

Abstract
Cisplatin is effective against solid tumors including ovarian cancer. However, inherent or acquired cisplatin resistance limits clinical success. We recently demonstrated that a combination of sodium arsenite (NaAsO2) and hyperthermia sensitizes p53-expressing ovarian cancer cells to cisplatin by modulating DNA repair pathway and enhancing platinum accumulation. However, it is not understood how this combination therapy modulates cell cycle following platinum-DNA damage. The goal of the present study was to determine if NaAsO2 and hyperthermia alter cisplatin-induced G2 arrest and cause mitotic arrest and mitotic catastrophe. Human epithelial ovarian cancer cells (A2780 and A2780/CP70) were treated with cisplatin ± 20 μM NaAsO2 at 37 or 39°C for 1 h. Cisplatin ± NaAsO2 at 37 or 39°C caused cells to accumulate in G2/M compartment at 36 h after treatment. Western blot analysis of cyclin A and cyclin B suggested that combined NaAsO2, hyperthermia, and cisplatin induced mitotic arrest. However, we observed < 3% mitotic index and phosphorylation of histone H3 on serine 10 was undetectable. These results did not confirm mitotic arrest. BUBR1 (BUB1B) also was not phosphorylated, suggesting disrupted mitotic checkpoint. Postmitotic cells accumulated in pseudo-G1 as demonstrated by cyclin E stabilization, CDKN1A induction, and hypophosphorylation of retinoblastoma protein. These cells also were positive for Annexin V binding indicating they were apoptotic. In summary, cisplatin plus NaAsO2 and hyperthermia induced pseudo-G1 associated apoptosis in ovarian cancer cells.
Keywords: ovarian cancer, sodium arsenite, hyperthermia, cisplatin, pseudo-G1, postmitotic
Abbreviations
- CDK1
cyclin-dependent kinase 1
- CDKN1A
cyclin-dependent kinase inhibitor 1A
- CP
cisplatin
- CPA
cisplatin plus sodium arsenite
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- H3Ser10P
histone H3 phosphorylated on serine 10
- NaAsO2
sodium arsenite
- pRBS807/811P
phosphorylation of retinoblastoma protein on Ser807/811
- Pt
platinum
Research highlights:
Cisplatin plus NaAsO2 and hyperthermia induces pseudo-G1 associated apoptosis.
Cisplatin plus NaAsO2 and hyperthermia promotes endoreduplication and mitotic exit.
Cisplatin plus NaAsO2 and hyperthermia causes cells to accumulate in G2/M compartment.
Cisplatin, NaAsO2, and hyperthermia stabilize cyclin E and induce CDKN1A.
Cisplatin, NaAsO2, and hyperthermia suppress Rb phosphorylation.
Cisplatin and its analogues are widely used to treat ovarian, testicular, head and neck, bladder, gastric, and lung cancer (Blackhall and Faivre-Finn, 2011; Norman et al., 2010; Pan et al., 2009; Vasey et al., 1999; Winter and Albers, 2011). The cytotoxicity of cisplatin is mediated through DNA damage. Cisplatin primarily forms 1,2-intrastrand crosslinks between adjacent purines and to a lesser extent 1,3-intrastrand crosslinks, monoadducts, and interstrand crosslinks (Basu and Krishnamurthy, 2010). In response to DNA damage, cells arrest in G1 or G2 to allow time for damage repair before the onset of DNA synthesis or entry into mitosis, respectively (Malumbres and Barbacid, 2009; Rieder, 2011). In addition, the mitotic spindle assembly checkpoint regulates mitotic progression in response to chromosomal or spindle irregularities (Tan et al., 2005).
The tumor suppressor protein p53 regulates cell cycle checkpoints, DNA repair, and apoptosis after DNA damage (Abraham, 2001; Horvath et al., 2007). P53 regulates cell cycle checkpoints by inducing transcription of cyclin-dependent kinase inhibitor CDKN1A, GADD45, and 14–3–3 σ (Basu and Krishnamurthy, 2010; Taylor and Stark, 2001). CDKN1A causes G1 cell cycle arrest by binding and inactivating cyclin E/CDK2 complex, which is required for retinoblastoma protein phosphorylation in order to release E2F for G1 to S cell cycle progression. Cisplatin is known to cause G2 arrest (Brozovic et al., 2009; He et al., 2011; Horvath et al., 2007). CDKN1A blocks G2 to M transition by binding and inactivating cyclin B/CDK1 complex (Taylor and Stark, 2001).
Cisplatin is used clinically in combination with hyperthermia to treat ovarian cancer (Helm et al., 2008; Helm, 2009), but complete response is not achieved (Dovern et al., 2010). We previously showed that combining sodium arsenite and hyperthermia with cisplatin sensitizes wild-type p53 expressing ovarian cancer cells to cisplatin by attenuating xeroderma pigmentosum group C protein (XPC) induction and increasing cellular and DNA platinum accumulation (Muenyi et al., 2012). However, the mechanism by which sodium arsenite and hyperthermia affect cell cycle regulation following cisplatin-DNA damage has not been investigated.
Sodium arsenite causes mitotic arrest and mitotic arrest associated apoptosis (mitotic catastrophe) (McNeely et al., 2006, 2008a; Taylor et al., 2008). P53 has been implicated in sodium arsenite induced mitotic arrest. The presence of functional p53 promotes mitotic exit (McNeely et al., 2006; Taylor et al., 2006), whereas cells with nonfunctional p53 are more susceptible to sodium arsenite-induced mitotic arrest (Taylor et al., 2006). In addition, a functional spindle checkpoint is required for arsenite-induced mitotic arrest and apoptosis (McNeely et al., 2008b; Wu et al., 2008). Similar to sodium arsenite, hyperthermia has been reported to induce mitotic catastrophe in cancer cells (Grzanka et al., 2008; Michalakis et al., 2005; Nakahata et al., 2002). Furthermore, hyperthermia increases mitotic catastrophe induced by arsenite (Taylor et al., 2008). Therefore, we hypothesized that adding sodium arsenite and hyperthermia to the cisplatin treatment would sensitize ovarian cancer cells to cisplatin by enhancing mitotic arrest associated apoptosis.
The purpose of the present studies is to determine if adding sodium arsenite and hyperthermia to the cisplatin treatment alters cisplatin-induced G2 cell cycle arrest and causes mitotic arrest and mitotic arrest associated apoptosis (mitotic catastrophe) in ovarian cancer cells treated with cisplatin. We show that cisplatin, sodium arsenite, and hyperthermia failed to activate spindle assembly checkpoint protein BUBR1, promoted endoreduplication, and caused cells to exit mitosis without undergoing cytokinesis. Post mitotic cells accumulated in pseudo-G1 with 2C DNA content and subsequently underwent apoptotic cell death.
MATERIALS AND METHODS
Chemicals
RNase A, cisplatin, and sodium arsenite were purchased from Sigma-Aldrich (St. Louis, MO). Propidium iodide was purchased from Invitrogen/Life Technologies (Grand Island, NY). Stock solutions (cisplatin in PBS [1 mg/ml] and sodium arsenite in water [10 mM]) were prepared freshly on the day of treatment and filter sterilized (0.22 µm) prior to use. Trisenox (arsenic trioxide dissolved in 1 M NaOH) and sodium arsenite both generate the same reactive pharmacological species (As[OH]3) in solution (Ramirez-Solis et al., 2004). Sodium arsenite was used for our studies because it is readily soluble in aqueous solution.
Cells and cell culture
Human ovarian cancer cells A2780 and A2780/CP70 were the kind gift of Dr. Eddie Reed. A2780/CP70 cells are derived from A2780 cells by selection in successively increasing concentrations of cisplatn (Behrens et al., 1987). Thus, A2780/CP70 cells are cisplatin resistant and are a model for acquired resistance. Cells were cultured in RPMI media supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified 5% CO2 incubator.
Flow cytometry analyses
Cells (1 × 106) were cultured in 10 cm dishes overnight and treated with half-maximal inhibitory (IC50) cisplatin (A2780 = 4 μM and A2780/CP70 = 40 μM) ± 20 μM sodium arsenite at 37 or 39°C for 1 h. Cells were washed twice with PBS and refed with fresh media and incubated at 37°C. Whole cells were trypsinized and collected at 0 and 36 h, washed twice with PBS, and fixed in 70% ethanol overnight at 4°C. Cells in 500 μl PBS were incubated at 37°C with RNase A (100 U/ml) for 30 min. After adding propidium iodide (5 μg/ml), cells were incubated at room temperature for 30 min protected from light. Propidium iodide fluorescence (DNA content) was determined by flow cytometry using FACScalibur (BD Biosciences, San Jose, CA). A minimum of 10,000 cells/sample were analyzed. Data were collected and analyzed using FLOWJO software (FLOWJO, Ashland, OR).
Western blot analyses
Cells (1 × 106) were cultured in 10 cm dishes overnight and treated with IC50 cisplatin (A2780 = 4 μM and A2780/CP70 = 40 μM) ± 20 μM sodium arsenite at 37 or 39°C for 1 h. Cells were washed twice with PBS and refed with fresh media and incubated at 37°C. Total cellular lysates were prepared at 0 (immediately), 24, and 36 h after treatment. Cells were lysed with lysis solution (10 mM Tris-HCl pH 7.4, 1mM EDTA, 1% sodium dodecyl sulfate, 180 μg/ml phenylmethylsulphonylfluoride). After removal of debris by centrifugation at 13,000 × g for 45 min at 4ºC, total protein concentration in supernatant was determined by Bradford assay (Bio-Rad, Hercules, CA), using bovine serum albumin as standard. Proteins were loaded (30–40 μg/lane) and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electro-transferred to nitrocellulose membranes. Membranes were probed with mouse monoclonal antibodies for ß-actin (Sigma, no. A 5441, 1:10,000 dilution), GAPDH (Ambion, no. AM4300, 1:10,000 dilution), cyclin A (Cell Signaling, no. 4656, 1:1000 dilution), cyclin B (BD Biosciences PharMingen, no. 55477, 1:1000 dilution), histone H3 phosphorylated on Ser10 (histone H3S10P) (Cell Signaling, no. 9706S, 1:1000 dilution) and cyclin E (BD Biosciences PharMingen no. 51–1459GR, 1:1000 dilution) or rabbit polyclonal antibodies for CDKN1A (H-164, Santa Cruz, no. SC 756, 1:1000 dilution), CDK1 (Upstate Cell Signaling Solutions, no. 06–141, 1:1000 dilution), and retinoblastoma protein phosphorylated on serines 807 and 811 (pRbSer807/811P) (cell signaling, no. 9308, 1:1000 dilution). Secondary antibodies (rabbit anti-mouse IgG, no. 81–6720 or goat anti-rabbit, no. 81–6120) conjugated to horseradish peroxidase (Zymed Laboratories, Inc. South San Francisco, CA) were bound to primary antibodies and protein bands detected using enhanced chemiluminescence (ECL) substrate (Pierce, Rockford, IL). Bands for BUBR1, cyclin E, histone H3Ser10P, and GAPDH were visualized using ECL plus Western blotting detection system (GE Healthcare, RPN2132) and bands were developed using Molecular Dynamics Storm 860 (GE Healthcare BioSciences) in blue fluorescence mode. ß-actin and GAPDH served as the loading control.
Mitotic index determination
Cells (5 × 105/ 6 cm dish) were treated with IC50 cisplatin ± 20 μM sodium arsenite at 37 or 39°C for 1 h. The cell monolayers were washed twice with PBS and incubated in drug-free media for 36 h. Cells were washed twice with cold PBS, trypsinized using 1x trypsin and collected by centrifugation at 500 × g for 5 min. Cells were resuspended in 150 μl serum free media and 2.5 ml of 0.4% KCl was added. The cell suspension was incubated for 10 min at 37ºC. Methanol:acetic acid (3:1 vol/vol) fixation solution was added to 2% vol/vol and cells collected by centrifugation at 500 × g for 5 min. Cells were resuspended in 2.5 ml fixation solution and fixed at room temperature for 20 min. Samples were centrifuged at 500 × g for 5 min and pellets resuspended in 0.5 ml fixation solution and chilled on ice for a minimum of 20 min. Aliquots of the suspensions were dropped onto microscope slides, air dried for about 1 min, and stained with Wright Giemsa solution (States et al., 2002). Slides were examined under a microscope and a minimum of 200 nuclei were counted on each slide for determination of mitotic index and mitotic catastrophe index. Chromosomal spreads with sharp features were scored as mitotic nuclei (Taylor et al., 2006).
Annexin V-FITC apoptosis assay
Cells (5 × 105/ 6 cm dish) were treated with IC50 cisplatin ± 20 μM sodium arsenite at 37 or 39°C for 1 h. The cell monolayers were washed twice with PBS and incubated in drug-free media for 36 h. Cells were trypsinized using 1× trypsin and collected by centrifugation at 500 × g for 5 min. Cells were resuspended in 500 μl of 1X binding buffer and 5 μl of Annexin V-FITC and 5 μl of propidium iodide (50 μg/ml) were added. Samples were incubated at room temperature in the dark for 5 min. FITC Annexin V/propidium iodide binding was analyzed by flow cytometry (excitation = 488 nm; emission = 530 nm) using FACScalibur (BD Biosciences, San Jose, CA). Minimum of 10,000 cells/sample were analyzed. Annexin V assay was performed following manufacturers’ instructions (Cat no.: K101–25, BioVision Research Products, Mountain View, CA).
Statistical analysis
Results were expressed as the mean ± SD of replicate experiments as indicated in figure legends. Statistical analyses were performed using one-way analysis of variance (ANOVA) and Student's t-test or Tukey's test with a significance level as p < 0.05, n = 3.
RESULTS
Flow Cytometry Determination of Cell Cycle Arrest
Cisplatin is a DNA damaging agent. Cellular response to DNA damage involves cell cycle arrest to allow time to repair damaged DNA (Basu and Krishnamurthy, 2010). Cisplatin is known to cause G2 arrest (Cepeda et al., 2007). The goal of this experiment was to determine if sodium arsenite and hyperthermia cotreatment altered the accumulation of cells in the G2 compartment of the cell cycle following cisplatin treatment. Flow cytometry data indicate that both A2780 and A2780/CP70 cells accumulated in the G2/M compartment at 36 h after cisplatin treatment (Fig. 1). A2780/CP70 cells accumulated in the G2/M compartment to a greater extent than A2780 cells. Accumulation of cells in the G2/M compartment was not altered by sodium arsenite and/or hyperthermia cotreatment with cisplatin.
FIG. 1.

Cell cycle analyses by flow cytometry. Plot of percentage of cells in each phase of the cell cycle. A2780 and A2780/CP70 cells were treated with their IC50 cisplatin (CP) (A2780, 4 μM; CP70, 40 μM) or CP plus 20 μM sodium arsenite at 37 or 39°C for 1 h. Cells were harvested at 0 and 36 h after treatment. DNA content was analyzed by flow cytometry after propidium iodide staining. Data are the means ± SD of samples from two independent experiments. Each experiment was performed in duplicate dishes. Statistical analysis was performed using one-way ANOVA and Student's t-test. p < 0.05, * = compared with G2/M partners.
Sodium Arsenite and Hyperthermia Cause Mitotic Arrest in Cisplatin Treated Ovarian Cancer Cells
Flow cytometry determination of DNA content using propidium iodide does not distinguish between G2 and M cells because these cells both have double the normal (2C) DNA content. In order to determine if cells are in the G2 or M phase of the cell cycle at 36 h after treatment, the expression of cyclins A and B and cyclin-dependent kinase CDK1 were determined. Furthermore, we determined if sodium arsenite and hyperthermia cotreatment altered the expression of cyclins A and B and CDK1 in response to cisplatin treatment. G2 to M progression requires degradation of cyclin A and accumulation of cyclin B (Malumbres and Barbacid, 2009). Data in Figure 2 indicate that cisplatin treatment at 37°C stabilized CDK1, cyclin A and cyclin B (Fig. 2, panel a), suggesting G2 arrest. Adding hyperthermia to cisplatin decreased the levels of both cyclin A and cyclin B in A2780 cells suggesting G1 arrest. In contrast, cyclins A and B were stabilized suggesting G2 arrest in A2780/CP70 cells (Fig. 2, panel b). Cotreatment with cisplatin and sodium arsenite decreased both cyclin A and cyclin B in A2780 cells suggesting G1 arrest. In contrast, cyclin A was undetectable and cyclin B was stabilized, suggesting mitotic arrest in A2780/CP70 cells (Fig. 2, panel c). Combined cisplatin, sodium arsenite, and hyperthermia stabilized cyclin B and CDK1 but attenuated the expression of cyclin A in both cell lines at 36 h after treatment (Fig. 2, panel d), suggesting mitotic arrest. These data suggest that sodium arsenite ± hyperthermia induced mitotic arrest in cisplatin treated cells.
FIG. 2.

Western blot analyses of G2/M cell cycle regulatory proteins. Representative Western blots of cyclin A and B and CDK1. Cells were treated with their respective IC50 cisplatin (CP) (A2780, 4μM; CP70, 40μM) or CP plus 20μM sodium arsenite (CPA) at 37 or 39°C (hyperthermia) for 1 h, then washed with PBS and refed with fresh media and incubated at 37°C. Cell lysates were prepared at 0, 24, and 36 h. ß-actin is the loading control. Blots shown are representative of three independent experiments.
Sodium Arsenite and Hyperthermia Do Not Enhance Mitotic Index in Cisplatin Treated Cells and Also Fail to Induce Histone H3Ser10 Phosphorylation
Data in Figure 2 suggest that sodium arsenite plus hyperthermia is causing cisplatin treated cells to arrest in mitosis. In order to confirm if indeed these cells are in mitosis, we determined mitotic index as described in the Materials and Methods section. Adding sodium arsenite and/or hyperthermia to cisplatin did not increase the mitotic index in either A2780 or A2780/CP70 cells (Figs. 3A and B). The observed less than 3% mitotic index suggests that sodium arsenite ± hyperthermia do not induce mitotic arrest in cisplatin treated cells. In addition, histone H3 phosphorylation on serine 10 (H3Ser10P), a mitotic marker, was undetected in both A2780 and A2780/CP70 cells treated with cisplatin ± sodium arsenite at 37 or 39°C (Fig. 3C). These data confirmed that a large fraction of cells are not undergoing mitotic arrest.
FIG. 3.

Mitotic index determination and Western blot analysis of protein marker of mitosis. (A) Representative picture of (a) mitotic spread and (b) interphase nuclei. (B) Plot of means of percent mitotic cells for duplicate slides. Cells were treated with their respective IC50 cisplatin (CP) (A2780, 4 μM; CP70, 40 μM) or CP plus 20 μM sodium arsenite (CPA) at 37 or 39°C (hyperthermia) for 1 h. Treated cells were washed with PBS and refed fresh media and incubated at 37°C for 36 h. Mitotic index was determined at 36 h after treatment. Data are single biological experiments performed in duplicate dishes. C. Western blot analysis of histone H3Ser10P. Protein lysates were prepared 36 h after treatment for Western blot analysis of histone H3Ser10P. Data are representative from duplicate biological experiments. A375 human melanoma cells were treated with 5μM sodium arsenite for 24 h served as positive control for mitotic cells (McNeely et al.,2008b). ß-actin served as loading control.
Cisplatin, Sodium Arsenite, and Hyperthermia Prevent BUBR1 Phosphorylation
A functional spindle assembly checkpoint is required for mitotic arrest (Tan et al., 2005; Wu et al., 2008; Zhou et al., 2002). The mitotic spindle checkpoint complex consists of MAD3/BUBR1, BUB3, and MAD2 (Tan et al., 2005). The mitotic spindle checkpoint induces mitotic arrest by inhibiting anaphase onset by associating with CDC20, a subunit of the anaphase promoting complex (APC). Therefore, we determined if A2780 and A2780/CP70 cells have functional spindle checkpoint by determining the phosphorylation of BUBR1 following 100 nM taxol treatment for 16 h. We observed that taxol treatment induced BUBR1 phosphorylation in A2780 and A2780/CP70 cells (Fig. 4), suggesting that these cells have a functional spindle assembly checkpoint. However, cisplatin or its cotreatment with sodium arsenite and hyperthermia failed to induce BUBR1 phosphorylation (Fig. 4); suggesting that mitotic spindle checkpoint is either disrupted or not activated.
FIG. 4.
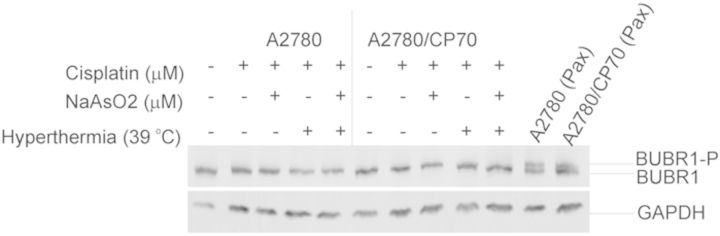
Western blot analysis of mitotic spindle assembly checkpoint protein. Western blot analysis of BUBR1 and phosphorylated BUBR1 in control lysates treated with 100nM paclitaxel 16 h. Cells were treated with their respective IC50 cisplatin (CP) (A2780, 4 μM; A2780/CP70, 40 μM) or CP plus 20 μM sodium arsenite (CPA) at 37 or 39°C (hyperthermia) for 1 h. Treated cells were washed with PBS and refed fresh media and incubated at 37°C. Protein lysates were prepared at 36 h after treatment for Western blot analysis. Cell treated with 100nM paclitaxel (Pax) as positive control. Data are representative of triplicate biological experiments. GAPDH served as loading control.
Cisplatin, Sodium Arsenite, and Hyperthermia Induced Pseudo-G1 Arrest in Ovarian Cancer Cells
Data in Figure 4 suggest that the mitotic spindle assembly checkpoint is not activated in A2780 or A2780/CP70 cells by any of the treatments. Absence of mitotic spindle checkpoint activation allows cells with damaged DNA to exit mitosis and progress through the cell cycle to G1. If cells exit mitosis without undergoing cytokinesis, they end up with 2C DNA content similar to G2/M cells (Lanni and Jacks, 1998), a phenomenon known as endoreduplication. Therefore, we determined if the cells accumulating in the G2/M compartment were pseudo-G1 cells by determining the expression of cyclin-dependent kinase inhibitor (CDKN1A), retinoblastoma protein phosphorylation on Ser807/811 (pRbSer807/811P), and cyclin E. Data in Figure 5 suggest that cisplatin stabilized CDKN1A over time and decreased pRbSer807/811P at 36 h after treatment (Fig. 5A panel a), suggesting G1 arrest. Adding sodium arsenite ± hyperthermia (Fig. 5A, panels c, b, and d, respectively) caused stronger CDKN1A induction and decreased levels of pRbSer807/811P compared with cisplatin alone at 37°C (Fig. 5, panels b, c, and d). These data confirmed that G1 arrest is taking place at 36 h after treatment. We also observed that cyclin E was stabilized in both A2780 and A2780/CP70 cells when compared with mitotic positive taxol treated A2780 and A2780/CP70 cells that did not express cyclin E (Fig. 5B). These data suggest that cisplatin ± sodium arsenite at 37 or 39ºC induces pseudo-G1 arrest in A2780 and A2780/CP70 cells.
FIG. 5.

Western blot analysis of protein markers of G1 arrest. A. Western blot analysis of CDKN1A and pRB Ser807/811P. Panel (a) is CP 37°C, (b) is CP 39°C, (c) is CPA 37°C, and (d) is CPA 39°C. B. Western blot analysis of cyclin E. Cells were treated with their respective IC50 cisplatin (CP; A2780, 4 μM; CP70, 40 μM) or CP plus 20 μM sodium arsenite (CPA) at 37 or 39°C (hyperthermia) for 1 h. Cells were washed with PBS and refed with fresh media and incubated at 37°C. Cell lysates were prepared at 0, 6, 12, 24, and 36 h. ß-actin and GAPDH are loading controls. A2780 and A2780/CP70 cells were treated with 100nM paclitaxel (Pax) for 16 h and served as negative control for cyclin E. Data are representative of triplicate biological experiments.
Cisplatin, Sodium Arsenite, and Hyperthermia Induce Apoptotic Cell Death in Pseudo-G1 Arrested Cells
We performed FITC Annexin V propidium iodide assay to determine if pseudo-G1 cells were undergoing apoptosis. Data in Figure 6 suggest that cisplatin alone or combined with sodium arsenite and/or hyperthermia significantly induced apoptotic cell death (∼15%) at 36 h after treatment.
FIG. 6.

Apoptotic cell death determination using FITC Annexin V propidium iodide assay. Cells were treated with their respective IC50 cisplatin (CP) (A2780, 4 μM; CP70, 40 μM), or CP plus 20 μM sodium arsenite (CPA) at 37 or 39°C (hyperthermia) for 1 h. Treated cells were washed with PBS and refed fresh media and incubated at 37°C for 36 h. Cell death was determined at 36 h after treatment. Data are means ± SD of triplicate biological experiments. Statistical analysis was performed using one-way ANOVA and Tukey test. p < 0.05, a = compared with untreated (UT) A2780 cells, b = compared with untreated A2780/CP70 cells.
DISCUSSION
Our previous studies demonstrated that combining sodium arsenite and hyperthermia with cisplatin sensitized ovarian cancer cells to the cisplatin treatment (Muenyi et al., 2012). The current study was aimed at determining how the cell cycle regulatory machinery responded to the combined treatment. We used well-characterized and widely used human ovarian cancer cell models for cisplatin-sensitive (A2780) and cisplatin-resistant subline (A2780/CP70) for this study. We showed that sodium arsenite and hyperthermia addition does not alter cisplatin-induced accumulation of cells in the G2/M compartment as determined by flow cytometry analysis of DNA content. However, examination of cell cycle regulatory protein expression indicated that these cells were not arrested in G2. Further investigation revealed that these cells with 2C DNA content also were not arrested in M phase and expressed proteins normally expressed in G1 arrested cells. Thus, a failure to activate the mitotic spindle assembly checkpoint in cells treated with cisplatin combined with sodium arsenite and/or hyperthermia allowed cells to exit mitosis and enter pseudo-G1 with 2C DNA content. These cells then underwent apoptotic cell death.
Standard chemotherapy for ovarian cancer includes treatment with both a taxane (e.g., paclitaxel) and platinum drugs (e.g., cisplatin or carboplatin). Cisplatin has long been known to induce G2 cell cycle arrest (Gibb et al., 1997). Arsenic induces mitotic arrest in a variety of tumor cell types (McNeely et al., 2008a,b; Taylor et al., 2008). Paradoxically, arsenic can suppress paclitaxel-induced mitotic arrest by causing arrest at other cell cycle checkpoints (Duan et al., 2009). Indeed, arsenic can cause delay at multiple points in the cell cycle (McCollum et al., 2005). Thus, it was of interest to investigate the impact of arsenic cotreatment with cisplatin to determine the effect on cell cycle checkpoints. Simple cell cycle analysis by DNA content (Fig. 1) indicated that all treatments caused an increase in the fraction of cells with 2C DNA content, normally interpreted as being in G2 or M phase. Therefore, we used Western blot analysis to determine the expression of G2/M regulatory proteins cyclin A and cyclin B. Cyclin A is usually degraded before cells enter mitosis, whereas cyclin B is stabilized during mitosis (Malumbres and Barbacid, 2009). Stabilization of cyclin A and cyclin B by cisplatin (CP 37°C) in both A2780 and A2780/CP70 cells suggests G2 arrest (Fig. 2, panel a), consistent with previous reports (Cepeda et al., 2007). However, the analysis revealed that cells cotreated with arsenite and/or hyperthermia clearly were not arrested in G2. Decreased expression of cyclin A and cyclin B by hyperthermia (CP 39°C) or arsenite cotreatment (CPA 37°C) with cisplatin in A2780 cells is consistent with G1 arrest (Fig. 2, panels b and c). However, arsenite cotreatment with cisplatin (CPA 37°C) decreased expression of cyclin A and stabilized cyclin B in A2780/CP70 cells (Fig. 2, panel c), consistent with mitotic arrest. Likewise, cotreatment with arsenite and hyperthermia (CPA 39°C) attenuated the expression of cyclin A and stabilized cyclin B in both cell lines (Fig. 2, panel d), consistent with mitotic arrest. However, despite the cyclin expression data suggesting mitotic arrest, mitotic arrest was not supported by mitotic index or histone H3Ser10P data (Figs. 3B and C). The lack of increased mitotic index in cisplatin treated cells and detectable histone H3 phosphorylation (H3Ser10P) clearly indicated that these cotreated cells are not arresting in mitosis.
Mitosis is the phase of the cell cycle where cells divide to produce two genetically identical cells from one cell. In order to ensure proper division, the mitotic spindle assembly checkpoint must ensure that all chromosomes are attached to the kinetochores by microtubules and that proper tension is exerted on the kinetochores before mitotic exit (Zhou et al., 2002). Mitotic arrest is a consequence of activation of the spindle assembly checkpoint. Cells with defective spindle assembly checkpoint will proceed through the cell cycle with aberrant chromosomes. The mitotic spindle assembly checkpoint complex consists of MAD3/BUBR1, BUB3, and MAD2 (Tan et al., 2005). The mitotic spindle assembly checkpoint inhibits anaphase onset by associating with CDC20, a subunit of the anaphase promoting complex., which is an E3-ubiqitin ligase that mediates degradation of securin and cyclin B. Degradation of securin and cyclin B is required for anaphase onset and subsequent mitotic exit. Treatment of A2780 and A2780/CP70 cells with a mitotic arrest inducing drug paclitaxel caused phosphorylation of BUBR1 in these cells (Fig. 4). Phosphorylation of BUBR1 indicates that these cells have functional spindle assembly checkpoint. However, cisplatin ± sodium arsenite treatment at 37 or 39°C failed to activate BUBR1 phosphorylation in ovarian cancer cells (Fig. 4). Disruption of mitotic spindle assembly checkpoint activation may allow cells to exit mitosis without undergoing cytokinesis (Lanni and Jacks, 1998). The resulting cells will enter pseudo-G1 with 2C DNA content.
In response to DNA damage, p53 is stabilized and activated and it transcriptionally activates CDKN1A expression. Induced CDKN1A binds to and inhibits CDK2/cyclin E complex, preventing the phosphorylation of retinoblastoma protein (pRb), thus blocking cells in G1. We observed strong induction of CDKN1A and suppression of pRbSer807/811P (Fig. 5A) at 36 h after treatment suggesting that G1 arrest is taking place. Accumulation of cells in G1 was supported by the stabilization of cyclin E (Fig. 5B). The data suggest that the cells accumulating in the G2/M compartment underwent endoreduplication and exited mitosis without undergoing cytokinesis and subsequently accumulated in pseudo-G1 with 2C DNA content. The phenomenon of cells with disrupted spindle assembly checkpoint undergoing endoreduplication and exiting mitosis without cytokinesis was reported by Lanni and Jacks (1998). These authors showed that when mouse embryo fibroblasts were treated with the spindle inhibitor nocodazole, the cells accumulated transiently in mitosis and progressed into G1 with 4N DNA content. These cells were classified as pseudo-G1 cells because they showed increased cyclin E and hypophosphorylated pRb, but had 4N DNA content similar to G2/M cells. Cells with disrupted spindle checkpoint are expected to be resistant to sodium arsenite induced mitotic arrest and apoptosis (Lanni and Jacks, 1998; Taylor and Stark, 2001). However, these pseudo-G1 cells underwent apoptotic cell death in response to cisplatin or its cotreatment with sodium arsenite and/or hyperthermia (Fig. 6).
CONCLUSIONS
We have demonstrated that cisplatin plus sodium arsenite at 37 or 39°C disrupts mitotic spindle assembly checkpoint activation and causes cells to exit mitosis without cytokinesis and subsequently accumulate in pseudo-G1. These pseudo-G1 cells with 2C DNA content stabilized cyclin E and induced CDKN1A and decreased pRbSer807/811P. In addition, these pseudo-G1 cells underwent apoptotic cell death. Thus, the mechanism by which cotreatment with arsenite and hyperthermia enhances cisplatin induced cell death is by disrupting the cell cycle regulatory machinery such that the G2 checkpoint and the spindle assembly checkpoint are bypassed and the cells accumulate in pseudo-G1 where they undergo apoptotic cell death. These findings suggest potential new mechanisms by which cisplatin in combination with sodium arsenite and hyperthermia induces cell death in wild-type p53 expressing ovarian cancer cells.
FUNDING
National Institutes of Health (grants P30ES014443, R01ES011314 to J.C.S.); dissertation completion award from University of Louisville (C.S.M.).
Supplementary Material
Acknowledgments
The authors thank Dr Chi Li for technical support with FACScalibur instrument and FlowJo software, Dr Hari Bodduluri for use of FACScalibur instrument, and Dr Jason Chesney for use of FlowJo software. Conflict of interest: The authors acknowledge that he/she had received a grant from the National Institutes of Health and the University of Louisville to do research in this area; the funding organizations do not have control over the resulting publication.
Footnotes
Present Address: Department of Biological Sciences, University of Memphis, Memphis, Tennessee 38152
Present Address: Department of Medicine, Washington University School of Medicine, 660 S. Euclid Ave., St Louis, Missouri 63110
REFERENCES
- Abraham R. T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Basu A., Krishnamurthy S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids. 2010;2010 doi: 10.4061/2010/201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens B. C., Hamilton T. C., Masuda H., Grotzinger K. R., Whang-Peng J., Louie K. G., Knutsen T., McKoy W. M., Young R. C., Ozols R. F. Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 1987;47:414–418. [PubMed] [Google Scholar]
- Blackhall F., Faivre-Finn C. Treatment of limited small cell lung cancer: An old or new challenge. Curr. Opin. Oncol. 2011;23:158–162. doi: 10.1097/CCO.0b013e328341ee4d. [DOI] [PubMed] [Google Scholar]
- Brozovic A., Damrot J., Tsaryk R., Helbig L., Nikolova T., Hartig C., Osmak M., Roos W. P., Kaina B., Fritz G. Cisplatin sensitivity is related to late DNA damage processing and checkpoint control rather than to the early DNA damage response. Mutat. Res. 2009;670:32–41. doi: 10.1016/j.mrfmmm.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Cepeda V., Fuertes M. A., Castilla J., Alonso C., Quevedo C., Perez J. M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007;7:3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- Dovern E., de Hingh I. H., Verwaal V. J., van Driel W. J., Nienhuijs S. W. Hyperthermic intraperitoneal chemotherapy added to the treatment of ovarian cancer. A review of achieved results and complications. Eur. J. Gynaecol. Oncol. 2010;31:256–261. [PubMed] [Google Scholar]
- Duan Q., Komissarova E., Dai W. Arsenic trioxide suppresses paclitaxel-induced mitotic arrest. Cell Prolif. 2009;42:404–411. doi: 10.1111/j.1365-2184.2009.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb R. K., Taylor D. D., Wan T., O’Connor D. M., Doering D. L., Gercel-Taylor C. Apoptosis as a measure of chemosensitivity to cisplatin and taxol therapy in ovarian cancer cell lines. Gynecol. Oncol. 1997;65:13–22. doi: 10.1006/gyno.1997.4637. [DOI] [PubMed] [Google Scholar]
- Grzanka D., Stepien A., Grzanka A., Gackowska L., Helmin-Basa A., Szczepanski M. A. Hyperthermia-induced reorganization of microtubules and microfilaments and cell killing in CHO AA8 cell line. Neoplasma. 2008;55:409–415. [PubMed] [Google Scholar]
- He G., Kuang J., Khokhar A. R., Siddik Z. H. The impact of S- and G2-checkpoint response on the fidelity of G1-arrest by cisplatin and its comparison to a non-cross-resistant platinum(IV) analog. Gynecol. Oncol. 2011;122:402–409. doi: 10.1016/j.ygyno.2011.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm C. W. The role of hyperthermic intraperitoneal chemotherapy (HIPEC) in ovarian cancer. Oncologist. 2009;14:683–694. doi: 10.1634/theoncologist.2008-0275. [DOI] [PubMed] [Google Scholar]
- Helm C. W., Bristow R. E., Kusamura S., Baratti D., Deraco M. Hyperthermic intraperitoneal chemotherapy with and without cytoreductive surgery for epithelial ovarian cancer. J. Surg. Oncol. 2008;98:283–290. doi: 10.1002/jso.21083. [DOI] [PubMed] [Google Scholar]
- Horvath V., Soucek K., Svihalkova-Sindlerova L., Vondracek J., Blanarova O., Hofmanova J., Sova P., Kozubik A. Different cell cycle modulation following treatment of human ovarian carcinoma cells with a new platinum(IV) complex vs cisplatin. Invest. New Drugs. 2007;25:435–443. doi: 10.1007/s10637-007-9062-7. [DOI] [PubMed] [Google Scholar]
- Lanni J. S., Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol. Cell Biol. 1998;18:1055–1064. doi: 10.1128/mcb.18.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M., Barbacid M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- McCollum G., Keng P. C., States J. C., McCabe M. J., Jr. Arsenite delays progression through each cell cycle phase and induces apoptosis following G2/M arrest in U937 myeloid leukemia cells. J. Pharmacol. Exp. Ther. 2005;313:877–887. doi: 10.1124/jpet.104.080713. [DOI] [PubMed] [Google Scholar]
- McNeely S. C., Belshoff A. C., Taylor B. F., Fan T. W., McCabe M. J., Jr, Pinhas A. R., States J. C. Sensitivity to sodium arsenite in human melanoma cells depends upon susceptibility to arsenite-induced mitotic arrest. Toxicol. Appl. Pharmacol. 2008a;229:252–261. doi: 10.1016/j.taap.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeely S. C., Taylor B. F., States J. C. Mitotic arrest-associated apoptosis induced by sodium arsenite in A375 melanoma cells is BUBR1-dependent. Toxicol. Appl. Pharmacol. 2008b;231:61–67. doi: 10.1016/j.taap.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeely S. C., Xu X., Taylor B. F., Zacharias W., McCabe M. J., Jr, States J. C. Exit from arsenite-induced mitotic arrest is p53 dependent. Environ. Health Perspect. 2006;114:1401–1406. doi: 10.1289/ehp.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalakis J., Georgatos S. D., Romanos J., Koutala H., Georgoulias V., Tsiftsis D., Theodoropoulos P. A. Micromolar taxol, with or without hyperthermia, induces mitotic catastrophe and cell necrosis in HeLa cells. Cancer Chemother. Pharmacol. 2005;56:615–622. doi: 10.1007/s00280-005-1002-7. [DOI] [PubMed] [Google Scholar]
- Muenyi C. S., Pinhas A. R., Fan T. W., Brock G. N., Helm C. W., States J. C. Sodium arsenite +/- hyperthermia sensitizes p53-expressing human ovarian cancer cells to cisplatin by modulating platinum-DNA damage responses. Toxicol. Sci. 2012;127:139–149. doi: 10.1093/toxsci/kfs085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata K., Miyakoda M., Suzuki K., Kodama S., Watanabe M. Heat shock induces centrosomal dysfunction, and causes non-apoptotic mitotic catastrophe in human tumour cells. Int. J. Hyperthermia. 2002;18:332–343. doi: 10.1080/02656730210129736. [DOI] [PubMed] [Google Scholar]
- Norman G., Soares M., Peura P., Rice S., Suh D., Wright K., Sculpher M., Eastwood A. Capecitabine for the treatment of advanced gastric cancer. Health Technol. Assess. 2010;14(Suppl. 2):11–17. doi: 10.3310/hta14suppl2/02. [DOI] [PubMed] [Google Scholar]
- Pan Q., Gorin M. A., Teknos T. N. Pharmacotherapy of head and neck squamous cell carcinoma. Expert. Opin. Pharmacother. 2009;10:2291–2302. doi: 10.1517/14656560903136754. [DOI] [PubMed] [Google Scholar]
- Ramirez-Solis A., Mukopadhyay R., Rosen B. P., Stemmler T. L. Experimental and theoretical characterization of arsenite in water: insights into the coordination environment of As-O. Inorg. Chem. 2004;43:2954–2959. doi: 10.1021/ic0351592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder C. L. Mitosis in vertebrates: The G2/M and M/A transitions and their associated checkpoints. Chromosome. Res. 2011;19:291–306. doi: 10.1007/s10577-010-9178-z. [DOI] [PubMed] [Google Scholar]
- States J. C., Reiners J. J. Jr, Pounds J. G., Kaplan D. J., Beauerle B. D., McNeely S. C., Mathieu P., McCabe M. J. Jr. Arsenite disrupts mitosis and induces apoptosis in SV40-transformed human skin fibroblasts. Toxicol. Appl. Pharmacol. 2002;180:83–91. doi: 10.1006/taap.2002.9376. [DOI] [PubMed] [Google Scholar]
- Tan A. L., Rida P. C., Surana U. Essential tension and constructive destruction: The spindle checkpoint and its regulatory links with mitotic exit. Biochem. J. 2005;386(Pt 1):1–13. doi: 10.1042/BJ20041415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor B. F., McNeely S. C., Miller H. L., Lehmann G. M., McCabe M. J., Jr, States J. C. p53 suppression of arsenite-induced mitotic catastrophe is mediated by p21CIP1/WAF1. J. Pharmacol. Exp. Ther. 2006;318:142–151. doi: 10.1124/jpet.106.103077. [DOI] [PubMed] [Google Scholar]
- Taylor B. F., McNeely S. C., Miller H. L., States J. C. Arsenite-induced mitotic death involves stress response and is independent of tubulin polymerization. Toxicol. Appl. Pharmacol. 2008;230:235–246. doi: 10.1016/j.taap.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor W. R., Stark G. R. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- Vasey P. A., Paul J., Birt A., Junor E. J., Reed N. S., Symonds R. P., Atkinson R., Graham J., Crawford S. M., Coleman R., et al. Docetaxel and cisplatin in combination as first-line chemotherapy for advanced epithelial ovarian cancer. Scottish Gynaecological Cancer Trials Group. J. Clin. Oncol. 1999;17:2069–2080. doi: 10.1200/JCO.1999.17.7.2069. [DOI] [PubMed] [Google Scholar]
- Winter C., Albers P. Testicular germ cell tumors: Pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2011;7:43–53. doi: 10.1038/nrendo.2010.196. [DOI] [PubMed] [Google Scholar]
- Wu Y. C., Yen W. Y., Yih L. H. Requirement of a functional spindle checkpoint for arsenite-induced apoptosis. J. Cell. Biochem. 2008;105:678–687. doi: 10.1002/jcb.21861. [DOI] [PubMed] [Google Scholar]
- Zhou J., Yao J., Joshi H. C. Attachment and tension in the spindle assembly checkpoint. J. Cell Sci. 2002;115(Pt 18):3547–3555. doi: 10.1242/jcs.00029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.