.2010.07.002. The latter was generated by the in situ one-electron oxidation of trityl 4b by excess potassium hexachloroiridate(IV) or hydrogen peroxide in the presence of peroxidase enzymes.
- 15.It is desirable, where possible, to deal with homogeneous solutions of both principal reagents, that is, trityl cation and nucleophile as homogeneity makes the control of the actual reagent concentrations much easier. In addition, homogeneity notably simplifies the critical procedure of mixing the reagents under anhydrous and anaerobic conditions.
- 16.The acid-catalyzed dehydration of triarylmethanols 1a,b to cations 2a,b inevitably produces an equimolar amount of water (see Schemes 1 and 2). Although the presence of a small amount of water cannot be prevented, fortunately this has little if any effect on the course of further transformations. We found that solutions of carbocations generated with the use of strong acids (neat trifluoroacetic acid, or 1.2–1.4 equiv. of CF3SO3H or HBF4) are very persistent. The cations remain intact even after aging their solutions for 4–96 h at room temperature under argon. For example, irrespective of the aging time, the quenching of cation 2a with dry diethylamine always gave trityls 8 and 4a as the only ultimate products of reaction. These TAMs are readily recognizable by chromatography on silica gel (see the Supporting Information for TLC analytical data), which separates them into two sharp-edged deep-green bands.[12] The quinoid 6a was never obtained, otherwise the presence of quinoid even in low concentration produces a very characteristic deep-purple band.[12] These data clearly indicate that small amounts of intrinsic water do not contribute appreciably to nucleophilic quenching of the carbocations as long as the reaction solutions remain strongly acidic. However, a priori, we cannot rule out the participation of water in the quenching of carbocations under the basic conditions typical of the further reaction of cations with nucleophiles, especially if weak nucleophiles are involved.
- 17.It was found in a series of experiments that the length of the carbocation-generating step (4–96 h) has no effect on the origin and distribution of the ultimate products.
- 18.When the storage time of the TFA-free solution of 2a was shortened from 96 to 4 h, the addition of diethylamine resulted in the production of a three-component mixture composed of monosubstituted TAM 8, symmetrical TAM 4a and quinoid 6a. This suggests that the CF3CO2− anion is a relatively weak nucleophile. It is not capable of quickly quenching the bulk of the carbocation over a few hours and then efficiently competing with diethylamine.
- 19.Banks BJ, Calverley MJ, Edwards PJ, Harley-Mason J. Tetrahedron Lett. 1981;22:1631–1634. [Google Scholar]
- 20.Experiments with the sodium salt of 11 prepared in THF and acetonitrile gave very similar results.
- 21.a) Tayyari SF, Milaninejad F. Spectrochim Acta, Part A. 2000;56:2679–2691. doi: 10.1016/s1386-1425(00)00304-8. [DOI] [PubMed] [Google Scholar]; b) Song JR, Lee SI. J Ind Eng Chem. 1997;3:198–202. [Google Scholar]
- 22.a) Hagel M, Liu J, Muth O, Rivera HJE, Schwake E, Sripanom L, Henkel G, Dyker G. Eur J Org Chem. 2007;2007:3573–3582. [Google Scholar]; b) Dyker G, Hagel M, Muth O, Schirrmacher C. Eur J Org Chem. 2006;2006:2134–2144. [Google Scholar]
- 23.a) Snyder SA, Sherwood TC, Ross AG. Angew Chem. 2010;122:5272–5276. doi: 10.1002/anie.201002264. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:5146–5150. doi: 10.1002/anie.201002264. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiao Y, Zhao ZK, Liu P. J Am Chem Soc. 2008;130:2164–2165. doi: 10.1021/ja710245d. [DOI] [PubMed] [Google Scholar]; c) Spears LG, Jr, Liao A, Minsek D, Lewis ES. J Org Chem. 1987;52:61–64. [Google Scholar]
- 24.Methanol and 2-propanol were applied in neat form and as homogeneous solutions of sodium alkoxides in the corresponding anhydrous alcohols. These changes did not affect the nature of the final products.
- 25.a) Rančić MP, Trisović NP, Milčić MK, Ajaj IA, Marinković AD. J Mol Struct. 2013;1049:59–68. [Google Scholar]; b) Kajimoto O, Kobayashi M, Fueno T. Bull Chem Soc Jpn. 1973;46:1422–1425. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.