

Abstract
Drug-induced phospholipidosis (PLD) is characterized by phospholipid accumulation within the lysosomes of affected tissues, resulting in lysosomal enlargement and laminar body inclusions. Numerous adverse effects and toxicities have been linked to PLD-inducing drugs, but it remains unknown whether drug-induced PLD represents a distinct toxicity or cellular adaptation. In silico and immortalized cellular models have been used to evaluate the PLD potential of new drugs, but these systems have some limitations. The aims of this study were to determine whether primary sandwich-cultured hepatocytes (SCH) can serve as a sensitive and selective model to evaluate hepatic drug-induced PLD, and to evaluate the impact of PLD on the uptake and biliary excretion of probe substrates, taurocholate (TC) and rosuvastatin (RSV). Rat SCH were cultured for 48 h with prototypic hepatic PLD-inducing drugs, amiodarone (AMD), chloroquine (CHQ), desipramine (DES), and azithromycin (AZI), as well as the renal PLD inducer gentamicin (GTM). LysoTracker Red localization and transmission electron microscopy indicated enlarged lysosomal compartments and laminar body inclusions in SCH treated with AMD, CHQ, DES, and AZI, but not GTM, relative to control. PLD resulted in a 51–92% decrease in the in vitro biliary clearance of both TC and RSV; the biliary excretion index significantly decreased for TC from 88 to 35–73%. These data suggested that PLD significantly reduced both organic anion transporting polypeptide-mediated uptake, and bile salt export pump-mediated biliary transport processes. The current study demonstrates that the rat SCH system is a promising model to study hepatic PLD in vitro. Altered hepatic transport of anionic substrates secondary to drug-induced PLD is a novel finding.
Abbreviations
- AMD
amiodarone
- AZI
azithromycin
- BCRP/Bcrp
breast cancer resistance protein
- BEI
biliary excretion index
- CADs
cationic amphiphilic drugs
- CHQ
chloroquine
- DES
desipramine
- GTM
gentamicin
- MRP/Mrp
multidrug resistance-associated protein
- OATP/Oatp
organic anion transporting polypeptide
- PLD
phospholipidosis
- RSV
rosuvastatin
- SCH
sandwich-cultured hepatocytes
- TC
taurocholate
Drug-induced phospholipidosis (PLD) manifests as lysosomal accumulation of phospholipids, resulting in development of concentric lamellar bodies visible via the gold standard method, electron microscopy (Reasor et al., 1989). Predictive computer models of PLD potential have suggested that most precipitant drugs are weakly basic and have amphiphilic features. These cationic amphiphilic drugs (CADs), including antiarrhythmics, antidepressants, and anti-infectives, are able to cross the lysosomal membrane where they become protonated at low physiologic pH (pH 3–5) and trapped due to an inability to exit the lysosomal compartment following protonation. Lysosomal trapping and PLD have been observed in many different tissue/cell types including lung, nerve, kidney, and liver cells (Kaloyanides and Pastoriza-Munoz, 1980; Logan et al., 2013; Ndolo et al. 2010; Oikawa et al., 2005; Reasor and Kacew, 1996; Ruben et al., 1989; Shahane et al., 2013). Several mechanisms have been proposed for CAD-induced PLD, including inhibition of lysosomal phospholipases, inhibition of H+-ATPases and direct binding to the phospholipids, resulting in inert phospholipid-drug complexes (Joshi et al., 1989; Reasor et al., 2006).
PLD has been associated with hepatic/alveolar fibrosis (amiodarone; AMD), renal tubular toxicity (gentamicin; GTM), and QT-wave prolongation secondary to inhibition of human ether-à-go-go-related gene (hERG) channels in the heart (tamoxifen) (Agoston et al., 2003; Kaloyanides and Pastoriza-Munoz, 1980; Oikawa et al., 2005; Sun et al., 2013; Xia et al., 2011). Accordingly, significant industrial and regulatory attention, including the formation of the Food and Drug Administration sponsored PLD Working Group in 2004, has been devoted to developing sensitive and specific models to detect PLD during preclinical drug development. In response, multiple computer, non-cell, and cell-based models have been evaluated. Computer models utilizing molecular structure, as well as non-cell-based and cell-based models focusing on altered gene expression or lysosomal uptake and retention of fluorescent probes (e.g., Nile red, LysoTracker Red), have shown modest sensitivity and specificity (Atienzar et al., 2007; Kazmi et al., 2013; Ndolo et al., 2010; Pelletier et al., 2007; Ploemen et al., 2004). Unfortunately, computer or non-cell based models may not be predictive of PLD induced by metabolites, and often fail to predict which tissue(s) will be affected due to the absence of relevant metabolizing enzymes and transport processes. Methods to visualize lysosomal compartments within the cell to screen for PLD potential are of particular interest to reduce the burden of confirmatory studies with electron microscopy.
Although most PLD-inducing drugs contain a cationic amphiphilic moiety, metabolically-deficient models may not accurately predict PLD liability of metabolites. Sandwich-cultured hepatocytes (SCH) are fully differentiated, express relevant metabolizing enzymes and hepatic transporters, and can be used to assess hepatic transport processes involved in the clearance of endogenous and exogenous substrates (Chandra et al., 2001; LeCluyse et al., 1996; Li, 1997; Liu et al., 1999; Swift et al., 2010). Therefore, this established system was selected for subsequent investigations to test the hypothesis that SCH represent a sensitive and selective model to screen for drug-induced hepatic PLD. Lysosomal dysfunction secondary to PLD may alter the cycling of transporters to and from the relevant membranes (basolateral or canalicular), resulting in altered hepatic transporter-mediated clearance of susceptible compounds.
The goal of the present study was to determine whether primary rat SCH can serve as a sensitive and selective model to evaluate hepatic drug-induced PLD and determine if PLD alters hepatic transport processes. Structurally diverse drugs that cause PLD to varying degrees by distinct mechanisms were selected for investigation. First, lysosomal localization was determined using LysoTracker Red. Second, the presence or absence of PLD was confirmed using transmission electron microscopy. Third, hepatic uptake by sodium-taurocholate cotransporting polypeptide (Ntcp) and organic anion transporting polypeptides (Oatps), and biliary excretion by the bile salt export pump (Bsep), multidrug resistance-associated protein 2 (Mrp2), and breast cancer resistance protein (Bcrp) were evaluated using taurocholate (TC) and rosuvastatin (RSV) as probes for Ntcp/Bsep and Oatps/Mrp2/Bcrp, respectively. Results demonstrated that rat SCH are a promising screening tool for drug-induced PLD.
MATERIALS AND METHODS
Chemicals and reagents
Dexamethasone, TC, AMD, chloroquine (CHQ), desipramine (DES), azithromycin (AZI), GTM, Hanks’ balanced salt solution (HBSS; standard buffer), Ca2+/Mg2+-free HBSS (Ca2+-free buffer), and thiazolyl blue tetrazolium bromide were purchased from Sigma-Aldrich (St. Louis, MO). RSV was purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). [3H]TC (1 mCi/ml; >97% purity) and [3H]RSV (1 mCi/ml; >97% purity) were purchased from PerkinElmer (Waltham, MA) and American Radiolabeled Chemicals (St. Louis, MO), respectively. Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, penicillin/streptomycin and insulin were purchased from Gibco (Grand Island, NY). Minimum essential medium, nonessential amino acids, and LysoTracker Red were purchased from Invitrogen (Carlsbad, CA). BioCoat culture plates, BioCoat glass inserts, Matrigel extracellular matrix, and insulin/transferrin/selenium culture supplement were purchased from BD Biosciences (Bedford, MA). All other chemicals and reagents were of analytical grade and readily available from commercial sources.
Animals
Male Wistar wild-type (WT) rats (250–350 g) from Charles River Labs (Wilmington, MA) were used as hepatocyte donors. Rats were allowed free access to water and food and were acclimated for a minimum of 1 week prior to experimentation. All animal procedures complied with the guidelines of the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill (Chapel Hill, NC). All procedures were performed while rats were under full anesthesia with ketamine/xylazine (140/8 mg/kg ip).
Culture of primary rat hepatocytes in sandwich configuration
Primary rat hepatocytes were isolated and cultured as described previously (Swift et al., 2010). Briefly, hepatocytes were seeded at a density of 1.75 × 106 cells/well onto six-well BioCoat plates. Approximately 24 h later, hepatocytes were overlaid with 2 ml of 0.25 mg/ml BD Matrigel basement membrane matrix in DMEM. Hepatocytes were cultured for 4 days to allow formation of canalicular networks before experimentation; medium was changed daily. For single (‘acute’) administration experiments, the 2–4 day feeding medium was not supplemented with DMSO or PLD-inducing drugs. For multiple (‘chronic’) administration experiments, the feeding medium used on days 2–4 (48 h) of culture was supplemented with 0.1% DMSO (vehicle control, used to solubilize PLD-inducing drugs), AMD (1, 10, or 50μM), CHQ (1 or 10μM), DES (1, 10, or 50μM), AZI (1, 10, or 50μM), or GTM (1mM). The concentrations of the PLD-inducing drugs selected for investigation were based on in vivo plasma concentrations and estimated hepatic concentrations (Micromedex, 2014).
MTT assay
Following incubation of rat SCH with the PLD inducers or vehicle control at 37°C for 48 h, the medium was aspirated, and 2 ml of 0.5 mg/ml thiazolyl blue tetrazolium bromide solution in plain DMEM was added. After 30 min, the medium was aspirated, and 1 ml of 0.04M HCl in isopropanol was added to solubilize the cells. Lysates were analyzed by absorbance at 570 and 650 nm. SCH treated for 48 h with 100μM CHQ and 0.1% DMSO served as positive and negative/vehicle controls, respectively.
LysoTracker Red localization in rat sandwich-cultured hepatocytes
On day 4, the medium from SCH treated with PLD-inducing drugs or vehicle control was aspirated and replaced with fresh medium containing 100nM LysoTracker Red. After 30 min at 37°C, wells were washed rapidly twice with warm Dulbecco's phosphate buffered saline, and both light and fluorescent (excitation/emission wavelengths of 577/590 nm) digital images were captured using a Zeiss Axiovert 200TV (Thornwood, NY) inverted phase contrast microscope. Images were analyzed using ImageJ software (free online at rsbweb.nih.gov).
Electron microscopy of rat sandwich-cultured hepatocytes
BioCoat glass inserts were placed in the BioCoat plates, and hepatocytes were cultured as described above. The medium was aspirated on day 4, and then SCH were fixed for 1 h with 2 ml of 2.5% glutaraldehyde/1% tannic acid in 0.1M sodium cacodylate (pH 7.3). The fixation solution was aspirated, and SCH were washed with 0.1M cacodylate solution three times for 5 min each. SCH were postfixed with 1% osmium tetroxide in 0.1M cacodylate for 20 min, then rinsed three times with distilled water and two times with 50% ethanol for 5 min each. SCH were stained further with 2% uranyl acetate in 50% ethanol for 30 min. Finally, the SCH were dehydrated with increasing concentrations of ethanol (two times with 70, 80, 90, and 95%, and then three times with 100%, for 5 min each), infiltrated with propylene oxide (1:1, 2:1, and 3:1 ratios of Epon 812 [Hatfield, PA] to propylene oxide for 1 h each), and finished with pure Epon for 1 h. The glass inserts were removed from the BioCoat plates, embedded in Epon 812, and allowed to cure at 60°C for at least 18 h. The glass inserts were dissolved with concentrated hydrofluoric acid and thinly sectioned on a Leica Ultracut ultramicrotome (Leica Microsystems, Buffalo Grove, IL). Sections were placed on copper grids and restained with uranyl acetate and lead citrate, after which digital images were captured using a Tecnai 12 electron microscope (Hillsboro, OR). Multiple SCH sections from each treatment were imaged using a Gatan Multiscan digital camera (Pleasanton, CA).
Probe substrate transport in rat sandwich-cultured hepatocytes
Determination of the uptake and biliary excretion of the probe substrates TC (Ntcp/Bsep) and RSV (Oatps/Mrp2/Bcrp) in rat SCH was described previously (Abe et al., 2009). Prior studies have established that these probes are metabolically stable in rat hepatocytes (Pfeifer et al., 2013; Schwarz et al., 1975). Briefly, day 4 SCH were washed twice with 2 ml of warm Ca2+-containing HBSS (standard; cells + bile) or Ca2+-free HBSS (cells) and then incubated with the same buffer for 10 min at 37°C. Subsequently, SCH were incubated with 1.5 ml of standard HBSS containing [3H]TC (1μM; 100 nCi/ml) or [3H]RSV (1μM; 100 nCi/ml) for 10 min. For acute dosing studies, PLD-inducing drugs (AMD 50μM, CHQ 10μM, DES 50μM, AZI 50μM, or GTM 1mM) or vehicle control (0.1% DMSO) also were added to the incubation medium along with the probe substrates. After 10 min, SCH were washed three times with ice-cold standard HBSS and lysed with 0.5% Triton X-100 in phosphate-buffered saline. Radioactivity in cell lysates was quantified by liquid scintillation spectroscopy (Packard Tricarb; PerkinElmer, Waltham, MA). Accumulation of TC and RSV was normalized to hepatocyte total protein using a BCA protein assay (Pierce Chemical, Rockford, IL). The biliary excretion index (BEI) and in vitro biliary clearance (CLbiliary; ml/min/kg) were calculated using B-CLEAR® technology (Qualyst Transporter Solutions, LCC, Durham, NC) based on the following equations:
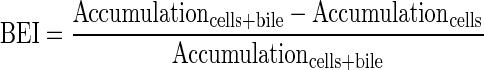 |
(1) |
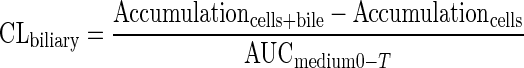 |
(2) |
where AUCmedium0−T represents the product of the incubation time (T) and the initial TC or RSV concentration in the medium (Abe et al., 2009; Pfeifer et al., 2013; Swift et al., 2010). CLbiliary units were converted to ml/min/kg body weight based on previously reported values for protein content in liver tissue (200 mg/g liver weight) and liver weight (40 g/kg body weight) (Davies and Morris, 1993).
Data analysis
The impact of chronic or acute incubation with PLD-inducing drugs on total (cells + bile) and cellular accumulation, BEI and CLbiliary was determined using one-way ANOVA with Dunnett's post hoc test. A p-value <0.05 was considered significant for all tests. All data are presented as mean ± standard error of the mean (SEM) unless otherwise indicated.
RESULTS
Rat Sandwich-Cultured Hepatocyte Toxicity
Initial pilot experiments showed that SCH incubated for 48 h with medium supplemented with 50–100μM CHQ exhibited complete toxicity based on the MTT assay. Thus, SCH incubated for 48 h with DMEM containing 100μM CHQ or 0.1% DMSO were selected as positive and negative (vehicle) controls, respectively. SCH treated with PLD-inducing drugs exhibited minimal to moderate toxicity (Fig. 1): 8–43% for AMD, 17–29% for CHQ, 2–55% for DES, 19–43% for AZI, and 26% for GTM. Toxicity increased as the dosing concentration increased for AMD, CHQ, DES, and AZI.
FIG. 1.

Toxicity of AMD, CHQ, DES, AZI, and GTM (incubation for 48 h) evaluated in day 4 rat SCH measured as percent inhibition of MTT formazan formation. SCH treated with CHQ at 100μM and 0.1% DMSO (vehicle) were used as the positive and negative controls, respectively. Data are presented as mean ± SEM, n = 3–7 replicates measured in triplicate.
LysoTracker Red Localization
LysoTracker Red localization was used to assess qualitatively the size and number of lysosomes in SCH treated with PLD-inducing drugs. Under standard conditions, LysoTracker Red (depicted as red/green fluorescence for contrast, Fig. 2) localized in vesicles close to the canalicular membrane. Exposure for 48 h to AMD, CHQ, DES, and AZI resulted in notably increased lysosomal size and/or number relative to control (Fig. 2), suggestive of PLD. Increased background fluorescence was observed only with CHQ treatment. Interestingly, GTM-treated SCH did not exhibit increased lysosomal number or size.
FIG. 2.
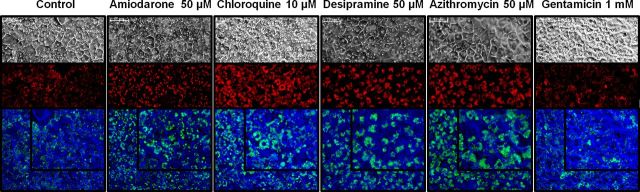
Representative images of day 4 rat SCH incubated for 30 min with 100nM LysoTracker Red following culture with PLD-inducing drugs or vehicle control (0.1% DMSO) for 48 h; only the highest incubation concentration is shown for clarity. Upper panels: light microscopy image; middle panels: LysoTracker Red fluorescence; lower panels: overlaid light and fluorescent images filtered to appear blue and green, respectively, for contrast (inset in upper right is enlarged image). Scale bar in upper left corner of each image represents 20 μm.
Transmission Electron Microscopy
All major cellular organelles including the nucleus, rough/smooth endoplasmic reticulum, Golgi apparatus, and tight junctions between hepatocytes were visualized in each SCH preparation. Inspection of SCH treated with 0.1% DMSO and 1mM GTM by electron microscopy showed a large number of mitochondria, glycogen granules, and small lysosomes; no pathologic lysosomes and only a few lipid droplets were observed. Inspection of SCH treated with AMD (50μM), CHQ (10μM), DES (50μM), and AZI (50μM) showed increased lipid droplet formation and enlarged lysosomal compartments with electron-dense deposits and membranous structures resembling laminar body inclusions, indicative of PLD (Fig. 3).
FIG. 3.

Representative electron micrographs of day 4 rat SCH after 48 h culture with PLD-inducing drugs or vehicle control (0.1% DMSO). Arrows indicate normal or enlarged laminar body-containing lysosomes. Scale bar represents 0.5 μm for all PLD-inducing drugs except control and gentamicin, for which the scale bar represents 2 and 1 μm, respectively.
Accumulation and Excretion of Taurocholate and Rosuvastatin in Rat SCH Following Acute (10-min) Exposure to PLD-Inducing Drugs
Total and cellular accumulation, BEI, and in vitro CLbiliary of TC and RSV were compared in SCH after acute exposure to PLD-inducing drugs. Following a 10-min incubation of 1μM [3H]TC or [3H]RSV with vehicle control (0.1% DMSO), AMD (50μM), CHQ (10μM), DES (50μM), AZI (50μM), or GTM (1mM), no significant differences in total accumulation (cells + bile) or cellular accumulation of TC or RSV were observed. Likewise, no statistically significant differences were observed for BEI or in vitro CLbiliary of either TC or RSV (Fig. 4).
FIG. 4.
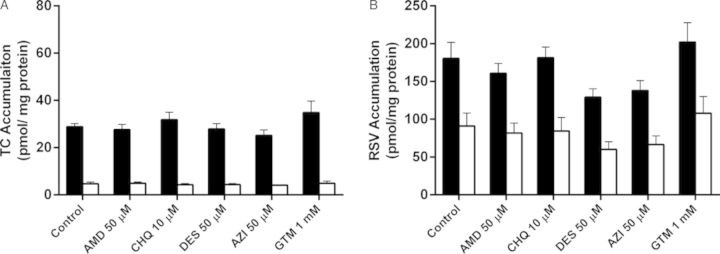
Accumulation of TC (A) and RSV (B) in cells + bile (black bars) and cells (white bars) of day 4 rat SCH following a 10-min co-incubation (“acute exposure”) with 1μM [3H]TC or [3H]RSV in the presence of vehicle control (0.1% DMSO) or PLD-inducing drugs: AMD, CHQ, DES, AZI, and GTM. Data represent the mean ± SEM of triplicate measurements in n = 3 hepatocyte preparations. No statistically significant differences were observed between control versus treated for total (cell + bile) accumulation or cellular accumulation.
Accumulation and Excretion of Taurocholate and Rosuvastatin in Rat SCH Following Chronic (48-h) Exposure to PLD-Inducing Drugs
Accumulation of [3H]TC and [3H]RSV in cells + bile and cells, as well as the in vitro CLbiliary, were compared between SCH treated for 48 h with vehicle control or PLD-inducing drugs following a 10-min incubation with 1μM [3H]TC or [3H]RSV. Total accumulation of TC (cells + bile) significantly decreased with increasing concentrations of AMD, CHQ, DES, and AZI (Fig. 5A). At the highest concentrations tested, TC total (cells + bile) accumulation decreased by 87.2 ± 1.8%, 71.8 ± 3.2%, 73.4 ± 0.9%, and 75.3 ± 3.6% compared with control for AMD, CHQ, DES, and AZI, respectively. TC BEI significantly decreased at the highest tested concentrations of AMD, DES, and AZI (Table 1). At the highest concentrations tested, the TC CLbiliary decreased by 92.3 ± 1.4%, 76.7 ± 3.4%, 81.0 ± 1.2%, and 89.3 ± 5.1% compared with control for AMD, CHQ, DES, and AZI, respectively. In contrast, TC cellular accumulation was not affected by any treatment (Fig. 5A).
FIG. 5.
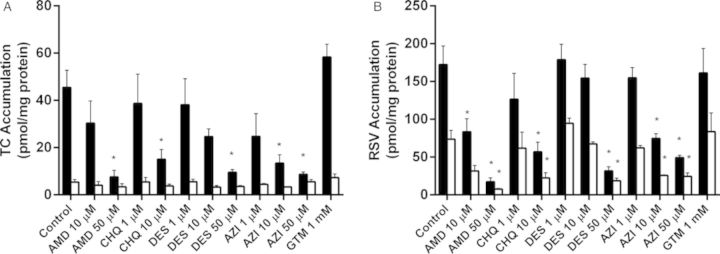
Accumulation of TC (A) and RSV (B) in cells + bile (black bars) and cells (white bars) of day 4 rat SCH following a 10-min incubation with 1μM [3H]TC or [3H]RSV. SCH were treated for 48 h (“chronic exposure”) with vehicle control (0.1% DMSO) or PLD-inducing drugs: AMD, CHQ, DES, AZI, and GTM. Data represent the mean ± SEM of triplicate measurements (n = 3–7 hepatocyte preparations); *p < 0.05, control versus treated.
TABLE 1. BEI and In Vitro CLbiliary of Taurocholate and Rosuvastatin Based on Data Presented in Figure 5 in Day 4 Rat SCH Following a 10-min Incubation With 1μM [3H]TC or [3H]RSV.
| Control | Amiodarone | Chloroquine | Desipramine | Azithromycin | Gentamicin | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10μM | 50μM | 1μM | 10μM | 1μM | 10μM | 50μM | 1μM | 10μM | 50μM | 1mM | ||
| Taurocholate | ||||||||||||
| BEI (%) | 88.3 ± 1.2 | 87.2 ± 0.8 | 52.9 ± 3.7* | 86.5 ± 1.9 | 73.4 ± 4.0 | 84.5 ± 1.5 | 87.0 ± 1.7 | 62.0 ± 3.2* | 75.3 ± 10.5 | 72.8 ± 4.7 | 34.8 ± 13.0* | 87.8 ± 1.5 |
| CLbiliary (ml/min/ kg) | 31.7 ± 5.0 | 20.8 ± 6.3 | 3.3 ± 1.2* | 26.3 ± 8.4 | 9.0 ± 2.9* | 25.7 ± 7.9 | 16.9 ± 2.1 | 4.7 ± 0.7* | 16.1 ± 7.4 | 8.0 ± 2.6* | 2.5 ± 1.0* | 40.4 ± 3.2 |
| Rosuvastatin | ||||||||||||
| BEI (%) | 56.7 ± 3.6 | 62.8 ± 4.2 | 50.1 ± 9.3 | 53.2 ± 4.4 | 62.7 ± 7.8 | 46.0 ± 5.7 | 55.6 ± 3.7 | 41.3 ± 0.9 | 59.4 ± 2.9 | 64.9 ± 3.7 | 50.6 ± 6.7 | 50.3 ± 5.4 |
| CLbiliary (ml/min/ kg) | 78.5 ± 13.5 | 41.4 ± 8.6 | 7.7 ± 3.7* | 51.5 ± 10.5 | 27.6 ± 6.2* | 66.8 ± 13.8 | 69.2 ± 12.5 | 10.3 ± 1.6* | 73.4 ± 9.8 | 38.8 ± 5.4 | 19.6 ± 2.3* | 61.9 ± 6.0 |
Notes. SCH were treated for 48 h (“chronic exposure”) with vehicle control (0.1% DMSO) or PLD-inducing drugs. Data represent the mean with SEM in parentheses of triplicate measurements (n = 3–7 hepatocyte preparations).
*p < 0.05, control versus treated.
Similarly, total (cells + bile) accumulation of RSV significantly decreased with increasing concentrations of AMD, CHQ, DES, and AZI (Fig. 5B). At the highest concentrations tested, RSV total (cells + bile) accumulation decreased by 89.2 ± 0.4%, 58.8 ± 2.2%, 85.2 ± 2.5%, and 77.1 ± 0.7% compared with control for AMD, CHQ, DES, and AZI, respectively. In contrast to TC, cellular RSV accumulation decreased with increasing concentrations of AMD, CHQ, DES, and AZI (Fig. 5B). At the highest concentrations tested, RSV cellular accumulation significantly decreased by 89.5 ± 1.8%, 68.9 ± 4.3%, 76.8 ± 1.3%, and 69.4 ± 2.5% compared with control for AMD, CHQ, DES and AZI, respectively. CLbiliary of RSV decreased whereas BEI was unaffected by increasing concentrations of AMD, CHQ, DES, and AZI (Table 1). At the highest concentrations tested, RSV CLbiliary decreased by 88.6 ± 2.8%, 50.5 ± 6.1%, 89.7 ± 2.5% and 81.4 ± 2.2%, compared with control for AMD, CHQ, DES, and AZI, respectively. The kidney-specific PLD-inducer, GTM, had no effect on total accumulation, cellular accumulation, BEI or in vitro CLbiliary of TC or RSV (Fig. 5, Table 1).
DISCUSSION
The primary goal of this study was to determine whether rat SCH could be used to detect hepatic drug-induced PLD. In addition, studies were designed to assess whether lysosomal dysfunction altered vectorial transport of two metabolically stable probe compounds (Pfeifer et al., 2013; Schwarz et al., 1975), TC and RSV. Five structurally different prototypic PLD-inducing drugs with distinct mechanisms were selected: AMD, CHQ, DES, AZI, and GTM. All of these drugs are known to cause hepatic PLD, with the exception of GTM, which induces PLD in kidney at human-relevant physiologic concentrations. Maximal plasma concentrations (Cmax) following standard doses of AMD (∼4.3μM), CHQ (∼3.7μM), DES (∼1.9μM), AZI (∼1.1μM) and GTM (∼12μM) were considered in selection of the lowest concentration of PLD-inducing drugs (1μM) for investigation in the SCH system (Micromedex, 2014). Concentrations that approximated 10- and 50-fold above the Cmax values also were examined because these drugs accumulate in hepatocytes. The high concentration of GTM was selected as a negative control to demonstrate that hepatic PLD would not be detected, and no changes in transport of anionic substrates would be observed. Experiments with the well-established lysosomal imaging probe, LysoTracker Red, demonstrated that a 48-h exposure of rat SCH to each of the four hepatic PLD inducers resulted in a qualitative increase in the number and/or size of lysosomal compartments, indicative of PLD (Fig. 2). The increased background noted in SCH treated with CHQ likely was due to an increase in lysosomal pH associated with CHQ's distinct mechanism of action (inhibition of H+-ATPase) (Goldman et al., 2009; Ndolo et al., 2010). Nonetheless, distinct and enlarged lysosomal compartments could be visualized in SCH treated with CHQ. As anticipated, GTM had no effect on either lysosomal number or size (Fig. 2).
Transmission electron microscopy of rat SCH exposed for 48 h to PLD-inducing drugs enabled visualization of all major organelles; enlarged lysosomal compartments with laminar figures diagnostic of PLD were observed in SCH treated with all PLD-inducing drugs except for GTM, which had no effect (Fig. 3). These results suggest that LysoTracker Red may be a useful tool to screen novel drug candidates in SCH for hepatic PLD potential (Kazmi et al., 2013; Figs. 2 and 3). Transmission electron microscopy also showed that both the cellular and organelle membranes were grossly intact. Membrane disruption has been reported in rats treated with high doses of AMD (150 mg/kg body weight per day) for 3 weeks (Agoston et al., 2003). However, at the concentrations investigated for all PLD-inducing drugs, there was no evidence of membrane disruption. Therefore, the mitochondrial toxicity observed in the current work, as measured by MTT formazan formation, does not appear to be due to membrane disruption.
An additional benefit of the rat SCH model is the ability to assess transporter-mediated vectorial flux of compounds through the hepatocyte into bile. Two probe substrates were selected to evaluate distinct transporters on the sinusoidal (blood side) and canalicular (bile side) membranes of hepatocytes. TC is a metabolically stable bile acid that is transported efficiently into the hepatocyte via Ntcp, and excreted into bile via Bsep; RSV is taken up by Oatps and possibly by Ntcp, and excreted into bile by Mrp2 and Bcrp (Abe et al., 2009; Byrne et al., 2002; Huang et al., 2006; Jemnitz et al., 2010; Kitamura et al., 2008; Trauner and Boyer, 2003). Changes in cellular accumulation and biliary excretion of probe compounds were examined to determine potential transporter-mediated alterations due to ‘acute’ (10-min exposure with no pretreatment) and ‘chronic’ (48-h pretreatment) administration of PLD inducers (Figs. 4 and 5). Alterations due to PLD-inducing drugs might be attributed to acute effects, or due to indirect effects, which would be present only after chronic administration. Following acute administration of each PLD-inducing drug, no significant changes were observed in TC or RSV total accumulation, cellular accumulation, BEI or CLbiliary (Fig. 4, Table 1). However, following chronic administration, all drugs that induced PLD in SCH decreased CLbiliary of TC and RSV, whereas cellular accumulation was reduced only for RSV. Reduced total accumulation, BEI and CLbiliary of TC with no change in cellular accumulation (in other words, reduced excretion of TC into the bile compartment) secondary to PLD is consistent with canalicular transporter dysfunction, specifically reduced Bsep-mediated biliary excretion in the case of TC (Annaert and Brouwer, 2005). Decreased RSV total (cells + bile) and cellular accumulation as well as CLbiliary, but no change in BEI, is consistent with decreased Oatp-mediated uptake (Annaert and Brouwer, 2005). Importantly, following chronic GTM administration, PLD was not observed in rat SCH, and no differences in TC or RSV disposition were noted (Fig. 5 and Table 1). Taken together, results from the acute- and chronic-exposure experiments indicated that PLD leads to functional changes in hepatic Oatp- and Bsep-mediated uptake and excretion, respectively. Results also suggest that Ntcp function is maintained and that RSV biliary excretion, mediated by both Mrp2 and Bcrp, does not appear to be altered by PLD. Lysosomal-associated alterations in the disposition and toxicity of drugs have been reported previously. For example, the impact of lysosomal pH-dependent entrapment on the cytotoxicity of chemotherapeutics with and without susceptibility to lysosomal sequestration was examined (Ndolo et al., 2010). When pH-dependent lysosomal sequestration was inhibited by CHQ, cellular toxicity of the lysosomotropic chemotherapeutic agent was enhanced, whereas no effect on toxicity was observed for the non-lysosomotropic chemotherapeutic agent. Additionally, exacerbation of the PLD phenotype with sucrose increased the uptake and lysosomal sequestration of CHQ, which was not observed when PLD induction was inhibited by bafilomycin A1 (Zheng et al., 2011). Therefore, lysosomal enlargement and sequestration appear to alter active anionic transport processes based on the present results, as well as the intracellular disposition of lysosomotropic drugs (Ndolo et al. 2010; Zheng et al., 2011).
Some limitations are associated with this work. First, compared with many published studies using immortalized cell lines to screen PLD potential, fewer drugs have been studied in rat SCH. Although this model is lower throughput than typical immortalized cell line screens, development of hepatic-specific PLD with drugs that exhibited a range of hepatic induction potency was demonstrated (AMD and CHQ are strong inducers whereas DES and AZI are weaker hepatic PLD inducers). Additionally, the PLD-inducing drugs investigated in this study affect different organ systems in vivo and have diverse postulated induction mechanisms in vivo (i.e,. inhibition of phospholipid metabolism with AMD, inhibition of H+-ATPase with CHQ). The present data indicate that rat SCH are both a sensitive and specific model for identification of drugs that cause hepatic PLD. All drugs tested, including GTM, caused mild to moderate toxicity based on the MTT assay (Fig. 1). Although drug-induced PLD has not been associated directly with cellular toxicity, the altered anionic transport observed may be related to toxicity. Interestingly, moderate toxicity was observed with GTM although no PLD or change in TC or RSV transport was apparent. This supports the hypothesis that altered TC and RSV transport secondary to PLD are not likely caused solely by the mild to moderate toxicity observed. Additionally, preliminary data indicate that streptomycin, an aminoglycoside included in the feeding medium that is structurally similar to GTM, did not induce PLD at the low concentrations utilized (172nM).
Drug-induced PLD leads to impaired phospholipid storage, metabolism, and excretion (Reasor et al., 1989). Several model systems have been evaluated to identify PLD-inducing drugs as early as possible during drug development. As such, PLD potential often is identified during early preclinical development and is handled on a case-by-case basis depending on the severity and target organ(s) affected (Chatman et al., 2009). This case-based assessment poses challenges due to a limited understanding of the (presumed) association between PLD and organ toxicity. Consequently, increasing attention has been placed on developing sensitive and specific models to predict drug-induced PLD, thereby avoiding late-stage failures due to unanticipated toxicities. A report by leading investigators encourages selection of lead drug candidates based on in silico and/or in vitro screening for PLD potential (Chatman et al., 2009). Multiple in silico quantitative structure-activity relationship (QSAR) and in vitro non-cell-based and cell-based models have been developed (Kazmi et al., 2013; LeCureux et al., 2011; Pelletier et al., 2007; Ploemen et al., 2004; Shahane et al., 2013; Vitovic et al., 2008; Zhou et al., 2011). These screening methods have identified primarily CADs as causal agents, with a few notable exceptions (i.e., GTM). Although invaluable for initial risk-benefit analyses, these models have limitations. In vitro (and by inference in silico) models that lack metabolic and transport processes may underestimate PLD potential, as evidenced by the observation that isolated, metabolically active murine macrophages exhibited greater sensitivity and specificity to predict PLD in vivo than immortalized cell lines (LeCureux et al., 2011). Additionally, organ-specific transporters and metabolizing enzymes are germane to assess PLD potential in vitro as well as facilitate accurate in vivo extrapolation to target organs. SCH are a well-established system expressing hepatic-relevant metabolizing enzymes and transport proteins, which may aid in increasing assay sensitivity to identify drugs that require metabolism or transport to induce hepatic PLD (Swift et al., 2010).
In conclusion, rat SCH are a viable system to evaluate drug-induced hepatic PLD. Initial screening with LysoTracker Red localization would increase throughput. PLD screening should be evaluated in a metabolically capable cell-based model such as SCH, which provide the additional benefit that transport processes can be evaluated concurrently. The present data reveal for the first time that drug-induced hepatic lysosomal dysfunction may be associated with altered Oatp- and Bsep-mediated anionic transport. Clearly, PLD-inducing drugs may exhibit many toxicologic endpoints that require further preclinical evaluation.
FUNDING
National Institute of General Medical Sciences, National Institutes of Health (R01 GM41935 to K.L.R.B); Amgen Predoctoral Fellowship in Pharmacokinetics and Drug Disposition (to B.C.F.).
Acknowledgments
The authors thank Hal Mekeel for expert technical assistance with transmission electron microscopy preparation and imaging, and Dr Mary Paine for thoughtful comments and review of this manuscript. Conflict of interest: Dr Kim Brouwer is coinventor of the sandwich-cultured hepatocyte technology for quantification of biliary excretion (B-CLEAR®) and related technologies, which have been licensed exclusively to Qualyst Transporter Solutions. Dr. Brouwer chairs the Scientific Advisory Board for Qualyst Transporter Solutions.
Footnotes
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- Abe K., Bridges A. S., Brouwer K. L. R. Use of sandwich-cultured human hepatocytes to predict biliary clearance of angiotensin II receptor blockers and HMG-CoA reductase inhibitors. Drug Metab. Dispos. 2009;37:447–452. doi: 10.1124/dmd.108.023465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoston M., Orsi F., Feher E., Hagymasi K., Orosz Z., Blazovics A., Feher J., Vereckei A. Silymarin and vitamin E reduce amiodarone-induced lysosomal phospholipidosis in rats. Toxicology. 2003;190:231–241. doi: 10.1016/s0300-483x(03)00188-4. [DOI] [PubMed] [Google Scholar]
- Annaert P. P., Brouwer K. L. R. Assessment of drug interactions in hepatobiliary transport using rhodamine 123 in sandwich-cultured rat hepatocytes. Drug Metab. Dispos. 2005;33:388–394. doi: 10.1124/dmd.104.001669. [DOI] [PubMed] [Google Scholar]
- Atienzar F., Gerets H., Dufrane S., Tilmant K., Cornet M., Dhalluin S., Ruty B., Rose G., Canning M. Determination of phospholipidosis potential based on gene expression analysis in HepG2 cells. Toxicol. Sci. 2007;96:101–114. doi: 10.1093/toxsci/kfl184. [DOI] [PubMed] [Google Scholar]
- Byrne J. A., Strautnieks S. S., Mieli-Vergani G., Higgins C. F., Linton K. J., Thompson R. J. The human bile salt export pump: Characterization of substrate specificity and identification of inhibitors. Gastroenterology. 2002;123:1649–1658. doi: 10.1053/gast.2002.36591. [DOI] [PubMed] [Google Scholar]
- Chandra P., LeCluyse E. L., Brouwer K. L. R. Optimization of culture conditions for determining hepatobiliary disposition of taurocholate in sandwich-cultured rat hepatocytes. In Vitro Cell. Dev. Biol. Anim. 2001;37:380–385. doi: 10.1007/BF02577575. [DOI] [PubMed] [Google Scholar]
- Chatman L. A., Morton D., Johnson T. O., Anway S. D. A strategy for risk management of drug-induced phospholipidosis. Toxicol. Pathol. 2009;37:997–1005. doi: 10.1177/0192623309352496. [DOI] [PubMed] [Google Scholar]
- Davies B., Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- Goldman S. D., Funk R. S., Rajewski R. A., Krise J. P. Mechanisms of amine accumulation in, and egress from, lysosomes. Bioanalysis. 2009;1:1445–1459. doi: 10.4155/bio.09.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Wang Y., Grimm S. ATP-dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab. Dispos. 2006;34:738–742. doi: 10.1124/dmd.105.007534. [DOI] [PubMed] [Google Scholar]
- Jemnitz K., Veres Z., Tugyi R., Vereczkey L. Biliary efflux transporters involved in the clearance of rosuvastatin in sandwich culture of primary rat hepatocytes. Toxicol. In Vitro. 2010;24:605–610. doi: 10.1016/j.tiv.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Joshi U. M., Rao P., Kodavanti S., Lockard V. G., Mehendale H. M. Fluorescence studies on binding of amphiphilic drugs to isolated lamellar bodies: Relevance to phospholipidosis. Biochim. Biophys. Acta. 1989;1004:309–320. doi: 10.1016/0005-2760(89)90078-7. [DOI] [PubMed] [Google Scholar]
- Kaloyanides G. J., Pastoriza-Munoz E. Aminoglycoside nephrotoxicity. Kidney Int. 1980;18:571–582. doi: 10.1038/ki.1980.175. [DOI] [PubMed] [Google Scholar]
- Kazmi F., Hensley T., Pope C., Funk R. S., Loewen G. J., Buckley D. B., Parkinson A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells) Drug Metab. Dispos. 2013;41:897–905. doi: 10.1124/dmd.112.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S., Maeda K., Wang Y., Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 2008;36:2014–2023. doi: 10.1124/dmd.108.021410. [DOI] [PubMed] [Google Scholar]
- LeCluyse E. L., Bullock P. L., Parkinson A., Hochman J. H. Cultured rat hepatocytes. Pharm. Biotechnol. 1996;8:121–159. doi: 10.1007/978-1-4899-1863-5_9. [DOI] [PubMed] [Google Scholar]
- LeCureux L., Cheng C. S., Herbst J., Reilly T. P., Lehman-McKeeman L., Otieno M. Evaluation and validation of multiple cell lines and primary mouse macrophages to predict phospholipidosis potential. Toxicol. In Vitro. 2011;25:1934–1943. doi: 10.1016/j.tiv.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Li A. P. Primary hepatocyte cultures as an in vitro experimental model for the evaluation of pharmacokinetic drug-drug interactions. Adv. Pharmacol. 1997;43:103–130. doi: 10.1016/s1054-3589(08)60203-3. [DOI] [PubMed] [Google Scholar]
- Liu X., LeCluyse E. L., Brouwer K. R., Lightfoot R. M., Lee J. I., Brouwer K. L. R. Use of Ca2+ modulation to evaluate biliary excretion in sandwich-cultured rat hepatocytes. J. Pharmacol. Exp. Ther. 1999;289:1592–1599. [PubMed] [Google Scholar]
- Logan R., Kong A., Krise J. P. Evaluating the roles of autophagy and lysosomal trafficking defects in intracellular distribution-based drug-drug interactions involving lysosomes. J. Pharm. Sci. 2013;102:4173–4180. doi: 10.1002/jps.23706. [DOI] [PubMed] [Google Scholar]
- Micromedex. Greenwood Village, Colo: Thomson Reuters (Healthcare); 2014. Micromedex Version 2.0. [Google Scholar]
- Ndolo R. A., Forrest M. L., Krise J. P. The role of lysosomes in limiting drug toxicity in mice. J. Pharmacol. Exp. Ther. 2010;333:120–128. doi: 10.1124/jpet.109.160226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndolo R. A., Jacobs D. T., Forrest M. L., Krise J. P. Intracellular distribution-based anticancer drug targeting: Exploiting a lysosomal acidification defect associated with cancer cells. Mol. Cell. Pharmacol. 2010;2:131–136. doi: 10.4255/mcpharmacol.10.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa H., Maesawa C., Sato R., Oikawa K., Yamada H., Oriso S., Ono S., Yashima-Abo A., Kotani K., Suzuki K., et al. Liver cirrhosis induced by long-term administration of a daily low dose of amiodarone: A case report. World J. Gastroenterol. 2005;11:5394–5397. doi: 10.3748/wjg.v11.i34.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier D. J., Gehlhaar D., Tilloy-Ellul A., Johnson T. O., Greene N. Evaluation of a published in silico model and construction of a novel Bayesian model for predicting phospholipidosis inducing potential. J. Chem. Inf. Model. 2007;47:1196–1205. doi: 10.1021/ci6004542. [DOI] [PubMed] [Google Scholar]
- Pfeifer N. D., Bridges A. S., Ferslew B. C., Hardwick R. N., Brouwer K. L. R. Hepatic basolateral efflux contributes significantly to rosuvastatin disposition II: Characterization of hepatic elimination by basolateral, biliary, and metabolic clearance pathways in rat isolated perfused liver. J. Pharmacol. Exp. Ther. 2013;347:737–745. doi: 10.1124/jpet.113.208314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer N. D., Yang K., Brouwer K. L. R. Hepatic basolateral efflux contributes significantly to rosuvastatin disposition. I. Characterization of basolateral versus biliary clearance using a novel protocol in sandwich-cultured hepatocytes. J. Pharmacol. Exp. Ther. 2013;347:727–736. doi: 10.1124/jpet.113.207472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploemen J. P., Kelder J., Hafmans T., van de Sandt H., van Burgsteden J. A., Saleminki P. J., van Esch E. Use of physicochemical calculation of pKa and CLogP to predict phospholipidosis-inducing potential: A case study with structurally related piperazines. Exp. Toxicol. Pathol. 2004;55:347–355. doi: 10.1078/0940-2993-00338. [DOI] [PubMed] [Google Scholar]
- Reasor M. J., Kacew S. An evaluation of possible mechanisms underlying amiodarone-induced pulmonary toxicity. Proc. Soc. Exp. Biol. Med. 1996;212:297–304. doi: 10.3181/00379727-212-44019. [DOI] [PubMed] [Google Scholar]
- Reasor M. J., Hastings K. L., Ulrich R. G. Drug-induced phospholipidosis: Issues and future directions. Expert Opin. Drug Saf. 2006;5:567–583. doi: 10.1517/14740338.5.4.567. [DOI] [PubMed] [Google Scholar]
- Reasor M. J., Ogle C. L., Kacew S. Amiodarone-induced pulmonary toxicity in rats: Biochemical and pharmacological characteristics. Toxicol. Appl. Pharmacol. 1989;97:124–133. doi: 10.1016/0041-008x(89)90061-6. [DOI] [PubMed] [Google Scholar]
- Ruben Z., Dodd D. C., Rorig K. J., Anderson S. N. Disobutamide: A model agent for investigating intracellular drug storage. Toxicol. Appl. Pharmacol. 1989;97(1):57–71. doi: 10.1016/0041-008x(89)90055-0. [DOI] [PubMed] [Google Scholar]
- Schwarz L. R., Burr R., Schwenk M., Pfaff E., Greim H. Uptake of taurocholic acid into isolated rat-liver cells. Eur. J. Biochem. 1975;55:617–623. doi: 10.1111/j.1432-1033.1975.tb02199.x. [DOI] [PubMed] [Google Scholar]
- Shahane S. A., Huang R., Gerhold D., Baxa U., Austin C. P., Xia M. Detection of phospholipidosis induction: A cell-based assay in high-throughput and high-content format. J. Biomol. Screen. 2013;19:66–76. doi: 10.1177/1087057113502851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Xia M., Shahane S. A., Jadhav A., Austin C. P., Huang R. Are hERG channel blockers also phospholipidosis inducers. Bioorg. Med. Chem. Lett. 2013;23:4587–4590. doi: 10.1016/j.bmcl.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift B., Pfeifer N. D., Brouwer K. L. R. Sandwich-cultured hepatocytes: An in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab. Rev. 2010;42:446–471. doi: 10.3109/03602530903491881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M., Boyer J. L. Bile salt transporters: Molecular characterization, function, and regulation. Physiol. Rev. 2003;83:633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- Vitovic P., Alakoskela J. M., Kinnunen P. K. Assessment of drug-lipid complex formation by a high-throughput Langmuir-balance and correlation to phospholipidosis. J. Med. Chem. 2008;51:1842–1848. doi: 10.1021/jm7013953. [DOI] [PubMed] [Google Scholar]
- Xia M., Shahane S. A., Huang R., Titus S. A., Shum E., Zhao Y., Southall N., Zheng W., Witt K. L., Tice R. R., et al. Identification of quaternary ammonium compounds as potent inhibitors of hERG potassium channels. Toxicol. Appl. Pharmacol. 2011;252:250–258. doi: 10.1016/j.taap.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N., Zhang X., Rosania G. R. Effect of phospholipidosis on the cellular pharmacokinetics of chloroquine. J. Pharmacol. Exp. Ther. 2011;336:661–671. doi: 10.1124/jpet.110.175679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Geraci G., Hess S., Yang L., Wang J., Argikar U. Predicting phospholipidosis: A fluorescence noncell based in vitro assay for the determination of drug-phospholipid complex formation in early drug discovery. Anal. Chem. 2011;83:6980–6987. doi: 10.1021/ac200683k. [DOI] [PubMed] [Google Scholar]