

Abstract
Cytometric studies utilizing flow cytometry or multi-well culture plate fluorometry are often limited by a deficit in temporal resolution and a lack of single cell consideration. Unfortunately, many cellular processes, including signaling, motility, and molecular transport, occur transiently over relatively short periods of time and at different magnitudes between cells. Here we demonstrate the multitrap nanophysiometer (MTNP), a low-volume microfluidic platform housing an array of cell traps, as an effective tool that can be used to study individual unattached cells over time with precise control over the intercellular microenvironment. We show how the MTNP platform can be used for hematologic cancer cell characterization by measuring single T cell levels of CRAC channel modulation, non-translational motility, and ABC-transporter inhibition via a calcein-AM efflux assay. The transporter data indicate that Jurkat T cells exposed to indomethacin continue to accumulate fluorescent calcein for over 60 minutes after calcein-AM is removed from the extracellular space.
Hematologic cancer is a disease of single cells. From initial transformation to drug resistance, the progression of malignancy depends upon the survival and proliferation of individual cells with unique genotypes expressing environmentally dependent phenotypes1,2,3. Therefore, a reliable method for time-dependent analysis of patients' single cancer cells in vitro may enhance early cancer detection, refine neoplastic cell characterization, and enable chemotherapeutic treatment customization4. Flow cytometry can measure single cell fluorescence, internal complexity, and volume5, but it cannot measure time-dependent, transient cell responses to stimuli. Multi-well culture plates containing large populations of cells can be observed over time but, for non-adherent cells, altering the composition of the extracellular media requires cumbersome centrifugation and resuspension that may induce unintended changes in gene expression6. Flow cytometry can measure the fluorescence of one cell at one moment in time, and multi-well plate fluorometry can measure the fluorescence of a large population of cells over time, but neither can adequately perform both tasks simultaneously. Microfluidic cell trapping devices are often cited as a solution to this problem, but those designed with only one cell trap are constrained by low throughput7,8. Devices with multitudinous single cell traps, however, offer a powerful alternative to traditional cell culture and analysis9.
Our group has developed a microfluidic cell-trapping device and characterization protocol that is able to overcome conventional limitations on microenvironment control, time sensitivity, and single cell analysis in vitro10,11,12,13,14,15. The small fluid volumes and low cell numbers required to conduct a microfluidic experiment enable the interrogation of small samples with precise control over cellular exposures16,17,18. Although other microfluidic devices have been developed to trap individual cells for analysis9, here we demonstrate expansion of this technology to include a gradient across the trap array and temporal control of cell state, and hence present a comprehensive cancer cell analysis tool to characterize the behavior of new and existing cell lines and ultimately help provide customized cancer treatment in a clinical setting.
Our platform consists of a proven class of physio-mimetic polydimethylsiloxane (PDMS) devices specifically engineered with tri-walled, U-shaped cell traps to reproduce and manipulate the relationship of the cell with its microenvironment10,11,13,14,15. The devices are able to quickly and efficiently trap hundreds to thousands of individual unattached cells in dimensionally customizable arrays that allow for rapid multivariate control of the cellular microenvironment over time. The internal volume of each device ranges from 20–200 nL, and typical flow through a loaded device is measured in nL/min. PDMS transparency permits the manipulation of individual cells through optical trapping19. Based on the large number of traps with nanoscale volumes and the supportive growing environment within the arrays, we call our device the multitrap nanophysiometer (MTNP). By coupling MTNPs with time-lapse bright field and fluorescence differential image analysis20, we have created an excellent platform for characterizing and quantifying single cell growth, locomotion, signaling, protein expression and molecular transport mechanisms.
We designed three experiments, each measuring a different aspect of single cell dynamics, to demonstrate the utility of the MTNP platform as a single cell characterization tool for non-adherent, hematologic cancers. Each experiment was conducted using T cells, either immortalized or primary, to ensure consistency within and between experiments. The first study addresses the finding that many cancer cells exhibit altered intracellular calcium concentrations in comparison to non-malignant cells of the same lineage21,22,23. Accordingly, we show that the MTNP platform can accurately induce and record calcium changes in single primary human T cells in a cyclical manner over time. Second, we explicate a method to optically quantify non-translational cell motility in the form of amoeboid extensions and retractions, an important dimension of single cell analysis that can predict the response of leukemia cells to chemotherapy24, and show its utility in measuring Jurkat T lymphocyte response to temperature change and cytotoxic agents. Finally, we use the MTNP modified with a chemical gradient mixer to perform a calcein-AM efflux assay and pharmacokinetic modeling of Jurkat cells exposed to the efflux pump inhibitor indomethacin. The calcein-AM efflux assay is commonly used to determine the ability of a cancer cell to resist certain chemotherapeutic drugs25 and has been shown to predict therapy response and survival rate in human leukemia patients26. The resulting data illustrate the significant but rarely considered time sensitivity of this efflux assay and highlight the importance of characterizing each cell individually to avoid imprecise results. Together, these three experiments provide a broad wealth of multi-dimensional, quantifiable data that are collected simultaneously for hundreds of cells. Used in this way, the MTNP has the potential to help characterize a cancer's unique behaviors and susceptibilities, eventually resulting in customized patient treatment and optimized individual care. In Supplementary Material we present additional applications of the MTNP, including observation and quantification of cell division and growth, membrane labeling, and pinocytic loading.
Results
Device fabrication and experimental setup
Fabrication of the MTNP was carried out as described in the literature11,27 using conventional SU-8/PDMS microfabrication methods. The MTNP depicted in Fig. 1 is a configurable 20–200 nL microfluidic device containing hundreds to thousands of precisely positioned cell-sized traps approximately one cell diameter high with individual volumes of 0.2 to 6.5 pL. The trap array is supplied by multiple inputs either flowing directly into the trap chamber or combined through a gradient mixer for the purpose of producing a chemical gradient transverse to the direction of flow, so that cells in different traps in the device can be exposed to varying known concentrations of solution. Cells are randomly loaded into the trap chambers from bulk cell-containing mixtures including cell culture samples or blood15. Trap arrays fill to near 100% occupancy, with each trap containing the desired number of cells (determined by trap size) after 5–10 min of loading with a solution containing 1 × 106 T cells per mL. The microenvironment is altered rapidly and with excellent reproducibility by switching between input syringes loaded with prepared solutions. From fluid dynamics modeling we estimate that the time constant for switching the MTNP's cellular microenvironment to be 0.25 s in an empty trap and 1.0 s in a fully loaded device in which cells are occluding flow through the traps (Fig. 1c). Automated acquisition of bright field and multiple fluorescent cell images during computer-controlled perfusion, followed by offline image processing and analysis, results in hundreds to thousands of single cell responses to precisely timed stimulants in the microenvironment.
Figure 1. Physical and hydrodynamic features of the microfluidic multitrap nanophysiometer (MTNP).

(a) Schematic showing typical MTNP setup. Pumps supply fluid to the device from left to right through input ports P1–P3. The Pc ports are used for cell loading and to rapidly introduce solutions to the entire array by bypassing the gradient mixer. An optional gradient mixer (i) combines fluids in 10 discrete volumetric ratios before directing them to the trap chamber (ii). Several trap styles (t1–t3) and alternate whole device configurations (b) are easily implemented to accommodate diverse cell types and tissue quantities. (c) COMSOL Multiphysics simulation of time taken for perfusate concentration to reach 100% in the center of the trapping area relative to the trap chamber entrance. Each curve represents the space either directly outside and adjacent to the trap, shown in the inset by the “out” arrow, or within the walls of the trap with two cells present, shown by the “in” arrow, at a flow rate of either 200 nL min−1 or 1000 nL min−1.
CRAC channel cycling
Calcium is a potent modulator of intracellular activity and T cell activation28,29. Accordingly, intracellular calcium levels in T cells fluctuate rapidly under conditions of simulated stimulation11. Ordinarily, calcium is kept at low levels in the cytosol of the cell due to its sequestration by the endoplasmic reticulum (ER). Cytosolic calcium levels are increased by ionomycin-mediated release of calcium from ER stores, which is followed immediately by opening of membrane calcium release activated calcium (CRAC) channels30,31,32,33. The resulting increase in intracellular calcium levels acts to modulate cellular metabolism and homeostasis28. It is important to note that malignant T cells have been shown to exhibit a ten-fold increase in CRAC channel expression over non-cancerous cells34,35, indicating that intracellular calcium level is a critical, differential measurement to obtain in the characterization of any possibly neoplastic cell population.
We used the MTNP platform to quantify and model this cellular phenomenon by introducing rapid, cyclical media exposures to primary human T cells while observing CRAC channel activity, measured with the calcium-sensitive fluorescent indicator Fluo-3, in response to induced calcium signaling by the ionophore ionomycin. In our experiment, three syringes in three separate syringe pumps were loaded with either A) media with supplemental calcium, B) media with supplemental calcium and ionomycin, or C) low calcium media, and cyclically perfused the loaded MTNP in the forward (A⇒B⇒C) direction while fluorescence was monitored over time. An 85-trap array was used to trap one to seven cells per trap, with a mode of three cells per trap and a total of over 250 trapped cells.
As expected, the fluorescence of cells rapidly increased with simultaneous exposure to ionomycin and calcium (A⇒B) but did not intensify with exposure to calcium alone (C⇒A). Concurrent removal of both ionomycin and calcium (B⇒C) effectively quenched all fluorescence (Fig. 2a). Cyclic introduction and removal of ionomycin and calcium caused the fluorescence and cytosolic calcium concentrations of the cells in the MTNP to oscillate in phase while the frequency of modulation of the microenvironment was controllable within the limits of diffusion in the device. Using the resulting data, we have modeled free cytosolic calcium (CC) cycling over time as a response to ionomycin-mediated membrane permeabilization (Fig. 2b). CC is normally maintained at low concentrations (50–100 nM) by the actions of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump and plasma membrane ATPase (kPMCA) pump, which transport calcium into the ER and extracellular space, respectively. The ER has a constitutive leak into the cytosol that is normally compensated for by the SERCA pump. Ionomycin binds calcium and chauffeurs it across the ER and plasma membrane with diffusion constant kI, thereby affecting CC relative to the extracellular calcium concentration (Co). CC values exceeding a threshold concentration (CTH), caused by depletion of the ER, open plasma membrane CRAC channels and allow a calcium influx (kC) if Co is elevated, as diagrammed in Fig. 2c. The change in CC is represented by the differential equation
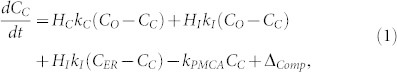 |
where Co and ionomycin are experimentally controlled, and their presence or absence is represented by the Heaviside functions HC (zero unless CC > CTH) and HI (zero unless ionomycin is present). CER is assumed to be a constant 600 μM. The initial rise in fluorescence that occurs before the first cycle is modeled as a permanent, finite compartment of capacity K for indicator-calcium conjugate with linear time dependence:
 |
There are a number of possible explanations for this rise, including equilibration with the calcium indicator acting as a buffer or mitochondrial loading of dye and calcium. Best-fit values for kC, kI, and kPMCA are 0.21 ± 0.04 min−1, 0.14 ± 0.01 min−1, and 4.03 ± 1.7 min−1, respectively. Overall the model, made possible by time-dependent data and precise media control in the MTNP, provides a detailed understanding of CRAC channel physiology and predicts the output parameters that one might expect to observe in a normal versus malignant T cell population.
Figure 2. Control and modeling of CRAC channels in individual T cells using extracellular Ca2+ and ionomycin.

(a) Depiction of 5 unique primary T cell responses to 4 induced calcium oscillations in the MTNP. Averaged values of all 5 cells are displayed in blue. Data were acquired at a rate of 1 image per min from randomly selected traps containing one cell and normalized to background illumination. A, B, and C show the composition of the perfusate flowing through the device at each point in time. A = media with supplemental calcium (Ca); B = media with calcium and ionomycin (IM); C = plain media. (b) Graphic rendering of the mathematical model in (c) superimposed on data from a single cell in experiment (a). (c) The model equations and schematic describe the change in free cytosolic calcium over time (dCC/dt), where the rate of ionomycin-mediated calcium diffusion (kI) is balanced by the cellular CRAC and PMCA calcium transporters pumping at rates kC and kPMCA, respectively. The rise in fluorescence before the first cycle is modeled as a permanent, finite compartment of capacity K for indicator-calcium conjugate with linear time dependence as defined in the integral. Best-fit parameters are reported in the text.
Cytokinetic toximetry and thermometry
Next we tested a non-invasive, non-fluorescent measurement to estimate cellular activity and energetics in response to external stimuli over time. Non-adherent cells, including the immortalized leukemic Jurkat T cell line, exhibit non-translational amoeboid motion in our traps that is evident in differential interference contrast or phase contrast time-lapse microscopy with acquisition periods of 20 s or less. The cells extend and retract membrane processes, often toward other cells, into the crevices in the trap structure or around the front of the trap side arms and into the faster flow streams of the device. Such motion is easily quantified using differential video microscopy20 and optical flow methods36,37,38,39.
To demonstrate the unique ability of the MTNP to measure this effect, we trapped Jurkat cells and perfused them with normal media for 245 min, followed by media containing 2% formaldehyde for 55 min. The non-translational motion of cells in the traps is evident in the bright field images (Fig. 3a) as well as the darker differential image frames (Fig. 3b). Integrated rectified difference (IRD) was calculated as the integration over x and y of the pixel-wise magnitude difference between successive images. The plot in Fig. 3c shows Jurkat cell IRD that has been divided by the IRD from an empty MTNP trap, sliding-window averaged over 10 frames and normalized so that average IRD during the first 45 min is unity. After 54 min, one of the Jurkat cells left the trap, causing a 41% decrease in IRD. The IRD of the remaining two cells stayed roughly constant until exposure to formaldehyde (a cell fixative) triggered a precipitous decrease in cell motion. This rapid decline in IRD was also seen with formaldehyde concentrations one and two orders of magnitude lower than in the data shown. A metabolic inhibitor with more subtle cytotoxic effects, NaF40, caused a dose-dependent decrease in IRD at 10–100 μM levels, with higher doses causing a faster IRD drop-off39.
Figure 3. Non-translational cell motion within the MTNP in response to noxious stimuli and temperature variation.

(a) Bright field image montage of trapped Jurkat T cells exhibiting non-translational motion over time. One cell exits the trap at 54 min (red arrow in 20 min frame). Final frame shows remaining two cells fixated in media containing 2% formaldehyde after 15 min of exposure. Scale bar, 20 μm. (b) Visual representation of non-translational motion by the integrated rectified differences (IRD) corresponding to the images in (a), calculated as the difference between the sum of all pixel intensities of adjacent images. (c) Plot of IRD vs. time for the entire stack of images represented by the subset above. Each data point was taken 20 s apart. Cell exits trap at vertical red line. Green vertical line indicates perfusion switch to formaldehyde. (d) Average IRD of a randomly selected sample of 5 cells from a population of Jurkat cells in the MTNP at several temperatures. The progression through 3 distinct temperatures (25°C, 37°C, 45°C) at 6 different times is shown. After temperature stabilization, data were collected every minute for 32 min at each temperature. Plotted are the IRD data of all trap regions of the MTNP normalized to IRD data of all non-trap regions (cellular IRD normalized to non-cellular IRD). Error bars represent a range of one standard deviation for the 32 mean data points at each temperature.
Cell motion in the MTNP as quantified by IRD measurements is sensitive to temperature in addition to the chemical composition of the media. We adjusted the temperature of the microscope incubator during an MTNP experiment to selected set points between 25°C and 45°C using a resistive heater and controller. The IRD of 5 cells randomly selected from a population of over 100 trapped Jurkat cells increased when temperature was raised and decreased when temperature was lowered (Fig. 3d). Cell motion at lower temperatures showed signs of hysteresis, as evidenced by heightened IRD after dropping the sample to 25°C from 37°C. The higher coefficient of variation in the data from temperature extremes (5.3% average) versus normal temperatures (2.5% average) suggests that the cells in the MTNP are actively adjusting to deviations from normal temperature, even over the course of the 32 min collection window. Therefore, assuming that IRD is reflecting changes in cellular activity caused by temperature change, it is likely that IRD may also reflect changes in cellular activity caused by drug exposure. Because cancerous cells respond to drugs and extracellular challenge with changes in motility24,41, we believe that simultaneous IRD quantification of non-translational motility using the MTNP platform will play a critical role in furthering our understanding of neoplastic single cell behavior and offer insight into the efficacy of potential cancer treatments.
Pharmacokinetic calcein-AM assay
Many cancer cells have high ATP-binding cassette (ABC) transporter activity and exhibit enhanced therapeutic resistance by effluxing chemotherapeutic drugs much faster than normal cells42. The substrates of two of these transporters, Pgp and MRP-1, include the hydrophobic chemotherapeutic drugs vincristine, doxorubicin, and etoposide43. Calcein acetoxymethyl ester (calcein-AM) shares substrate homology with these drugs and is used as a marker to test a cell's propensity for drug-extrusion resistance44. Calcein-AM is a hydrophobic molecule that passively crosses the cell membrane and fluoresces within the cytosol upon hydrolysis by intracellular esterases into the hydrophilic calcein-F45. Calcein-AM is a substrate of both PgP and MRP-1, while calcein-F, the form of the dye that remains trapped intracellularly, is only a substrate of MRP-146. Indomethacin, a non-steroidal anti-inflammatory drug (NSAID), has been shown to enhance chemotherapeutic drug toxicity through competitive inhibition MRP-1 efflux pumps47,48. Accordingly, our third experimental aim was to demonstrate how the MTNP platform can be used to characterize and model drug resistance and efflux inhibition efficacy in cancer cells by using it to perform a calcein-AM efflux assay in the presence of indomethacin.
We used the gradient mixer motif on the MTNP (Fig. 1a) to perfuse a trapped population of over 300 calcein-AM-loaded Jurkat cells with an indomethacin concentration gradient and measured the effect of varying drug concentrations on MRP-1 function. Fluorescence images (Fig. 4a) were acquired every 5 min and analyzed after subdivision into ten zones of increasing drug concentration. Ranging from 0–20 μM, each zone provided an indomethacin concentration 2.2 μM greater than the one below it and housed an average of 30 trapped cells. During initial calcein-AM loading in the MTNP, fluorescence of all live cells began to steadily increase at approximately the same rate. Once the calcein-AM syringe (Pc in Fig. 1) was stopped and the transverse indomethacin gradient was established (using P1 with 20 μM indomethacin and P3 with plain media), we observed markedly different trends of calcein-F fluorescence in each of the ten zones. Cells experiencing lower levels of indomethacin (0–6.7 μM) achieved peak fluorescence within 5 min of calcein-AM discontinuation and steadily lost fluorescence, likely due to continuous, uninhibited MRP-1 efflux of calcein-F from the intracellular space. Unexpectedly, at indomethacin concentrations of 8.9 μM and above, cell fluorescence continued to increase and, in the most inhibited cell zones, never peaked during the course of the 70 min indomethacin exposure (Fig. 4b). The results suggest that a significant amount of non-hydrolyzed calcein-AM remained trapped within the cell after blockade of MRP-1 pumps and removal of calcein-AM from the extracellular space. This residual calcein-AM, in an amount greater than that which would be expected to remain intracellularly under conditions of passive diffusion, would have been gradually converted to calcein-F over the course of the experiment in those zones with sufficiently blocked transporters.
Figure 4. Calcein-AM efflux assay in the presence of an MTNP-generated indomethacin gradient.

(a) Fluorescence image of trapped Jurkat T cells taken 90 min after start of experiment. For analysis, the trap chamber was divided into 10 equal-area radial zones based on laminar flow modeling, with each zone having 2.22 μM greater indomethacin concentration than the one below. Zone 10 contained a maximum indomethacin concentration of 20 μM. Zone 1 had no drug, only plain media. Scale bar, 110 μm. (b) Fluorescence intensity vs. time of cells exposed to the indomethacin gradient for 70 min after loading with calcein-AM dye for 20 min. Images were taken every 5 min. Fluorescence was normalized to background illumination. Z1–Z10 represent the average fluorescence in arbitrary units of all stably trapped cells in each zone with an average of 30 cells per zone. The 95% confidence interval was derived after zeroing all cell traces at t = 0 and is given for the three bolded zones (Z9, Z5, and Z3), although the values were similar for all zones. The maximum 95% confidence intervals of all 10 zones are equal to or less than those given for Z3. Z9* (green line) represents a single outlier cell trapped in zone 9 whose fluorescence was more than 4 standard deviations lower than the other cells in zone 9 for the majority of its data points. (c) Plot of fluorescent intensity vs. time derived from the model based on the cellular enzymatic and transport kinetics depicted in the inset. Here, CAM is intracellular calcein-AM concentration, k1 is the rate of calcein-AM uptake by cells, k2 is the rate of calcein-AM conversion into calcein-F (CF), and k3f and k4f are the rates of calcein-AM and calcein-F efflux from cells, respectively. Both k3f and k4f are decreased in the presence of indomethacin. Best-fit parameters are reported in the text.
Based on these findings, we created a calcein kinetics model to predict the intracellular levels of each molecular form in relation to efflux pump activity over time. A corresponding graph of the model outputs and a visual representation of the proposed cellular dynamics are shown in Fig. 4c. Here, calcein-AM in the media is taken up by the cells at rate k1, where it accumulates intracellularly with concentration CAM and is slowly diminished by conversion into intracellular calcein-F (CF) at rate k2. Uptake of calcein-AM is inversely proportional to the pre-existing intracellular concentration of calcein-AM, as would be expected with passive diffusion or facilitated transport mechanisms. Enzymatic conversion of calcein-AM to calcein-F is proportional to the amount of calcein-AM present. Calcein-AM is effluxed from the cell at rate k3. CF produced inside the cell by conversion from CAM is effluxed at rate k4. The model equations are
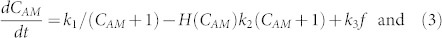 |
 |
where H(CAM) is the Heaviside function (0 unless CAM > 0). The rates of export of both CAM and CF are affected by the concentration of indomethacin (CI) outside the cell in a manner described by the Fermi-Dirac distribution (also called the Fermi function)
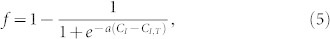 |
a sigmoidal function with slope parameter a and threshold CI,T. We constrain CAM to be non-negative. Best-fit values for k1, k2, k3, and k4 are 13.2 min−1, 0.041 min−1, 0.06 min−1, and 1.03 min−1, respectively. Fermi function values of CI,T and a are 9 and 0.55, respectively. The sigmoidal dependence is commensurate with the belief that indomethacin itself is exported from the cell47 and must achieve a threshold concentration before it can exert its competitive inhibition on the export machinery in the cytosol.
Notably, the dimensions of the MTNP traps in this experiment were specifically designed to trap only one cell per well, enabling fluorescence measurements of each cell individually over time. Although most cells followed the same general rate of change in fluorescence for their zone, one outlier cell in zone 9 (green trace) exhibited a fluorescence increase similar to that which would be expected from a cell in zone 5 (Fig. 4b). The majority of the outlier cell's data points are more than four standard deviations lower than the mean for zone 9, suggesting that this particular cell is able to extrude more or accumulate less fluorescence than its counterparts. Such variation highlights the necessity of examining cellular characteristics on the single cell level, especially in diseases like cancer where subtle up- and down-regulation of proteins in a small number of cells may mean the difference between treatment success and failure.
Discussion
We have shown that our microfluidic MTNP provides a broadly customizable platform for characterizing and quantifying the dynamic behavior of individual non-adherent cells as they respond to changes in the chemical microenvironment. The convection-diffusion system of the MTNP simultaneously guards cells from excessive shear and hydrodynamically maintains their close contact with precisely formulated convective flow streams, enabling high-bandwidth manipulations of the microenvironment of living single cells. Appropriate media formulations, flow rates, and experimental protocols have been determined that allow cells to grow and thrive in the physio-mimetic MTNP conditions where waste is constantly removed and nutrients are constantly replenished. Rapid, computer-controlled switching of media induces experimental perturbations of cells with drugs, toxins, dyes, and chemicals in a pre-determined, repeatable manner for high-content, unattended studies of biological cells, requiring only small quantities of starting material. Taking advantage of these capabilities, we have shown that the MTNP platform is a unique and versatile tool capable of characterizing hematologic cancer cell levels of calcium attenuation, cytoskeletal motility, and ABC-transporter activity in response to various stimuli. Clinically, these measures might help identify cancer cells, indicate prognosis, and predict drug resistance potential in a blood leukocyte sample from a leukemia patient.
Although the outcomes of the CRAC channel and optical motility experiments were predictable, the calcein data were unexpected. Specifically, to the best of our knowledge, the phenomenon of intracellular fluorescence steadily increasing in cells that were removed from calcein-AM in the presence of the MRP-1 inhibitor indomethacin has not previously been described in the literature. Our data suggest a calcein-AM trapping effect that only becomes apparent when the calcein-AM efflux pump, MRP-1, is sufficiently inhibited. This effect seems at odds with the currently held belief that calcein-AM rapidly diffuses across the cell membrane25. Passive diffusion down its concentration gradient would imply that calcein-AM diffuses out of the cell at a similar rate as that with which it is taken up after extracellular calcein-AM is removed. Our cells were incubated with calcein-AM for 20 min, but the effect of increasing intracellular fluorescence after extracellular calcein-AM was removed persisted for longer than 20 min in efflux-inhibited zones. This suggests that a complementary mechanism of calcein-AM uptake or retention, possibly active internal transport or intracellular protein binding, may exist alongside concentration-dependent passive diffusion.
Alternatively or additionally, calcein-AM may diffuse out of the cell at a faster rate than it diffuses into the cell. Intracellular calcein-AM concentration may not reach the level of that found outside the cell during the loading phase when calcein-AM transporters are uninhibited. Therefore, upon removal of extracellular calcein-AM, the concentration gradient across the cell membrane would be smaller in the reverse direction. Fick's law of diffusion states that the rate of diffusion is dependent upon the magnitude of the concentration gradient. A slower rate of outward diffusion may explain why calcein-AM appears to linger within cells beyond 20 min after extracellular calcein-AM removal. Clearly more research is necessary to fully understand the cause of this phenomenon and to confirm that it holds true for other MRP-1 inhibitors besides indomethacin.
Many researchers who utilize the calcein-AM assay do not measure the change in fluorescence over time with high enough temporal resolution to be able to account for the effect seen in our data. Studies often test inhibition of calcein efflux pumps by obtaining a single fluorescence data point for each cell via flow cytometry 60 or 90 min after removal of the dye46,49 or use multi-well plate fluorometers that take nearly 30 min to analyze all wells on a single culture plate25. If the time between ABC-transport inhibitor application and fluorescence measurement acquisition is not strictly controlled, the resulting proportional data may be deceptively inaccurate, overestimating the effect of a given MRP-1 inhibitor versus a non-inhibitor. Utilization of the MTNP platform in these types of experiments can help ensure the accuracy of ABC-transporter activity results.
The calcein data also emphasize the importance of analyzing every experimental cell individually, especially when assessing a cancer cell population for selectable drug resistance. The outlier cell in zone 9 would not have been noticed in a large multi-well culture and likely would have been discarded in a flow cytometry analysis as a deviant data point or one outside of the given fluorescence parameters. Yet the implications of this single cell's behavior are enormous. As noted, the cell fluoresces over four standard deviations less than the rest of the cells in zone 9 experiencing the same concentration of indomethacin-induced MRP-1 inhibition. The cause of this attenuated fluorescence may include decreased intracellular esterase activity, increased MRP-1 and/or Pgp activity, resistance to indomethacin-induced MRP-1 inhibition, or a combination of all three. Enhanced transporter activity in a cancer cell, especially a cancer stem cell, would spell a poor prognosis for drug treatment with substrates of that same transporter.
The ultimate advantage of the microfluidic MTNP platform is its broad customizability to diverse in vitro experimental requirements. The MTNP has tremendous potential to explore the poorly understood behaviors of heterogeneous cancer cells, but it also may be used in any study utilizing flow cytometry or multi-well plate culture to bolster the relevancy of outcomes with temporal and single cell-specific data. Single cell capture and analysis techniques are of growing interest50, and the multitrap nanophysiometer is an easily mastered and broadly applicable implementation11,13,14,15,19,51,52,53,54 that provides both dynamic control of cellular state and the quantitative measurements to specify parameters of mathematical rate models. Few other methods are able to manipulate and sustain dynamic microenvironments with such accuracy and ease of use. Single cells are highly complex, highly varied biological systems and require analytical technology of matching prowess in order to be fully understood.
Methods
Device fabrication
Fabrication of the MTNP and other microfluidic devices has been described elsewhere in detail11,12. Reusable masters were created by chrome masked UV photolithography of SU-8 on clean silicon wafers. The developed and hardened bas-relief structure on the master was transferred by replica molding of liquid polydimethylsiloxane (PDMS), which was cured at 60–65°C for several hours before being removed from the master. The solidified PDMS was peeled from the master, punched with access holes to accommodate intubation by input and output tubing, and plasma bonded to a glass slide, cover slip, or other PDMS.
Experimental setup
The MTNP was mounted in a small (50 mm/16 mL) Petri dish with a viewport and several access ports for media and gas tubing cut in the bottom. Moistened filter paper was used to surround the device and maintain ambient humidity near 100%. A 5% CO2, 95% air gas mixture was piped into the Petri dish from pressurized gas cylinders. The MTNP was supplied with one or more lengths of polyetheretherketone (PEEK) tubing attached to glass Hamilton syringes mounted on Harvard Apparatus Pico Plus syringe pumps, as described elsewhere15. All experiments were conducted at a flow rate of 200 nL min−1 unless stated otherwise.
Microscopy
Imaging of the experimental MTNP was carried out on a motorized, computer-controlled Zeiss Axiovert 200 confocal microscope. The microscope stage was completely enclosed within a Plexiglas incubator and maintained at a constant temperature with a controlled convective heating system. The microscope was controlled with MetaMorph software that also automated image acquisition and switching of the syringe pumps with customized serial communications sub-routines. Image focus was maintained with a MetaMorph autofocus procedure run at regular intervals. Bright field and fluorescence images were collected with a cooled CCD camera. The motorized stage was used to collect images from multiple fields of view within the MTNP trap chamber region. Images collected by the automated microscope were indexed, aligned55, and registered using ImageJ software before they were segmented to correct errors introduced by the filter sets of the microscope and motion artifacts.
Cell processing
Primary human T cells were obtained following isolation from human blood donors and thawed from liquid nitrogen aliquots 24 hours prior to use in RPMI with 10% FBS. Jurkat T cells were cultured in RPMI with 10% FBS and split every 2–3 days, according to standard ATCC culture methods. All cells used were below their ninth passage. The protocol for loading T cells was the same in all experiments and has been described in great detail elsewhere11,12,15. Briefly, after perfusing the device with media to remove all air bubbles, the PEEK tubing from the inlet was removed and placed in a suspension of cells diluted to 1 × 106 cells per mL media. Flow was reversed at 200 nL min−1 for 5–10 min, depending on trap array size, to aspirate cells. Flow was again reversed in the forward direction as PEEK tubing was reinserted into its MTNP, allowing cell expulsion and trapping at a flow rate that remained at 200 nL min−1 throughout the length of the experiment.
CRAC channel cycling
Primary human T cells were pre-incubated for 30 min with a calcium-sensitive fluorescent indicator (Fluo-3) in complete RPMI media before loading into the MTNP. Cells were then cyclically perfused with three media solutions prepared from base RPMI media: A) RPMI-C supplemented to 1.8 mM CaCl2 with no ionomycin, B) RPMI-C with 1 μM ionomycin supplemented to 1.8 mM CaCl2 or C) RPMI-C with neither ionomycin nor supplemental calcium. Perfusion (A⇒B⇒C), as shown in Fig. 2a, began with 18 min of A followed by 6 min B, 7 min C, and 7 min A, for a total of 4 complete automated cycles. Bright field and fluorescence images of several fields of view were collected every 60 s as the perfusate was switched. Fluorescence cell traces were extracted from the aligned and transformed image stacks in ImageJ by finding the desired trap or cell in the bright field images, extracting the pixel values of the same region in each fluorescence image, and repeating for each time point of the series. Background fluorescence was subtracted from the cell fluorescence, and final values were reported in arbitrary fluorescence units.
Toximetry and thermometry
Cytotoxicity and fixation experimental protocols were performed as described elsewhere39. Temperature set points within the microscope-mounted incubator were programmed and controlled using an AirTherm (World Precision Instruments, Sarasota, FL) heating unit. To compute IRD from a series of bright field images, the magnitude of the time derivative of the 8-bit gray scale images was calculated in ImageJ as the pixel-wise absolute difference between the Nth frame and the (N + j)th frame where j is an integer. If there was no motion between frames, the Nth and (N + j)th images were identical and all pixels of the difference image were zero. Gray pixels represented differences in the images, including those due to cell motion (device motion was compensated by image alignment). Integration of the absolute difference images over X and Y resulted in a single IRD value that was calculated at each time point for which a difference image was calculable. Background IRD calculated in areas with no cells was used to correct cellular IRD for artifacts such as changes in illumination intensity.
Pharmacokinetic calcein-AM assay
Jurkat cells were loaded according to the standard protocol into an MTNP modified with a gradient mixer, as depicted in Fig. 1. The gradient extremes (P1 and P3) were connected to syringes containing either 20 μM of indomethacin in RPMI 10% FBS or plain RPMI 10% FBS, respectively. Another syringe containing 2 μM calcein-AM in 1X DPBS was connected to a separate inlet (Pc) that pumped straight into the trap chamber. Bright field and fluorescence images of the trap chamber were taken every 5 min. COMSOL simulations were used to divide the trap chamber into the 10 distinct zones of indomethacin concentration provided by the MTNP gradient mixer for image analysis. In ImageJ, each zone of fluorescence was cut into its own separate image stack for alignment and brightness calibration. The “analyze particles” function was used to draw a region of interest (ROI) around each cell. All ROIs were expanded by the same small, non-overlapping amount to ensure that they would completely encompass the amoeboid movements of trapped cells from image to image. The mean gray value of each ROI was extracted for each time point, and the mean of these values for each zone is reported in Fig. 4b. Data points from cells that disappeared from view, contacted other cells, or lay on the border between zones were excluded from data analysis. Background fluorescence, subtracted from cell fluorescence, was obtained by averaging the mean gray values of three empty spaces outside the ROIs in the front, middle, and back of each trap zone. The numbers of data-producing cells in each zone were as follows: Z10 = 39, Z9 = 29, Z8 = 30, Z7 = 38, Z6 = 37, Z5 = 31, Z4 = 28, Z3 = 24, Z2 = 24, Z1 = 24.
Author Contributions
T.F.B. collected and analyzed the calcein-AM/indomethacin data, helped develop the calcein efflux model, and created the final manuscript and figures. L.T.H. conducted temperature-dependent cell motion experiments. E.G.K. performed IRD experiments including cellular fixation. M.E.P. performed CRAC channel experiments. K.T.S. supervised SyBBURE personnel, helped develop experimental concepts, derived mathematical models, provided imaging macros and wrote portions of the manuscript. E.M.W. provided COMSOL simulation data. J.P.W. guided the project, provided inspiration for each model, and contributed to development of the manuscript. All of these authors contributed to the editing of the manuscript. S.E.A., J.W.C., J.J.H., B.A.N., E.J.S., L.L.W. and J.S.W. were involved in the early stages of the development of the MTNP, contributed to the development of the experimental protocols, and conducted the experiments described in the Supplementary Material.
Supplementary Material
Supplementary Materials
Acknowledgments
We are indebted to Brian Koss and Wolfgang Losert for their work on developing optical trapping in the MTNP. We gratefully acknowledge contributions and assistance from Randall Peterson in the Department of Physics at Sewanee: The University of the South. Additionally, we would like to thank Dr. Dana Marshall at Meharry Medical College for her ongoing support. We also thank Matt Houston, Ronald S. Reiserer, David K. Schaffer, Philip C. Samson, Shannon Faley, Mary Marschner, Amy Ostrowski, Dalis Collins, Michael Irvin, Dr. Carl Johnson, and the students, staff, and faculty of the Systems Biology and Bioengineering Undergraduate Research Experience funded by Gideon Searle (SyBBURE Searle) at Vanderbilt University. This work was supported in part by SyBBURE Searle and the Vanderbilt Institute for Integrative Biosystems Research and Education (VIIBRE). J.P.W. acknowledges support from the National Institutes of Health (5U01AI061223-05 and, through the American Recovery and Reinvestment Act, 5RC2DA028981-01) and the Defense Threat Reduction Agency (HDTRAI-09-1-0013).
References
- Cohen Y., Gutwein O., Garach-Jehoshua O., Bar-Haim A. & Kornberg A. The proliferation arrest of primary tumor cells out-of-niche is associated with widespread downregulation of mitotic and transcriptional genes. Hematology Advance Article, 10.1179/1607845413Y.0000000125 (2013). [DOI] [PubMed] [Google Scholar]
- Steele A. J. et al. The JAK3-selective inhibitor PF-956980 reverses the resistance to cytotoxic agents induced by interleukin-4 treatment of chronic lymphocytic leukemia cells: Potential for reversal of cytoprotection by the microenvironment. Blood 116, 4569–4577 (2010). [DOI] [PubMed] [Google Scholar]
- Konopleva M., Tabe Y., Zeng Z. H. & Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: Mechanisms and approaches. Drug Resist. Update 12, 103–113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barteneva N. S., Ketman K., Fasler-Kan E., Potashnikova D. & Vorobjev I. A. Cell sorting in cancer research-diminishing degree of cell heterogeneity. BBA-Rev. Can. 1836, 105–122 (2013). [DOI] [PubMed] [Google Scholar]
- Herzenberg L. A. et al. The history and future of the fluorescence activated cell sorter and flow cytometry: A view from Stanford. Clin. Chem. 48, 1819–1827 (2002). [PubMed] [Google Scholar]
- Soto T. et al. Transduction of centrifugation-induced gravity forces through mitogen-activated protein kinase pathways in the fission yeast Schizosaccharomyces pombe. Microbiol. -SGM 153, 1519–1529 (2007). [DOI] [PubMed] [Google Scholar]
- Wheeler A. R. et al. Microfluidic device for single-cell analysis. Anal. Chem. 75, 3581–3586 (2003). [DOI] [PubMed] [Google Scholar]
- Werdich A. et al. A microfluidic device to confine a single cardiac myocyte in a sub-nanoliter volume on planar microelectrodes for extracellular potential recordings. Lab Chip 4, 357–362 (2004). [DOI] [PubMed] [Google Scholar]
- Di Carlo D., Wu L. Y. & Lee L. P. Dynamic single cell culture array. Lab Chip 6, 1445–1449 (2006). [DOI] [PubMed] [Google Scholar]
- Prokop A. et al. NanoLiterBioReactor: Long-term mammalian cell culture at nanofabricated scale. Biomed. Microdevices 6, 325–339 (2004). [DOI] [PubMed] [Google Scholar]
- Faley S. et al. Microfluidic platform for real-time signaling analysis of multiple single T cells in parallel. Lab Chip 8, 1700–1712 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faley S. L. et al. Microfluidic single cell arrays to interrogate signalling dynamics of individual, patient-derived hematopoietic stem cells. Lab Chip 9, 2659–2664 (2009). [DOI] [PubMed] [Google Scholar]
- Wlodkowic D., Faley S., Zagnoni M., Wikswo J. P. & Cooper J. M. Microfluidic single-cell array cytometry for the analysis of tumour apoptosis. Anal. Chem. 81, 5517–5523 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodkowic D., Faley S., Skommer J., McGuinness D. & Cooper J. M. Biological implications of polymeric microdevices for live cell assays. Anal. Chem. 81, 9828–9833 (2009). [DOI] [PubMed] [Google Scholar]
- Seale K. T., Faley S. L., Chamberlain J. & Wikswo J. P. Jr Macro to nano: A simple method for transporting cultured cells from milliliter scale to nanoliter scale. Exp. Biol. Med. 235, 777–783 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lion N., Reymond F., Girault H. H. & Rossier J. S. Why the move to microfluidics for protein analysis? Curr. Opin. Biotechnol. 15, 31–37 (2004). [DOI] [PubMed] [Google Scholar]
- Wikswo J. P. et al. Engineering challenges of BioNEMS: The integration of microfluidics, and micro- and nanodevices, models, and external control for systems biology. IEE Proc. -Nanobiotechnol. 153, 81–101 (2006). [DOI] [PubMed] [Google Scholar]
- Wikswo J. P. et al. Engineering challenges for instrumenting and controlling integrated organ-on-a-chip systems. IEEE Trans. Biomed. Eng. 60, 682–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S. et al. Automated cell transport in optical tweezers-assisted microfluidic chambers. IEEE T. Autom. Sci. Eng. 10, 980–89 (2013). [Google Scholar]
- Allen R. D. & Allen N. S. Video-enhanced microscopy with a computer frame memory. J. Microsc. 129 Pt.1, 3–17 (1983). [DOI] [PubMed] [Google Scholar]
- Durham A. C. H. & Walton J. M. Calcium-ions and the control of proliferation in normal and cancer-cells. Bioscience Rep. 2, 15–30 (1982). [DOI] [PubMed] [Google Scholar]
- Joseph P. et al. Cadmium-induced cell transformation and tumorigenesis are associated with transcriptional activation of c-fos, c-jun, and c-myc proto-oncogenes: Role of cellular calcium and reactive oxygen species. Toxicol. Sci. 61, 295–303 (2001). [DOI] [PubMed] [Google Scholar]
- Michel F. et al. Defective calcium response in B-chronic lymphocytic-leukemia cells - alteration of early protein tyrosine phosphorylation and of the mechanism responsible for cell calcium influx. J. Immunol. 150, 3624–3633 (1993). [PubMed] [Google Scholar]
- Forster A. H. et al. Use of moving optical gradient fields for analysis of apoptotic cellular responses in a chronic myeloid leukemia cell model. Anal. Biochem. 327, 14–22 (2004). [DOI] [PubMed] [Google Scholar]
- Ansbro M. R., Shukla S., Ambudkar S. V., Yuspa S. H. & Li L. W. Screening compounds with a novel high-throughput ABCB1-mediated efflux assay identifies drugs with known therapeutic targets at risk for multidrug resistance interference. PloS One 8, Article e060334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaszi E. et al. Calcein assay for multidrug resistance reliably predicts therapy response and survival rate in acute myeloid leukaemia. Br. J. Haematol. 112, 308–314 (2001). [DOI] [PubMed] [Google Scholar]
- Brittain S., Paul K., Zhao X. M. & Whitesides G. Soft lithography and microfabrication. Phys. World 11, 31–36 (1998). [Google Scholar]
- Fracchia K. M., Pai C. & Walsh C. M. Modulation of T cell metabolism and function through calcium signaling. Front. Immunol. 4, Article 324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A., Camacho P., Lechleiter J. D. & Herman B. Measurement of intracellular calcium. Physiol. Rev. 79, 1089–1125 (1999). [DOI] [PubMed] [Google Scholar]
- Dolmetsch R. E., Xu K. L. & Lewis R. S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936 (1998). [DOI] [PubMed] [Google Scholar]
- Lewis R. S. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 19, 497–521 (2001). [DOI] [PubMed] [Google Scholar]
- Luik R. M. & Lewis R. S. New insights into the molecular mechanisms of store-operated Ca2+ signaling in T cells. Trends Mol. Med. 13, 103–107 (2007). [DOI] [PubMed] [Google Scholar]
- Wu M. M., Luik R. M. & Lewis R. S. Some assembly required: Constructing the elementary units of store-operated Ca(2+) entry. Cell Calcium 42, 163–172 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschbaum H. H. & Cahalan M. D. Single-channel recording of a store-operated Ca2+ channel in Jurkat T lymphocytes. Science 283, 836–839 (1999). [DOI] [PubMed] [Google Scholar]
- Fomina A. F., Fanger C. M., Kozak J. A. & Cahalan M. D. Single channel properties and regulated expression of Ca2+ release-activated Ca2+ (CRAC) channels in human T cells. J. Cell Biol. 150, 1435–1444 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojima M., Tamagawa H., Hayashi N., Hanioka T. & Shizukuishi S. Semi-automated measurement of motility of human subgingival microflora by image analysis. J. Clin. Periodontol. 25, 612–616 (1998). [DOI] [PubMed] [Google Scholar]
- Ojima M., Tamagawa H., Nagata H., Hanioka T. & Shizukuishi S. Relation of motility of subgingival microflora as a clinical parameter to periodontal disease status in human subjects. J. Clin. Periodontol. 27, 405–410 (2000). [DOI] [PubMed] [Google Scholar]
- Harris M. P., Kim E., Weidow B., Wikswo J. P. & Quaranta V. Migration of isogenic cell lines quantified by dynamic multivariate analysis of single-cell motility. Cell Adh. Migr. 2, 127–136 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. G. R., Sivasubramaniam P., Wikswo J. P. & Seale K. T. Differential optical flow for automated cell motility. In: Micro Total Analysis Systems 2008: Proceedings of the μTAS 2008: Twelfth International Conference on Miniaturized Systems for Chemistry and Life Sciences, Locascio, L. E., ed. Chemical and Biological Microsystems Society, San Diego, CA, 1831–1833 (2008). [Google Scholar]
- Eklund S. E. et al. Metabolic discrimination of select list agents by monitoring cellular responses in a multianalyte microphysiometer. Sensors 9, 2117–2133 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J., Singer R. H. & Segall J. E. The great escape: When cancer cells hijack the genes for chemotaxis and motility. Annu. Rev. Cell Dev. Biol. 21, 695–718 (2005). [DOI] [PubMed] [Google Scholar]
- Bradshaw D. M. & Arceci R. J. Clinical relevance of transmembrane drug efflux as a mechanism of multidrug resistance. J. Clin. Oncol. 16, 3674–3690 (1998). [DOI] [PubMed] [Google Scholar]
- Gottesman M. M., Fojo T. & Bates S. E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 (2002). [DOI] [PubMed] [Google Scholar]
- Glavinas H., Krajcsi P., Cserepes J. & Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 1, 27–42 (2004). [DOI] [PubMed] [Google Scholar]
- Lichtenfels R., Biddison W. E., Schulz H., Vogt A. B. & Martin R. CARE-LASS (calcein-release-assay), an improved fluorescence-based test system to measure cytotoxic T-lymphocyte activity. J. Immunol. Methods. 172, 227–239 (1994). [DOI] [PubMed] [Google Scholar]
- Legrand O., Simonin G., Perrot J. Y., Zittoun R. & Marie J. P. Pgp and MRP activities using calcein-AM are prognostic factors in adult acute myeloid leukemia patients. Blood 91, 4480–4488 (1998). [PubMed] [Google Scholar]
- Rosenbaum C., Rohrs S., Muller O. & Waldmann H. Modulation of MRP-1-mediated multidrug resistance by indomethacin analogues. J. Med. Chem. 48, 1179–1187 (2005). [DOI] [PubMed] [Google Scholar]
- Ye C. G. et al. Indomethacin and SC236 enhance the cytotoxicity of doxorubicin in human hepatocellular carcinoma cells via inhibiting P-glycoprotein and MRP1 expression. Cancer Lett. 304, 90–96 (2011). [DOI] [PubMed] [Google Scholar]
- Dogan A. L., Legrand O., Faussat A. M., Perrot J. Y. & Marie J. P. Evaluation and comparison of MRP1 activity with three fluorescent dyes and three modulators in leukemic cell lines. Leuk. Res. 28, 619–622 (2004). [DOI] [PubMed] [Google Scholar]
- Di Carlo D. & Lee L. P. Dynamic single-cell analysis for quantitative biology. Anal. Chem. 78, 7918–7925 (2006). [DOI] [PubMed] [Google Scholar]
- Faley S., Copland M., Reboud J. & Cooper J. M. Intracellular protein trafficking kinetics in chronic myeloid leukemia stem cells using a microfluidic platform. Integr. Biol. 4, 368–373 (2012). [DOI] [PubMed] [Google Scholar]
- Faley S. L., Copland M., Reboud J. & Cooper J. M. Cell chip array for microfluidic proteomics enabling rapid in situ assessment of intracellular protein phosphorylation. Biomicrofluidics 5, 024106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodkowic D. et al. Chip-based dynamic real-time quantification of drug-induced cytotoxicity in human tumor cells. Anal. Chem. 81, 6952–6959 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodkowic D., Skommer J., Faley S., Darzynkiewicz Z. & Cooper J. M. Dynamic analysis of apoptosis using cyanine SYTO probes: From classical to microfluidic cytometry. Exp. Cell Res. 315, 1706–1714 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenaz P., Ruttimann U. E. & Unser M. A pyramid approach to subpixel registration based on intensity. IEEE T. Image Process. 7, 27–41 (1998). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials