

Abstract
Aryl hydrocarbon receptor (AHR) activation by xenobiotic ligands such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is key to their toxicity. Following activation and nuclear translocation, AHR heterodimerizes with the AHR nuclear translocator (ARNT) and binds to AHR response elements (AhREs) in the enhancer of target genes, of which Cyp1a1 is the prototype. Previously, we showed that concomitant with AHR binding, histone H3 in the Cyp1a1 enhancer-promoter AhRE cluster became phosphorylated in serine-10 (H3S10), suggesting that the ligand-activated AHR recruited one or more kinases to the enhancer chromatin to phosphorylate this residue. To test this hypothesis, we used mouse hepatoma Hepa-1c1c7 cells and their c35 mutant derivative, lacking a functional AHR, to search for candidate kinases that would phosphorylate H3S10 in an AHR dependent manner. Using chromatin immunoprecipitation with antibodies to a comprehensive set of protein kinases, we identified three kinases, IκB kinase α (IKKα), mitogen and stress activated protein kinase 1 (MSK1), and mitogen and stress activated protein kinase 2 (MSK2), whose binding to the Cyp1a1 enhancer was significantly increased by TCDD in Hepa-1c1c7 cells and absent in control c35 cells. Complexes of AHR, ARNT, and IKKα could be coimmunoprecipitated from nuclei of TCDD treated Hepa-1c1c7 cells and shRNA-mediated IKKα knockdown inhibited both H3S10 phosphorylation in the Cyp1a1 enhancer and the induction of Cyp1a1, Aldh3a1, and Nqo1 in TCDD-treated cells. We conclude that AHR recruits IKKα to the promoter of its target genes and that AHR-mediated H3S10 phosphorylation is a key epigenetic requirement for induction of AHR targets. Given the role of H3S10ph in regulation of chromosome condensation, AHR-IKKα cross-talk may be a mediator of chromatin remodeling by environmental agents.
Keywords: aryl hydrocarbon receptor, chromatin histone modifications, epigenetics, gene regulation, protein kinases, inhibitor of nuclear factor kappa-B kinase subunit alpha
Abbreviations
- AHR
aryl hydrocarbon receptor
- AhRE
AHR responsive element
- Aldh3a1
aldehyde dehydrogenase 3a1
- ARNT
aryl hydrocarbon receptor nuclear translocator
- B[a]P
benzo(a)pyrene
- CBP
cAMP response element-binding protein (CREB)-binding protein
- ChIP
chromatin immunoprecipitation
- DNMT1
DNA methyltransferase 1
- ERα
estrogen receptor α
- ERK1/2
extracellular-regulated kinases 1 and 2
- H3K14
histone H3 lysine14
- H3K9
histone H3 lysine9
- H3S10ph
histone H3 phosphoserine-10
- HDAC
histone deacetylase
- HP1
heterochromatin protein1
- IGF2
insulin-like growth factor 2
- IKKα/β/γ
IκB kinase α/β/γ
- IL-17
interleukin-17
- IL-6
interleukin-6
- IκB
inhibitor of NF-κB
- JNK
c-Jun n-Terminal kinase
- MSK1/2
mitogen and stress activated protein kinases 1 and 2
- NF-κB
nuclear factor κB
- Nqo1
NADPH-dependent quinone oxydoreductase 1
- PAH
polycyclic aromatic hydrocarbon
- PIM1
proviral insertion in murine 1
- RSK1/2
p90 ribosomal S6 kinases 1 and 2
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TFF1
trefoil factor 1
- TGFα
transforming growth factor-α
- TNF-α
tumor necrosis factor-α
- TSS
transcription start site
- VRK1
vaccinia-related kinase 1
Alterations of chromatin structure by mechanisms such as DNA methylation and histone modifications are critical epigenetic components of the cellular transcription factor repertoire that regulates gene expression (Goldberg et al., 2007). Changes in chromatin structure can disrupt transcription factor functions and vice versa, transcription factors can cause significant changes in chromatin structure. Many studies have shown that posttranslational modifications on histone tails are involved in the dynamics of chromosome functions (Fischle et al., 2003b). Thus, although euchromatin and heterochromatin have opposite effects on the accessibility of transcription factors to DNA (Jenuwein and Allis, 2001), transcription factors may recruit histone modification enzymes to their target binding sites and initiate cascades of histone modification events that change euchromatin into heterochromatin, or vice versa, as transcription may require (Kouzarides, 2007).
During the cell cycle, three kinds of histones (H1, H2A, and H3) are extensively phosphorylated. In contrast to H1 phosphorylation, which occurs throughout the cell cycle, phosphorylation of histone H3 is low in interphase cells and occurs almost exclusively during mitosis, due to phosphorylation of the serine residue in position 10 (H3S10ph). For this reason, H3S10ph is commonly used as a marker of mitosis (Cerutti and Casas-Mollano, 2009; Tang et al., 2012) and chromosome condensation (Van et al., 1998). Phosphorylated in serine-10 (H3S10) can also be phosphorylated in some gene promoters to activate transcription (Cerutti and Casas-Mollano, 2009).
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that plays a critical role in the metabolic detoxification of xenobiotic agents (Nebert et al., 2004). In addition to its role in xenobiotic metabolism, the AHR cross-talks with multiple signal transduction pathways and has essential functions in cell proliferation (Chang et al., 2007; Ma and Whitlock, 1996; Puga et al., 2009), development (Crews and Fan, 1999), immunity (Quintana and Sherr, 2013), and circadian rhythm (Mukai et al., 2008). Activation by ligand induces AHR nuclear translocation and heterodimerization with AHR nuclear translocator (ARNT) followed by binding of AHR-ARNT complexes to AHR response elements (AhREs) in the promoter region of target genes, of which Cyp1a1 is the prototype, to activate their transcription (Puga et al., 2009). In recent times, it has become evident that the transcription activity of the AHR is also regulated at the epigenetic level, not only in the control of transcriptional induction but also in the maintenance of the silent state of AHR-regulated genes. Thus, binding of AHR-ARNT complexes to AhRE motifs recruits transcription cofactors and associated chromatin remodeling proteins that signal initiation of gene transcription (Hestermann and Brown, 2003). AHR-ARNT heterodimers recruit histone acetyltransferase p300 to the target promoter to function as a coactivator that increases the level of histone acetylation and enhances transcription activity (Kobayashi et al., 1997). Conversely, HDAC1-DNMT1 complexes, reversibly bound in naive cells at the Cyp1a1 promoter, maintain this gene in a transcriptionally silent facultative heterochromatic state, poised for removal upon induction (Schnekenburger et al., 2007a). It is evident from these studies that epigenetic mechanisms are strongly involved not only in regulating transcription activation but also in maintaining the silent status of AHR-regulated genes.
Previous work from our laboratory has shown that following activation by ligand and concomitant with binding of AHR-ARNT heterodimers to the Cyp1a1 enhancer region, the level of phosphorylated H3S10 increased significantly, an increase that can be blocked by specific inhibitors of AHR-mediated gene induction (Ovesen et al., 2011; Schnekenburger et al., 2007a). These observations suggested the possibility that AHR recruited one or more protein kinases to phosphorylate this serine residue in the neighborhood of its binding targets. To test this hypothesis, we used mouse hepatoma Hepa-1c1c7 cells and their c35 mutant derivative, lacking a functional AHR, to search for candidate kinases that would phosphorylate H3S10 in an AHR dependent manner. We find that three kinases, IκB kinase α (IKKα), mitogen, and stress activated protein kinase 1 (MSK1) and mitogen and stress activated protein kinase 2 (MSK2), are recruited by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-activated AHR to the Cyp1a1 AhRE motif cluster in the enhancer domain in Hepa-1c1c7 cells but absent in control c35 cells. It is likely that the cross-talk between AHR and IKKα mediates chromatin remodeling resulting from exposure to environmental agents and ultimately plays a role in environmentally driven AHR-related toxicity.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatments
Mouse hepatoma Hepa-1c1c7 (hereafter referred to as Hepa-1) from the American Type Culture Collection and its mutant derivative c35, expressing a DNA-binding defective AHR (Sun et al., 1997), were cultured in α-minimal essential medium (Gibco) supplemented with 5% (vol/vol) fetal bovine serum (Sigma) and 1% (vol/vol) penicillin/streptomycin (Gibco) under 5% CO2 at 37°C. Cells were treated with TCDD at 5nM or benzo(a)pyrene (B[a]P) at 5μM or with dimethyl sulfoxide (DMSO) vehicle control never to exceed 0.1%.
ChIP-qPCR Analysis
Chromatin immunoprecipitation (ChIP) was performed with minor modifications by procedures previously described (Schnekenburger et al., 2007a; Wells and Farnham, 2002). Cells were incubated for 10 min at room temperature with 1% formaldehyde. After cross-linking, the reaction was quenched with 0.125M glycine for 5 min at room temperature. After washing twice with ice-cold 1× PBS, cells were scraped from the dishes, and 1.0 × 106 cells were pelleted by centrifugation, resuspended in cell lysis buffer (0.5mM PIPES [pH 8.0], 85mM KCl, 0.5% IGEPAL CA-630 and protease inhibitor cocktail), and incubated on ice for 10 min. The nuclei were pelleted, resuspended in nucleus lysis buffer (50mM Tris-HCl [pH 8.1], 10mM EDTA, 1% SDS and protease inhibitor cocktail), and incubated on ice for 10 min. Chromatin was sheared by sonication in a crushed-ice–water bath with 30 cycles of 30-s bursts of 200 W with a 30-s interval between bursts, using a Bioruptor (Diagenode). After centrifugation to remove cell debris, chromatin was precleared for 1 h at 4°C with a 1:1 mixture of protein A/G-agarose beads (a 50% gel slurry saturated with salmon sperm DNA and bovine serum albumin). The precleared chromatin was diluted six times in immunoprecipitation (IP) dilution buffer (16.7mM Tris-Cl [pH 8.1], 167mM NaCl, 1.2mM EDTA, 1.1% Triton X-100, 0.01% SDS and protease inhibitor cocktail), and 2.5% of the supernatants was used as input. The diluted chromatin was incubated overnight on a rotating platform at 4°C with antibodies specific for the proteins of interest. A list of antibodies used can be found in Supplementary table 1. The immune complexes were recovered by 3-h incubation at 4°C with a 1:1 mixture of protein A/G-agarose beads. The agarose beads were pelleted and washed three times with dialysis buffer (50mM Tris-HCl [pH 8.0], 2mM EDTA, 0.2% Sarkosyl and protease inhibitor cocktail) and sequentially three times with IP wash buffer (100mM Tris-HCl [pH 9.0], 500mM LiCl, 1% IGEPAL CA-630, 1% deoxycholic acid and protease inhibitor cocktail). Precipitated chromatin complexes were removed from the beads by incubation with elution buffer (50mM NaHCO3, 1% SDS) with mild vortexing. Cross-linking was reversed by adding NaCl to a final concentration of 0.3M and incubating overnight at 65°C in the presence of RNase A. Samples were then digested with proteinase K at 45°C for 2 h. DNA was purified using a PCR Purification kit (QIAGEN) and eluted in nuclease-free water. An aliquot was used for analysis by qRT PCR. The sequences of primers used for PCR are listed in Supplementary table 2. The amount of ChIP DNA was calculated as the percent of total input DNA.
RNA Isolation and Reverse Transcription
Total RNA was extracted using Trizol reagent (Invitrogen) following the manufacturer's protocols. cDNA was prepared with reverse transcription mix containing 1× reverse transcriptase buffer, 15μM dNTP, 10mM dithiothreitol (DTT), 500 units of Superscript III Reverse Transcriptase (Invitrogen), and 20 units of RNAsin (Promega) at 42°C for 2 h. RNA was removed by treatment in 0.05N NaOH, and after neutralization, cDNA was precipitated with ethanol in the presence of 10 mg glycogen carrier and 300mM sodium acetate pH 5.2. Precipitates were dissolved in 200 μl ddH2O and 1-μl aliquots were used for subsequent quantification by real-time PCR amplification.
Real-Time Quantitative RT-qPCR
Real-time quantitative RT-qPCR was performed with a PCR mixture containing 1×Power SYBR Master Mix (Applied Biosystems) and 2.5μM of each forward and reverse primer for target mRNA. The sequences of primers used for mRNA quantitation can be found in Supplementary table 2. Amplification was performed on a Stratagene Mx3000P thermocycler (Agilent Biotechnologies) with 1 cycle of 95°C for 10 min and 40 cycles of 95°C for 30 s and 60°C for 1 min. PCR efficiencies were verified by examination of the corresponding melting curves for each primer pair. Amplification of β-actin cDNA in the same sample was used as an internal control for all PCR amplification reactions. Data were calculated by using the ΔΔCt method, where
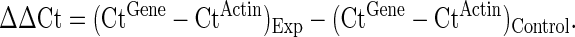 |
Protein Extraction, SDS-PAGE, and Western Blotting
Total or nuclear and cytosolic proteins were extracted from 80% confluent cells grown in 100-mm dishes. Cells were lysed in NTEN buffer (20mM Tris-HCl [pH 8.0], 100mM NaCl, 1mM EDTA, 0.5% IGEPAL CA-630 and protease inhibitor cocktail) with sonication to reduce viscosity. Cell lysates were centrifuged and supernatants were used for total protein. For separation of nuclear and cytosol, cells were lysed in cell lysis buffer (10mM HEPES [pH 7.9], 10mM KCl, 0.1mM EGTA, 1.5mM MgCl2, 0.2% IGEPAL CA-630, 1mM DTT and protease inhibitor cocktail), centrifuged and the supernatants recovered for the cytosolic fraction. The pellets were dissolved in nuclear lysis buffer (20mM HEPES [pH 7.9], 420mM NaCl, 0.1mM EGTA, 1.5mM MgCl2, 25% glycerol, 1mM DTT and protease inhibitor cocktail), and after removing cell debris by centrifugation, the supernatant was saved as the nuclear fraction. Protein concentrations were determined by using Pierce BCA Protein Assay kit (Thermo Scientific). Loading buffer was added to all fractions and proteins denatured by boiling. Fifteen micrograms of protein were separated in 7.5 or 10% polyacrylamide gel and transferred to Immobilon-P (Millipore). Membranes were blocked in 1× TBS containing 0.1% (vol/vol) Tween 20 (TBS-T) and 5% fat-free milk at room temperature for 2 h. Membranes were probed with the pertinent primary antibodies (see list in Supplemental table 3) followed by species-specific horseradish peroxidase-conjugated secondary antibodies. Proteins of interest were visualized with a Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific).
Protein Coimmunoprecipitation
A 500 μg aliquot of nuclear or cytosolic proteins was diluted with co-IP buffer (20mM Tris-HCl [pH 7.4], 150mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X-100, 0.5% IGEPAL CA-630, and proteinase inhibitor cocktail). Protein lysates were precleared by 1-h incubation with 30 μl of Protein A/G agarose beads at 4°C. Precleared lysates were incubated with 5 μg of either rabbit polyclonal anti-AHR, mouse monoclonal anti-IKKα, mouse monoclonal anti-ARNT, or control rabbit IgG at 4°C overnight. To collect the antibody-antigen complexes, 30 μl of Protein A/G agarose beads were added and the reaction was incubated at 4°C for 3 h. Beads were washed six times with co-IP buffer and the antibody-antigen complexes were removed from beads by incubation with elution buffer. Concentrated gel loading buffer was added and the eluted samples were boiled for 5 min and subjected to SDS-PAGE (polyacrylamide gel electrophoresis) for Western blotting.
shRNA Knockdown
The validated lentiviral shRNA constructs for IKKα (TRCN0000012348 and TRCN0000012349) and scrambled control shRNA (SH01) from the Mission ShRNA Lentiviral Collection (Sigma) were purchased from the Lentivirus-shRNA Core of the Cincinnati Children's Hospital Medical Center. Hepa-1 cells were infected with these viruses in the presence of 10 μg/ml polybrene and IKKα-knockdown cells were selected by 3 μg/ml of puromycin. The efficiency of knockdown was greater than 90%, as determined by quantitative real-time RT-qPCR and Western blotting.
Statistical Analysis
All results are expressed as the mean ± standard error of biological replicates. Statistical analysis was performed using IBM SPSS Statistics version 19.0. A two-way ANOVA followed by Bonferroni's post hoc test was performed to compare means of mRNA expression, histone modification, and protein binding. A p-value of less than 0.05 was considered to be statistically significant.
RESULTS
Binding of Activated AHR to the Cyp1a1 Enhancer Domain Promotes Histone H3S10 Phosphorylation
In the previous work, we reported that AHR activation by B[a]P, its prototypical PAH ligand, caused the elevation of serine-10 phosphorylation in the histone H3 associated with the cluster of AhREs corresponding to the enhancer region of the target Cyp1a1 gene, located between coordinates −1.2 and −0.8 kb from the transcription start site (TSS) (Ovesen et al., 2011; Schnekenburger et al., 2007a,b). To determine whether H3S10 phosphorylation could be linked to AHR binding at the Cyp1a1 enhancer, we used chromatin immunoprecipitation with anti-AHR and anti-H3S10ph antibodies to examine the kinetics of appearance of H3 phosphoserine-10 and AHR binding to this region of the Cyp1a1 gene. Our results indicated that relative to control cells, B[a]P treatment of Hepa-1 cells caused parallel increases in AHR binding and H3S10 phosphorylation, both reaching levels 20- to 25-fold above control 60 min after induction (Fig. 1A). The effect on serine-10 phosphorylation was not unique to B[a]P-dependent AHR activation, the AHR ligand used in those experiments because it happened equally well when the cells were treated with TCDD, a second AHR ligand (Fig. 1B). These results suggest the possibility that the AHR recruits and brings to its cognate binding motifs one or more protein kinases responsible for H3S10 phosphorylation or that, alternatively, kinase and AHR are independently but simultaneously recruited to the binding motif.
FIG. 1.

H3S10 phosphorylation is linked to AHR binding at the Cyp1a1 enhancer. The extent of anti-H3S10ph and anti-AHR antibodies binding to the Cyp1a1 enhancer region (from −1.5 to −0.9 kb) was determined by ChIP-qPCR. (A) Hepa-1 cells were treated with B[a]P or control vehicle and binding was determined at 0, 15, 30, and 60 min after treatment. (B) Hepa-1 cells were treated with B[a]P, TCDD, or control DMSO vehicle and binding was determined 90 min after treatment. Statistically significant differences were determined by one-way ANOVA. A p-value <0.05 (*) was considered as significant in comparison to the 0-min time-point (A) or the control DMSO treatment group (B).
IKKα, MSK1 and MSK2 are Recruited to the Cyp1a1 Enhancer as a Result of AHR Activation
Several serine/threonine protein kinases are known to phosphorylate H3S10, either globally or in the promoters of certain genes. These kinases include Aurora-A and Aurora-B (Crosio et al., 2002), p38 (Zhong et al., 2000), JNK (Tiwari et al., 2012), ERK1/2 (Zhong et al., 2000), RSK2 (Sassone-Corsi et al., 1999), PIM1 (Zippo et al., 2007), VRK (Kang et al., 2007), PKB/AKT (Davies et al., 2010), MSK1 (Lee et al., 2011; Li et al., 2011b), MSK2 (Soloaga et al., 2003), and IKKα (Anest et al., 2003; Yamamoto et al., 2003). To determine if treatment with AHR ligands would recruit to the Cyp1a1 enhancer any of these known protein kinases, we used ChIP-qPCR with specific antibodies to the 12 kinases mentioned above, plus Aurora-C and RSK1, two kinases not previously tested for H3S10 phosphorylation. Of the 14 kinases tested, 11 showed increased binding to the tested Cyp1a1 enhancer region, located between coordinates −1.2 and −0.8 kb from the TSS, when compared with a control ChIP with nonimmune rabbit IgG. However, only the increases observed with three of them, IKKα, MSK1, and MSK2, were significantly different between TCDD- and DMSO-treated cells (Fig. 2), narrowing down to these three the number of candidate kinases that were likely to phosphorylate H3S10 in the Cyp1a1 enhancer.
FIG. 2.
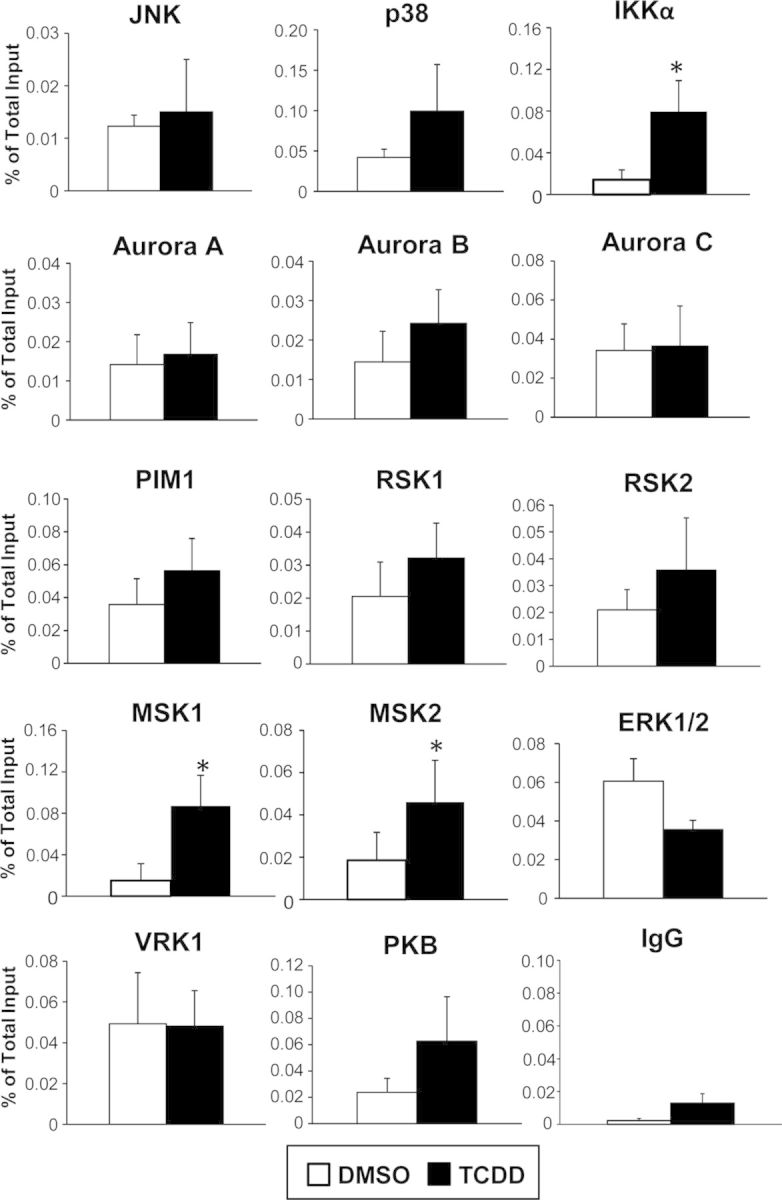
TCDD treatment significantly increased binding of IKKα, MSK1, and MSK2 to the Cyp1a1 enhancer region. Of the 14 protein kinases tested (see text) only IKKα, MSK1, and MSK2 showed a significant increase of binding to the Cyp1a1 enhancer region after TCDD treatment relative to control, as determined by ChIP qPCR 90 min after treatment. Data are presented as the means ± SEM of at least three independent determinations. Statistically significant differences were determined by Student's t-test. The asterisk (*) denotes a p-value of <0.05.
H3S10 Phosphorylation Requires Ligand-Dependent Recruitment of IKKα, MSK1, or MSK2 by the Activated AHR
As indicated earlier, the results described above could be the consequence of either independent binding of AHR and kinase to the enhancer site or of targeted recruitment of the kinase by the AHR. To distinguish between these two possibilities and assess whether recruitment was AHR-dependent, we used ChIP-qPCR to measure the binding of IKKα, MSK1, and MSK2 in TCDD-treated Hepa-1 cells and its derivative, c35, which express an AHR mutant that can translocate to the nucleus when activated by ligand but is DNA-binding defective (Sun et al., 1997). In addition, to determine whether the kinases could bind to other domains of the extended Cyp1a1 promoter, we tiled ca. 4 kb of the entire Cyp1a1 promoter, using primer sets for the ChIP analyses covering from −3.2 to +0.6 kb from the TSS. Compared with DMSO control, treatment of Hepa-1 cells with TCDD led to a significant elevation of the binding of AHR, IKKα, MSK1, and MSK2, and phosphorylation of H3S10 mostly in the enhancer domain between coordinates −1.5 and −0.9 kb, with lesser increases in the adjacent areas (Fig. 3). In contrast, TCDD treatment of the DNA-binding defective c35 cells did not cause a significant increase relative to DMSO control in either the level of H3S10ph, or the binding of AHR, IKKα, MSK1, or MSK2 throughout the extended promoter region tested, leading to significant binding differences in all the parameters measured between the two cell lines (Fig. 3). Immunoprecipitation with control IgG showed no significant binding anywhere in the tested region (Fig. 3). The protein levels of AHR, ARNT, IKKα, MSK1, and MSK2 in whole cell extracts and nuclear and cytosolic fractions were not changed by TCDD treatment within the time frame of our experimental conditions (Supplementary fig. 1), indicating that the increase of IKKα, MSK1, and MSK2 binding to the Cyp1a1 enhancer region was not due to an increase in the amount of these proteins. These results suggest that the AHR is directly involved in the recruitment of the kinases to the Cyp1a1 enhancer because no such recruitment takes place in cells with a DNA-binding defective AHR.
FIG. 3.
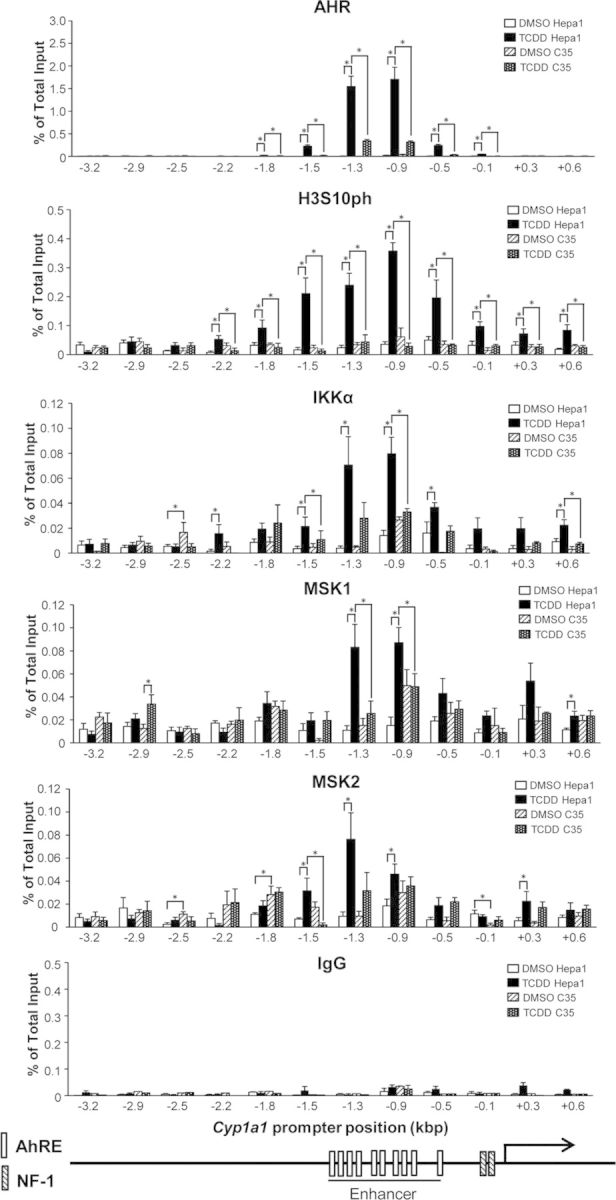
The DNA-binding competent AHR in Hepa-1 cells but not the DNA-binding defective AHR in c35 cells, recruits IKKα, MSK1, and MSK2 to the Cyp1a1 enhancer to promote phosphorylation of H3S10 in that region. The level of H3S10ph, AHR, MSK1, MSK2, and IKKα binding in the extended Cyp1a1 promoter region from −3.2 to +0.6 kb, was determined in Hepa-1 and c35 cells by ChIP qPCR at 90 min after TCDD or DMSO control treatments. Data are shown as the means ± SEM of at least three independent experimental determinations. Statistically significant differences at each promoter position were determined by two-way ANOVA followed by Bonferroni's post hoc test. A p-value<0.05 (*) was considered as significant.
To confirm the ChIP data, we focused our attention in more detail on the characterization of IKKα-AHR interactions and examined whether after TCDD treatment, IKKα formed nuclear complexes with AHR-ARNT heterodimers. Coimmunoprecipitation experiments indicated that antibodies to any of the three proteins precipitated the other two as well from nuclear but not from cytosolic extracts, as determined by Western immunoblotting (Fig. 4), suggesting that IKKα, the ligand-activated AHR, and ARNT are likely to form tripartite nuclear complexes on the Cyp1a1 enhancer.
FIG. 4.
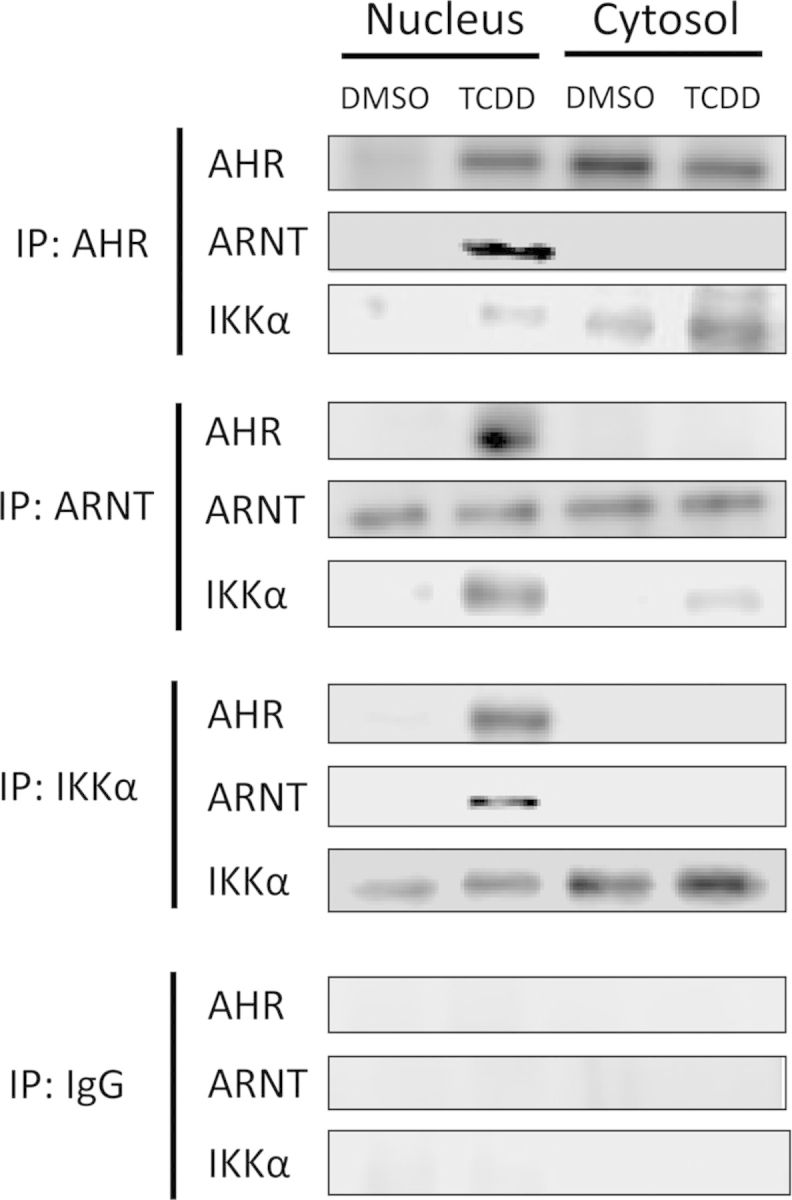
IKKα coimmunoprecipitates with nuclear AHR-ARNT complexes in TCDD-treated Hepa-1 cells. Cells were treated with TCDD or control DMSO for 90 min and thereafter nuclear and cytosolic fractions were separated and immunoprecipitated with the antibodies indicated in the left-hand side (IP) followed by Western blot detection with the antibodies indicated next to each dataset. A nonspecific rabbit IgG was used as an immunoprecipitation control.
Phosphorylation of H3S10 by IKKα Regulates TCDD-Dependent Induction of AHR Battery Genes
Several genes encoding phase I and phase II detoxification enzymes, including CYP1A1, NQO1, and ALDH3A1 among others, are coordinately regulated by AHR activation, forming the so-called “Ah gene battery” (Robertson et al., 1987). To examine whether IKKα and the phosphorylation of H3S10 played a key regulatory epigenetic role on the TCDD-dependent induction of Ah battery genes, we generated a stable IKKα knockdown Hepa-1 cell derivative by infection with an Ikkα shRNA lentivirus, and a corresponding control cell line, infected with a lentivirus carrying a scrambled shRNA. In knockdown cells, but not in controls, expression of IKKα was significantly silenced at both the protein and mRNA levels (Supplementary figs. 2A and B). As expected, IKKα knockdown abolished IKKα binding to the target enhancer chromatin and significantly reduced, but did not completely eliminate, H3S10 phosphorylation, without affecting AHR binding (Fig. 5). mRNA induction of Cyp1a1, Aldh3a1, and Nqo1 by TCDD relative to DMSO vehicle was significantly inhibited in IKKα knockdown cells compared with controls, to a greater extent for Cyp1a1 and Aldh3a1 mRNA and less, but still statistically significant, for Nqo1 (Fig. 6). These results indicate that IKKα is recruited by AHR to the Cyp1a1 enhancer region and that H3S10 phosphorylation by IKKα is a critical component of the Ah receptor signaling pathway. By controlling H3S10 phosphorylation, IKKα exerts an epigenetic regulatory role on the expression of these, and possibly other Ah battery genes.
FIG. 5.
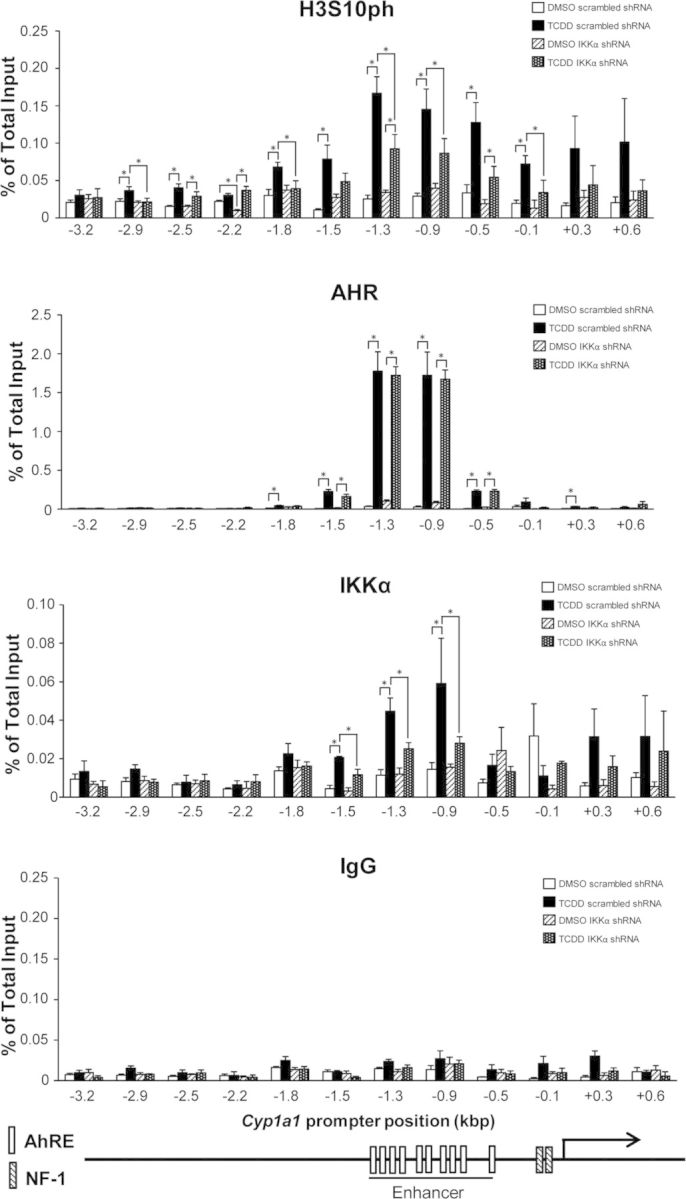
Phosphorylation of H3S10 in the Cyp1a1 enhancer is largely dependent on IKKα. The extent of H3S10ph, AHR, and IKKα binding in the entire Cyp1a1 promoter region, from −3.2 to +0.6 kb, was determined in scrambled shRNA and IKKα shRNA knockdown cells by ChIP qPCR at 90 min after TCDD or DMSO control treatment. Data are presented as the means ± SEM of at least three independent experiments. Statistically significant differences were determined by two-way ANOVA followed by Bonferroni's post hoc test at each promoter position. The asterisk (*) denotes a p-value of <0.05.
FIG. 6.
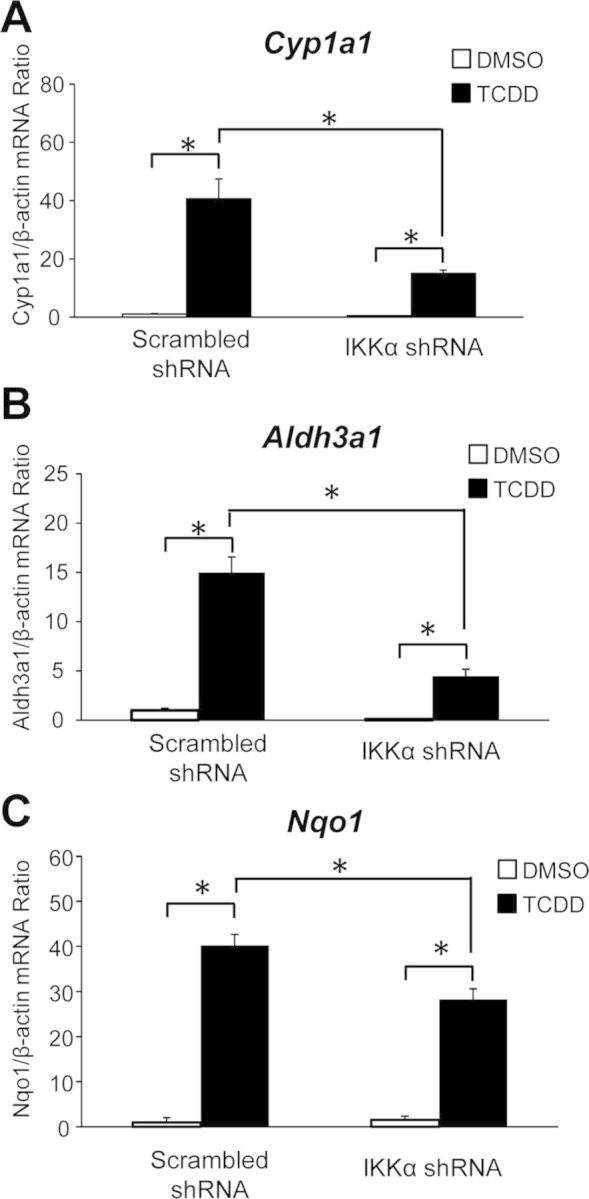
Induction of AHR battery genes, Cyp1a1, Aldh3a1, and Nqo1 is repressed in IKKα knockdown cells. The mRNA levels of Cyp1a1, Aldh3a1, and Nqo1 were determined by real-time RT-qPCR in IKKα knockdown and scrambled control cells after an 8-h treatment with TCDD or DMSO control. Statistically significant differences were determined by two-way ANOVA followed by Bonferroni's post hoc test at each promoter position. The asterisk (*) denotes a p-value of <0.05.
DISCUSSION
The results from the studies reported here show that the ligand-activated Ah receptor recruits three kinases, IKKα, MSK1, and MSK2, to the AHR binding motif in the enhancer chromatin of its target gene, Cyp1a1. IKKα, and possibly the other two kinases as well, occupy the AhRE motif chromatin, forming a tripartite complex with AHR and ARNT that phosphorylates serine-10 in histone H3. H3S10 phosphorylation is a requirement for maximal Cyp1a1 mRNA induction because IKKα knockdown significantly reduces the induction level, not only of Cyp1a1, but of other AHR battery genes as well, including Aldh3a1 and Nqo1. Inhibition of H3S10 phosphorylation and induction of these target genes are incomplete in IKKα knockdown cells, which show a low but significant residual level of both, suggesting that all three kinases may participate in the phosphorylation of this site, even though IKKα appears to be the major factor. Our experiments only provide direct evidence of the role of IKKα on H3S10 phosphorylation and do not rule out the possibility that, even though they bind to histone H3 in nucleosomes associated with AhRE motifs, MSK1 and MSK2 may be involved in phosphorylating residues other than H3S10, as perhaps, H3S28. Conversely, IKKα may also be involved in phosphorylation of additional histone residues.
The components of the epigenetic machinery that regulate the expression of AHR battery genes are not well understood. Our findings on the phosphorylation of histone H3 expand a knowledge base initiated by earlier work by others showing that the recruitment to the Cyp1a1 promoter of the histone acetyl transferase p300/CBP by AHR-ARNT heterodimers was required to induce transcription (Kobayashi et al., 1997), as was the recruitment of transcriptional coactivators (Beischlag et al., 2002) and chromatin remodeling proteins Brahma and Mediator (Wang and Hankinson, 2002; Wang et al., 2004). Previous studies from our laboratory have addressed the mechanisms that regulate the basal state of AHR target genes, and showed that a DNMT1-HDAC1/2 complex occupies the Cyp1a1 promoter in unstimulated Hepa-1 cells and maintains the gene in a silent state (Schnekenburger et al., 2007a,b). Work in vivo has shown that exposure of preimplantation mouse embryos to TCDD changes the DNA methylation status of the imprinted genes Igf2 and H19 (Wu et al., 2002, 2004) and that exposure during gestational days 8 and 14 promotes epigenetic transgenerational inheritance of adult onset disease and sperm epimutations (Manikkam et al., 2012). Epigenetic regulation of the AHR pathway, including the Ahr gene itself, is likely to play an important role in the control of the expression of its target genes, and thus, in its biological functions and its mechanistic role in xenobiotic metabolism and developmental toxicity.
Of the three protein kinases detected, MSK1/2 has been shown to induce the expression of early stress response genes, such as c-fos and c-jun, by phosphorylation of H3S10 after mitogen stimulation (Soloaga et al., 2003), and to regulate TFF1 transcription by phosphorylating H3S10 in the TFF1 promoter in an ERα-dependent manner (Li et al., 2011b). In addition, MSK1/2 has been associated with the activation of the AHR pathway for quite some time. We and others have shown that AHR ligands activate the ERK1/2 pathway within minutes of treatment, indicating that gene induction is not required for induction of this pathway (Park et al., 2005; Tan et al., 2002, 2004). Pharmacological inhibitors of the ERK1/2 pathway repress AHR-mediated Cyp1a1 induction (Reiners et al., 1998; Shibazaki et al., 2004a,b; Tan et al., 2002, 2004), providing a potential link between the AHR pathway and MSK1/2 activation, which is modulated by ERK1/2 (Cargnello and Roux, 2011). Possibly, induction of ERK1/2 by TCDD triggers the activation of MSK1/2, which in turn may phosphorylate H3S10 or some other target and promote Cyp1a1 induction. We believe this to be the simplest explanation linking MSK1/2-dependent H3S10 phosphorylation and the induction of AHR gene targets.
On the other hand, there are no reports in the literature of an association between IKKα and AHR. It has been well established that IKKα forms a cytosolic complex with IKKβ and IKKγ that phosphorylates IκB and marks it for proteasomal degradation. As a consequence, NF-κB transcription factor subunits are free to translocate to the nucleus to drive expression of target genes (Hacker and Karin, 2006). Growing evidence, however, has accumulated that IKKα has a nuclear function that is independent of its association with the other IKKs and the regulation of NF-κB nuclear translocation, but still related to NF-κB-dependent gene expression. Nuclear IKKα phosphorylates H3S10 in the promoters of the IκBα and IL-6 genes in mouse embryo fibroblasts stimulated with TNF-α, a function critical for cytokine-induced gene expression (Anest et al., 2003; Yamamoto et al., 2003). Intriguingly, however, nuclear IKKα is also a main cofactor in a Smad4-independent TGFβ-Smad2/3 pathway in keratinocyte differentiation, but in this role, neither its kinase activity nor NF-κB signaling is involved in the process (Descargues et al., 2008a,b), unlike in the differentiation of Th17 cells, where, independent of NF-κB, nuclear IKKα associates with the promoter of the IL17a gene and phosphorylates H3S10 (Li et al., 2011a). Our results are consistent with a role for a functional kinase activity of nuclear IKKα independent of NF-κB signaling. For the first time, however, this activity appears to be intrinsically associated with AHR activation.
The binary switch hypothesis (Fischle et al., 2003a) has proposed the concept of “methylation/phosphorylation” to address the alternative biological functions of posttranslational modifications taking place in the adjacent amino residues, S10 and K9, of histone H3. Phosphorylation of H3S10 and methylation of H3K9 modulate the binding of HP1 to histone H3. During interphase, binding of HP1 to di- or trimethylated H3K9 maintains a constitutive heterochromatin status. When cells enter mitosis, however, phosphorylation of H3S10, acetylation of H3K14, and possibly methylation of H3K4, hinder methylation of H3K9, causing HP1 to be released from the histone (Fischle et al., 2005; Hirota et al., 2005; Teperek-Tkacz et al., 2010), bringing a change to the higher structure of chromatin. This mechanism, which appears to be fundamental to the role of H3S10 phosphorylation in chromosome condensation during mitosis, has been suggested to apply as well to H3S10 phosphorylation in the promoters of certain genes to regulate transcription during interphase (Winter et al., 2008). Our findings revealing an AHR-dependent and AhRE-specific epigenetic mechanism, point at the likelihood that AHR cross-talk with the IKKα signaling pathway may provide clues to understand at the molecular level how certain environmental agents may mediate chromatin remodeling and affect the resulting phenotype.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (5R01 ES006273, 5R01 ES010807).
Supplementary Material
Acknowledgments
We thank Qin Wang, Chia-I Ko, Yunxia Fan, Vinicius Carreira, and Francisco-Javier Sánchez-Martín for a critical reading of the manuscript.
Footnotes
Present address: Laboratoire de Biologie Moléculaire et Cellulaire du Cancer, Hôpital Kirchberg, 9, rue Edward Steichen, L-2540 Luxembourg, Luxembourg.
REFERENCES
- Anest V., Hanson J. L., Cogswell P. C., Steinbrecher K. A., Strahl B. D., Baldwin A. S. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- Beischlag T. V., Wang S., Rose D. W., Torchia J., Reisz-Porszasz S., Muhammad K., Nelson W. E., Probst M. R., Rosenfeld M. G., Hankinson O. Recruitment of the NCoA/SRC-1/p160 family of transcriptional coactivators by the aryl hydrocarbon receptor/aryl hydrocarbon receptor nuclear translocator complex. Mol. Cell. Biol. 2002;22:4319–4333. doi: 10.1128/MCB.22.12.4319-4333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnello M., Roux P. P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti H., Casas-Mollano J. A. Histone H3 phosphorylation: Universal code or lineage specific dialects? Epigenetics. 2009;4:71–75. doi: 10.4161/epi.4.2.7781. [DOI] [PubMed] [Google Scholar]
- Chang X., Fan Y., Karyala S., Schwemberger S., Tomlinson C. R., Sartor M. A., Puga A. Ligand-independent regulation of transforming growth factor beta1 expression and cell cycle progression by the aryl hydrocarbon receptor. Mol. Cell. Biol. 2007;27:6127–6139. doi: 10.1128/MCB.00323-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews S. T., Fan C. M. Remembrance of things PAS: Regulation of development by bHLH-PAS proteins. Curr. Opin. Genet. Dev. 1999;9:580–587. doi: 10.1016/s0959-437x(99)00003-9. [DOI] [PubMed] [Google Scholar]
- Crosio C., Fimia G. M., Loury R., Kimura M., Okano Y., Zhou H., Sen S., Allis C. D., Sassone-Corsi P. Mitotic phosphorylation of histone H3: Spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol. 2002;22:874–885. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G. F., Ross A. R., Arnason T. G., Juurlink B. H., Harkness T. A. Troglitazone inhibits histone deacetylase activity in breast cancer cells. Cancer Lett. 2010;288:236–250. doi: 10.1016/j.canlet.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Descargues P., Sil A. K., Karin M. IKKalpha, a critical regulator of epidermal differentiation and a suppressor of skin cancer. EMBO J. 2008a;27:2639–2647. doi: 10.1038/emboj.2008.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descargues P., Sil A. K., Sano Y., Korchynskyi O., Han G., Owens P., Wang X. J., Karin M. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc. Natl. Acad. Sci. U. S. A. 2008b;105:2487–2492. doi: 10.1073/pnas.0712044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W., Tseng B. S., Dormann H. L., Ueberheide B. M., Garcia B. A., Shabanowitz J., Hunt D. F., Funabiki H., Allis C. D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- Fischle W., Wang Y., Allis C. D. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003a;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- Fischle W., Wang Y., Allis C. D. Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 2003b;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- Goldberg A. D., Allis C. D., Bernstein E. Epigenetics: A landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Hacker H., Karin M. Regulation and function of IKK and IKK-related kinases. Sci. STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Hestermann E. V., Brown M. Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Mol. Cell. Biol. 2003;23:7920–7925. doi: 10.1128/MCB.23.21.7920-7925.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T., Lipp J. J., Toh B. H., Peters J. M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- Jenuwein T., Allis C. D. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kang T. H., Park D. Y., Choi Y. H., Kim K. J., Yoon H. S., Kim K. T. Mitotic histone H3 phosphorylation by vaccinia-related kinase 1 in mammalian cells. Mol. Cell. Biol. 2007;27:8533–8546. doi: 10.1128/MCB.00018-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A., Numayama-Tsuruta K., Sogawa K., Fujii-Kuriyama Y. CBP/p300 functions as a possible transcriptional coactivator of Ah receptor nuclear translocator (Arnt) J. Biochem. (Tokyo) 1997;122:703–710. doi: 10.1093/oxfordjournals.jbchem.a021812. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lee C. C., Lin Y. H., Chang W. H., Lin P. C., Wu Y. C., Chang J. G. Squamocin modulates histone H3 phosphorylation levels and induces G1 phase arrest and apoptosis in cancer cells. BMC Cancer. 2011;11:58. doi: 10.1186/1471-2407-11-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Ruan Q., Hilliard B., Devirgiliis J., Karin M., Chen Y. H. Transcriptional regulation of the Th17 immune response by IKK(alpha) J. Exp. Med. 2011a;208:787–796. doi: 10.1084/jem.20091346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Sun L., Zhang Y., Wang D., Wang F., Liang J., Gui B., Shang Y. The histone modifications governing TFF1 transcription mediated by estrogen receptor. J. Biol. Chem. 2011b;286:13925–13936. doi: 10.1074/jbc.M111.223198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Whitlock J. P. J. The aromatic hydrocarbon receptor modulates the Hepa 1c1c7 cell cycle and differentiated state independently of dioxin. Mol. Cell. Biol. 1996;16:2144–2150. doi: 10.1128/mcb.16.5.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manikkam M., Tracey R., Guerrero-Bosagna C., Skinner M. K. Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS One. 2012;7:e46249. doi: 10.1371/journal.pone.0046249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai M., Lin T. M., Peterson R. E., Cooke P. S., Tischkau S. A. Behavioral rhythmicity of mice lacking AhR and attenuation of light-induced phase shift by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Rhythms. 2008;23:200–210. doi: 10.1177/0748730408316022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert D. W., Dalton T. P., Okey A. B., Gonzalez F. J. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- Ovesen J. L., Schnekenburger M., Puga A. Aryl hydrocarbon receptor ligands of widely different toxic equivalency factors induce similar histone marks in target gene chromatin. Toxicol. Sci. 2011;121:123–131. doi: 10.1093/toxsci/kfr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. J., Yoon W. K., Kim H. J., Son H. Y., Cho S. W., Jeong K. S., Kim T. H., Kim S. H., Kim S. R., Ryu S. Y. 2,3,7,8-Tetrachlorodibenzo-p-dioxin activates ERK and p38 mitogen-activated protein kinases in RAW 264.7 cells. Anticancer Res. 2005;25:2831–2836. [PubMed] [Google Scholar]
- Puga A., Ma C., Marlowe J. L. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana F. J., Sherr D. H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013;65:1148–1161. doi: 10.1124/pr.113.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiners J. J., Jr, Lee J. Y., Clift R. E., Dudley D. T., Myrand S. P. PD98059 is an equipotent antagonist of the aryl hydrocarbon receptor and inhibitor of mitogen-activated protein kinase kinase. Mol. Pharmacol. 1998;53:438–445. doi: 10.1124/mol.53.3.438. [DOI] [PubMed] [Google Scholar]
- Robertson J. A., Hankinson O., Nebert D. W. Autoregulation plus positive and negative elements controlling transcription of the genes in the [Ah] battery. Chem. Scr. 1987;27A:83–87. [Google Scholar]
- Sassone-Corsi P., Mizzen C. A., Cheung P., Crosio C., Monaco L., Jacquot S., Hanauer A., Allis C. D. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285:886–891. doi: 10.1126/science.285.5429.886. [DOI] [PubMed] [Google Scholar]
- Schnekenburger M., Peng L., Puga A. HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation. Biochim. Biophys. Acta. 2007a;1769:569–578. doi: 10.1016/j.bbaexp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnekenburger M., Talaska G., Puga A. Chromium cross-links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol. Cell. Biol. 2007b;27:7089–7101. doi: 10.1128/MCB.00838-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibazaki M., Takeuchi T., Ahmed S., Kikuchi H. Blockade by SB203580 of Cyp1a1 induction by 2,3,7,8-tetrachlorodibenzo-p-dioxin, and the possible mechanism: Possible involvement of the p38 mitogen-activated protein kinase pathway in shuttling of Ah receptor overexpressed in COS-7 cells. Ann. N. Y. Acad. Sci. 2004a;1030:275–281. doi: 10.1196/annals.1329.034. [DOI] [PubMed] [Google Scholar]
- Shibazaki M., Takeuchi T., Ahmed S., Kikuchi H. Suppression by p38 MAP kinase inhibitors (pyridinyl imidazole compounds) of Ah receptor target gene activation by 2,3,7,8-tetrachlorodibenzo-p-dioxin and the possible mechanism. J. Biol. Chem. 2004b;279:3869–3876. doi: 10.1074/jbc.M305880200. [DOI] [PubMed] [Google Scholar]
- Soloaga A., Thomson S., Wiggin G. R., Rampersaud N., Dyson M. H., Hazzalin C. A., Mahadevan L. C., Arthur J. S. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W., Zhang J., Hankinson O. A mutation in the aryl hydrocarbon receptor (AHR) in a cultured mammalian cell line identifies a novel region of AHR that affects DNA binding. J. Biol. Chem. 1997;272:31845–31854. doi: 10.1074/jbc.272.50.31845. [DOI] [PubMed] [Google Scholar]
- Tan Z., Chang X., Puga A., Xia Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons: Role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochem. Pharmacol. 2002;64:5–6. 771–780. doi: 10.1016/s0006-2952(02)01138-3. [DOI] [PubMed] [Google Scholar]
- Tan Z., Huang M., Puga A., Xia Y. A critical role for MAP kinases in the control of Ah receptor complex activity. Toxicol. Sci. 2004;82:80–87. doi: 10.1093/toxsci/kfh228. [DOI] [PubMed] [Google Scholar]
- Tang D., Yuan H., Vielemeyer O., Perez F., Wang Y. Sequential phosphorylation of GRASP65 during mitotic Golgi disassembly. Biol. Open. 2012;1:1204–1214. doi: 10.1242/bio.20122659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teperek-Tkacz M., Meglicki M., Pasternak M., Kubiak J. Z., Borsuk E. Phosphorylation of histone H3 serine 10 in early mouse embryos: Active phosphorylation at late S phase and differential effects of ZM447439 on first two embryonic mitoses. Cell Cycle. 2010;9:4674–4687. doi: 10.4161/cc.9.23.14023. [DOI] [PubMed] [Google Scholar]
- Tiwari V. K., Stadler M. B., Wirbelauer C., Paro R., Schubeler D., Beisel C. A chromatin-modifying function of JNK during stem cell differentiation. Nat. Genet. 2012;44:94–100. doi: 10.1038/ng.1036. [DOI] [PubMed] [Google Scholar]
- Van H. A., Goodrich D. W., Allis C. D., Brinkley B. R., Mancini M. A. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J. Cell Sci. 1998;111(Pt 23):3497–3506. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- Wang S., Ge K., Roeder R. G., Hankinson O. Role of mediator in transcriptional activation by the aryl hydrocarbon receptor. J. Biol. Chem. 2004;279:13593–13600. doi: 10.1074/jbc.M312274200. [DOI] [PubMed] [Google Scholar]
- Wang S., Hankinson O. Functional involvement of the Brahma/SWI2-related gene 1 protein in cytochrome P4501A1 transcription mediated by the aryl hydrocarbon receptor complex. J. Biol. Chem. 2002;277:11821–11827. doi: 10.1074/jbc.M110122200. [DOI] [PubMed] [Google Scholar]
- Wells J., Farnham P. J. Characterizing transcription factor binding sites using formaldehyde crosslinking and immunoprecipitation. Methods. 2002;26:48–56. doi: 10.1016/S1046-2023(02)00007-5. [DOI] [PubMed] [Google Scholar]
- Winter S., Fischle W., Seiser C. Modulation of 14-3-3 interaction with phosphorylated histone H3 by combinatorial modification patterns. Cell Cycle. 2008;7:1336–1342. doi: 10.4161/cc.7.10.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q., Ohsako S., Baba T., Miyamoto K., Tohyama C. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on preimplantation mouse embryos. Toxicology. 2002;174:119–129. doi: 10.1016/s0300-483x(02)00047-1. [DOI] [PubMed] [Google Scholar]
- Wu Q., Ohsako S., Ishimura R., Suzuki J. S., Tohyama C. Exposure of mouse preimplantation embryos to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the methylation status of imprinted genes H19 and Igf2. Biol. Reprod. 2004;70:1790–1797. doi: 10.1095/biolreprod.103.025387. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Verma U. N., Prajapati S., Kwak Y. T., Gaynor R. B. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- Zhong S. P., Ma W. Y., Dong Z. ERKs and p38 kinases mediate ultraviolet B-induced phosphorylation of histone H3 at serine 10. J. Biol. Chem. 2000;275:20980–20984. doi: 10.1074/jbc.M909934199. [DOI] [PubMed] [Google Scholar]
- Zippo A., De R. A., Serafini R., Oliviero S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Cell Biol. 2007;9:932–944. doi: 10.1038/ncb1618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.