

Abstract
Septic shock is a life threatening condition that can develop subsequent to infection. Mortality can reach as high as 80% with over 150000 deaths yearly in the United States alone. Septic shock causes progressive failure of vital homeostatic mechanisms culminating in immunosuppression, coagulopathy and microvascular dysfunction which can lead to refractory hypotension, organ failure and death. The hypermetabolic response that accompanies a systemic inflammatory reaction places high demands upon stored nutritional resources. A crucial element that can become depleted early during the progression to septic shock is glutathione. Glutathione is chiefly responsible for supplying reducing equivalents to neutralize hydrogen peroxide, a toxic oxidizing agent that is produced during normal metabolism. Without glutathione, hydrogen peroxide can rise to toxic levels in tissues and blood where it can cause severe oxidative injury to organs and to the microvasculature. Continued exposure can result in microvascular dysfunction, capillary leakage and septic shock. It is the aim of this paper to present evidence that elevated systemic levels of hydrogen peroxide are present in septic shock victims and that it significantly contributes to the development and progression of this frequently lethal condition.
Keywords: Septic shock, Hydrogen peroxide, Hypermetabolic, Sepsis, Systemic inflammatory response syndrome
Core tip: For decades septic shock has been attributed to an over-active immune response. However, immune modulation has failed to reduce mortality, casting doubt on a direct causal role for the immune response in the development of septic shock. A closer look suggests that septic shock is the result of a generalized build-up of hydrogen peroxide, a toxic cellular by-product generated as a consequence of the hypermetabolic state that accompanies a systemic immune response. This finding points to the systemic accumulation of hydrogen peroxide as a significant risk factor for the development of septic and non-septic shock syndromes.
INTRODUCTION
Sepsis is a life threatening condition that is associated with a systemic inflammatory response to a microbial infection[1]. Sepsis is the most common cause of mortality in the intensive care unit with a fatality rate that can rise to 80% for those developing multiple organ failure. The progression of an exaggerated systemic inflammatory response is thought to be responsible for the eventual development of septic shock and death[2]. However, multiple therapeutic efforts aimed at controlling the immune response with the intent of interrupting the process leading to organ failure have been uniformly unsuccessful[1]. This simple fact has prompted a reappraisal of the role played by the immune system in the development of this condition that kills more than 150000 Americans yearly; more than breast, colon, prostate and brain cancer combined[3,4].
Although immune activation is clearly evident, recent evidence suggests that the immune response may not be the direct mediator of the pathologic process that leads to septic shock. Studies conducted to define the circulating leukocyte transcriptome have revealed that there is no qualitative difference in the immunogenetic response when comparing burn or blunt trauma patients with complicated or uncomplicated outcomes. In other words, severely injured patients who die from their injuries have the same immunogenetic response as patients who recover; the only difference being the duration and intensity of systemic inflammation[5].
The lack of a unique immunogenetic response suggests that septic shock is the phenotypic expression of a separate process that is initiated simultaneously with systemic immune activation. The multiple organ involvement, which can lead to death within a few days, suggests that this concomitant process is systemic in nature and initiated in parallel with inflammatory response. Moreover, the microvascular edema associated with multiple organ failure, which persists despite efforts at immunosuppression, suggests that a non-immune mediated angiopathic agent is being released into the systemic circulation[1].
The high (8%) increased mortality rate for each hour of delay before instituting antibiotics after the onset of hypotension suggests that the duration of this parallel process is closely linked with a greater risk of an adverse outcome and down regulating the immune response with successful therapy simply allows this parallel process to turn off[6].
In other words, survival is closely correlated with the early down regulation of a systemic process closely linked to systemic immune activation suggesting depletion of a crucial biochemical element that is critical for survival. Put differently, if catabasis (immune down regulation) is achieved by successful antibiotic therapy prior to depletion of this critical element the patient will survive, if not the patient is at high risk for organ failure, septic shock and death.
HYPERMETABOLIC RESPONSE
A key systemic process that is turned on and up-regulated with systemic inflammation is cellular metabolism, which becomes hypermetabolic from the onset of sepsis[7]. The sustained high fever, highly amplified protein synthesis, tachycardia and tachypnea characteristic of a septic immune response requires supra-physiological energy supplies. It is estimated that basal energy requirements for a septic patient can reach up to 10000 calories daily[8]. This hypermetabolic state not only requires increased nutrient intake but also generates a large amount of toxic cellular by-products as a result of increased electron transport chain (ETC) activity required to synthesize sufficient adenosine triphosphate (ATP) to support a prolonged hypermetabolic state. This critical need for supplemental nutrients often cannot be met as it occurs at a time when caloric intake is curtailed as a result of the severe illness afflicting the patient[8]. This suggests the progressive depletion of an element whose principal function is to metabolize a toxic cellular waste product that, upon accumulation, leads to organ dysfunction, microangiopathic edema and refractory hypotension, the characteristic pathologic findings in septic shock.
An important toxic product that is continuously generated as a result of cellular metabolism is hydrogen peroxide (H2O2), which is formed as a result of several metabolic activities including protein synthesis (disulfide bond formation), DNA recycling (Xanthine oxidase), ATP synthesis (ETC activity) and fatty acid oxidation (peroxisomal metabolism)[9-12]. Most H2O2 is degraded to water via the enzymatic action of glutathione peroxidase (GPx), a selenium containing enzyme that has an obligate requirement for the co-factor glutathione (GSH) in order to metabolize H2O2. The biochemical reaction is: 2 GSH + H2O2 → GS-SG + 2H2O in which two molecules of GSH are converted to one molecule of glutathione disulfide (GS-SG) and two molecules of water. Glutathione is consumed during this process and must be replenished in order for the cell to prevent accumulation of H2O2 to toxic levels[13,14].
Replenishment of glutathione, however, is not favored during periods of sustained hypermetabolism and caloric insufficiency, which frequently accompany critical illnesses such as sepsis leading to depletion of glutathione reserves.
Within 48 h of diagnosis critically ill children with sepsis were found to have a 60% decrease in whole blood GSH synthesis, suggesting depletion of whole body GSH stores[15,16]. Systemic GSH depletion is supported by studies showing over 50% decrease in lung and skeletal muscle GSH in septic and critically ill patients[17,18]. The critical importance of glutathione was demonstrated by a study which documented significantly decreased erythrocyte glutathione in septic non-survivors vs survivors (P < 0.0001)[19]. This suggests high levels of circulating H2O2 capable of permeating erythrocyte cell membranes and oxidizing (and depleting) intracellular glutathione in septic shock non-survivors. Elegant studies have also demonstrated a significantly higher mitochondrial respiratory rate in non-survivors at three months following sepsis suggesting that failure to down regulate the hypermetabolic state (and excess H2O2 production) is independently associated with higher mortality even after surviving the initial infectious insult[20].
Generalized depletion of body stores can result in cellular deficiency of GSH leading to a toxic accumulation of H2O2. A highly toxic oxidizing agent, H2O2 is the principal mediator of cellular oxidative damage. It does so by generating hydroxyl radical (OH*), the most potent reactive oxygen radical known in biological systems. Hydroxyl radical will indiscriminately disintegrate proteins, peroxidize lipids and oxidatively damage DNA leading to cell death[21,22].
Compounding the cellular cytotoxicity of H2O2 is its ability to freely diffuse through biological membranes allowing it to permeate other cellular compartments and diffuse to the extracellular space from where it can pass through the capillary endothelium into the blood stream[16,23]. Thus, the end result of a systemic GSH deficiency is the systematic discharge of excess H2O2 by all organs of the body into the bloodstream where it can damage distant capillary beds leading to systemic microcirculatory dysfunction, microangiopathic edema and refractory hypotension, a hallmark of septic shock.
This is supported by studies showing decreased human endothelial cell levels of GSH and eventual death after in vitro exposure to plasma from septic shock patients[24]. This implies a membrane diffusible agent capable of oxidizing intracellular GSH suggesting that a toxic level of plasma H2O2 was the offending oxidizing agent mediating this effect. This is consistent with the well documented oxidative damage and dose dependent cytotoxicity that occurs during human endothelial cell exposure to H2O2[25,26].
In other studies high levels of urinary H2O2 were found to correlate with a fatal outcome in patients with sepsis and adult respiratory distress syndrome suggesting an important role for H2O2 in the pathogenesis of septic shock[27]. Taken together the evidence suggests that H2O2 exerts a significant microangiopathic effect contributing to the development of microcirculatory dysfunction and the progression to refractory hypotension and fatal septic shock.
MECHANISM OF DISEASE
The above evidence supports a pathogenesis of septic shock which is initiated by the systemic depletion of glutathione as the crucial event responsible for the accumulation of H2O2 in tissues. Subsequent diffusion of H2O2 into the blood stream leads to systemic elevation of this highly toxic oxidizing agent resulting in the microvascular dysfunction and organ failure observed in septic shock (Figure 1).
Figure 1.
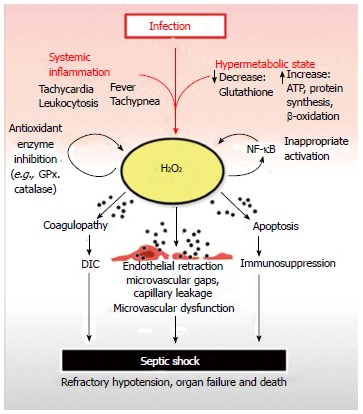
Septic shock begins with a systemic inflammatory reaction to an infection. A contemporaneous increase in metabolism is initiated, which can deplete reserves of critical nutrients such as glutathione. Glutathione is crucial for the neutralization of H2O2, a toxic, membrane-permeable oxidizing agent generated as a by-product of cellular metabolism. Depletion of cellular glutathione results in elevation of H2O2 which can diffuse out of organ parenchymal cells and into capillary endothelium before reaching the bloodstream. Once in the systemic circulation, excess H2O2 is distributed throughout the body resulting in systemic oxidative damage to plasma components, organs and blood vessels. The net result is H2O2 induced coagulopathy, immunocyte apoptosis and microvascular dysfunction leading to disseminated intravascular coagulation, immunosuppression, organ failure and septic shock respectively. H2O2 inhibits GPx and catalase, which are critical anti-oxidant enzymes required for H2O2 neutralization. This prevents restoration of normal plasma and tissue redox balance while exacerbating oxidative tissue damage. H2O2 can also activate nuclear factor-κB (NF-κB) contributing to the inappropriate activation of this master pro-inflammatory transcription factor observed in septic shock. The pathologic activation of NF-kB contributes to elevated tumor necrosis factor-alpha levels, another potent generator of intracellular H2O2. GPx: Glutathione peroxidase.
At the onset, a systemic inflammatory response is accompanied by a generalized hypermetabolic state which provides the energy needed to sustain the highly upregulated immune response switched on by the presence of a pathogen. The abrupt global increase of cellular bioenergetic reactions to several times their normal basal state presents the cell with a surge of toxic metabolic by-products that must be neutralized to avoid accumulation and cell death. Hydrogen peroxide, a toxic reactive oxygen species, is a significant metabolic by-product that is generated in increased amounts when cellular processes such as protein synthesis, DNA recycling and ATP production are upregulated during periods of hypermetabolism that accompany systemic inflammation.
The majority of cellular H2O2 is neutralized by GPx, a selenium containing enzyme, which utilizes the tri-peptide co-factor glutathione as a donor of reducing equivalents during the enzymatic conversion of H2O2 to water. GSH is consumed in this reaction and must be replenished in order to prevent accumulation of H2O2 within the cell. However, during periods of high H2O2 production the availability of glutathione may be insufficient to keep up with demand leading to net H2O2 accumulation and glutathione depletion resulting in severe cellular dysfunction and organ failure.
Excess H2O2 can easily diffuse out of pericapillary parenchymal cells through capillary endothelium and into the blood stream. This augments endothelial generated H2O2 resulting in oxidative damage and microangiopathic dysfunction. The inability to buffer cellular H2O2 signals a systemic failure of reductive (anti-oxidant) capacity as the excess oxidant load is discharged into the blood stream. Over time plasma reductive capacity is exhausted leading to severe disruption in plasma redox potential, which studies have shown is strongly associated with an unfavorable outcome[28].
HYDROGEN PEROXIDE CAN REPRODUCE CLINICAL ABNORMALITIES OBSERVED IN SEPTIC SHOCK
Microcirculatory dysfunction
The capillary bed is not simply a conduit for the passage of cells. It is a highly dynamic and integrated system of endothelial cells that continuously interacts with its surrounding environment through a variety of displayed receptors and elaborated mediators whose functions includes vasoregulation, coagulation factors, barrier maintenance, immune cell recruitment and oxygen transport[29]. Microcirculatory dysfunction is now considered to play a central role in the pathogenesis of sepsis and microvascular leakage has a defining role in its outcome[1,29].
Histological analysis of microvasculature in a baboon model of lethal Escherichia coli (E. coli) sepsis revealed large gaps between endothelial cells accompanied by a significant increase in endothelial permeability[30,31]. These changes are also observed upon exposure of human umbilical vein endothelial cells (HUVEC) to H2O2. Studies have demonstrated an 18x increase (from 20 to 360 gaps/mm2) in inter-endothelial cell gaps within 30 min of HUVEC exposure to H2O2. A time and dose dependent H2O2 induced endothelial contraction to about 60% of normal planar surface area was also observed[32,33]. This provides a microanatomical basis by which excess H2O2 can account for the life threatening massive edema observed both in humans and experimental models of sepsis[29,30].
Accompanying endothelial cell retraction during H2O2 exposure is the loss of tight junction proteins at the sites of gap formation, which strongly correlated with increased paracellular permeability[34-36]. Extensive cytoskeletal disruption and rearrangement was also shown to occur after endothelial cell exposure to H2O2[37-39]. Endothelial shape changes have been observed to occur in experimental models of sepsis and several studies have reported these pathological changes upon endothelial cell exposure to H2O2[30,40-43].
The net effect of continuous H2O2 exposure on the systemic microvasculature is severe disruption. Barrier function is compromised, intercellular communication is blunted and signal transduction is abrogated. This leads to microvascular edema, arteriovenous shunts and vaso-dysregulation as a result of cumulative oxidative damage sustained from continued penetration of H2O2 into endothelial cells. This is supported by studies of low dose H2O2 perfusion into isolated rat lung, which increased pulmonary vascular bed permeability and capillary filtration coefficient[41].
Studies of bovine brain microvascular endothelial cells exposed to H2O2 revealed increased paracellular permeability of the blood brain barrier (BBB) with loss of tight junctional proteins (44)[44]. H2O2 can by-pass the normally protective BBB by simply diffusing into tissues and cells[15]. This can result in dysfunction of cerebral microvasculature and could account for the early mental changes observed in patients with sepsis as a result of impaired synaptic transmission[8].
Immune activation
Numerous genes are activated during a systemic immune response in a critically ill or septic individual. Studies in healthy human volunteers receiving low dose endotoxin identified over 4500 activated genes, most of which were involved in the innate or adaptive immune response (5)[5]. The simultaneous activation of this many genes is facilitated by preformed cytoplasmic signal transcription factors that serve as rapid response mediators to injury and infection. Nuclear factor kappa B (NF-κB) is a transcription factor that plays a central role in the activation and regulation of multiple genes that control immune and inflammatory reactions[45]. NF-κB is significantly elevated in adults and children with sepsis[46-48]. NF-κB is also a highly redox sensitive transcription factor capable of being activated by low levels of H2O2[49,50] and has been proposed as a biomarker for oxidative stress[51]. This suggests that high levels of ambient H2O2 may be involved in the inappropriate activation of NF-κB observed in septic shock[45].
A central role for the innate immune system is suggested by the neutrophilic infiltration into multiple organs observed in septic shock[45]. H2O2 is a highly potent neutrophilic chemo-attractant that can establish a chemotactic gradient as it diffuses out of parenchymal cells into the adjacent microvasculature. Circulating neutrophils can track this H2O2 gradient and enter the organ parenchyma via diapedesis. The net result is neutrophil infiltration into the parenchyma of multiple organs[52-54].
Coagulopathy
Intravascular activation of the coagulation cascade with generation of fibrin and formation of diffuse microvascular thrombi is a pathologic and physiologic hallmark of sepsis[55]. This presents clinically as disseminated intravascular coagulation (DIC) and is found in up to 50% of patients with sepsis[56]. DIC leads to abnormal bleeding and intravascular clotting, obstructing limb and organ blood flow, and is a strong predictor of mortality[56].
Endothelial derived tissue factor (TF) is the major physiological route by which fibrin generation is initiated in sepsis. Importantly, this process is triggered only at sites of vascular injury or endothelial disruption where plasma clotting factors can encounter the TF protein that activates this extrinsic clotting pathway[56,57]. Studies utilizing immunohistochemistry in a lethal E. coli baboon sepsis model preferentially localized TF and TF mRNA at arterial branch areas, which is compatible with enhanced contact by a plasma derived oxidizing agent (e.g., H2O2) at these sites of altered blood flow[30].
H2O2 can induce vascular injury by peroxidation of cell membrane lipids and studies have shown a marked increase in endothelial cell TF and TF mRNA after 1 and 5 min exposure to Xanthine oxidase, a H2O2 generating enzyme[10,58]. This indicates that TF is highly sensitive to H2O2 induced upregulation, which suggests with a contributory role for H2O2 in sepsis- associated DIC. Consistent with this mechanism is a case report describing a fatal case of sepsis with DIC and multiorgan failure in a previously healthy 37-year-old man after receiving several intravenous infusions of H2O2[59].
Immunosuppression
Septic patients experience a considerable decline in lymphocyte numbers through apoptosis in the latter stages of sepsis and this is a significant contributing factor to the immunosuppression experienced by septic individuals[60]. Studies have shown that H2O2 is a potent apoptosis inducing agent[61]. B lymphocytes treated with agents that inhibit GSH synthesis experience a 95% decline in GSH concentration in 12 h. This is followed by a rise in intracellular H2O2 after which apoptosis occurs. By 72 h nearly 50% of B cells have died via apoptosis[62]. T cells are also highly sensitive to the effects of GSH depletion. Studies have recorded a 30% decline in circulating T lymphocytes within 4 wk after glutathione levels declined to suboptimal levels in healthy volunteers[63]. This supports a role for H2O2 in the development of sepsis induced immunosuppression.
Erythrocyte rigidity
Red blood cell deformability is markedly reduced in sepsis and studies have demonstrated a significant reduction in red blood cell deformability upon exposure to H2O2[64]. A direct relationship was found between oxidant induced changes in erythrocyte deformability and severity of multi-organ failure in septic individuals[65]. This suggests that plasma derived H2O2 is a source of oxidant-induced RBC membrane damage.
Circulating endothelial cells
Circulating endothelial cells (CEC) are a reliable, sensitive and specific indicator of vascular damage[66]. These cells rarely exist in the peripheral blood of healthy individuals[67]. Patients with s evere sepsis and septic shock have significantly higher numbers of CECs indicating widespread vascular damage[68,69]. Studies have shown that human endothelial cell detachment is produced by exposure to H2O2[43]. The presence of CECs in patients with sepsis but without shock suggests that endothelial damage precedes the development of organ damage[68]. This is compatible with H2O2 release from organ parenchymal cells into the capillary vascular bed causing microvascular dysfunction and edema with subsequent development of organ failure.
Sepsis associated encephalopathy
Sepsis associated encephalopathy (SAE) is a diffuse cerebral dysfunction occurring in the setting of sepsis but without direct infection of the central nervous system[70]. SAE is characterized by alterations in mental status and motor activity that can range from inattention, disorientation and delirium to agitation, hypoactivity and coma[71,72]. Delirium is frequently the first manifestation of sepsis and often precedes organ failure[73,74]. SAE is reported to occur in up to 70% of septic patients (71).
Neurons are especially sensitive to H2O2 induced oxidative damage. Studies have shown a concentration dependent cell death starting at 10 μmol/L when neurons are exposed to H2O2[75]. The tripeptide glutathione is critically important in order to prevent oxidative damage of the brain due to H2O2[76]. Glutathione is composed of amino acids glycine, cysteine and glutamate. Cysteine is the rate limiting substrate for neuronal glutathione synthesis and transsulfuration of homocysteine is a major source of cysteine in most cells. However, the brain’s neuronal transsulfuration pathway is thought to be a negligible source of cysteine due to low activity of neuronal cystathionine-gamma-lyase (EC 4.4.1.1), a crucial enzyme in the transsulfurationn pathway leading to the synthesis of cysteine[77,78]. Neurons, therefore, rely mainly on the absorption of extracellular cysteine provided by astrocytes for the synthesis of glutathione[77]. Thus, the dependence of brain neurons on extracellular cysteine in order to synthesize glutathione severely limits their ability to upregulate antioxidant defenses in response to H2O2 mediated oxidative stress. This makes brain neurons highly vulnerable to H2O2 oxidative damage and dysfunction. This is consistent with the encephalopathy that is reported to occur after accidental ingestion of H2O2[79]. Encephalopathy was also a manifestation after intravenous administration of H2O2 during alternative medicine therapy[59].
The main interaction site of neurons and astrocytes is the synaptic cleft[80]. Astrocytes export glutathione directly into the synaptic cleft. Ectoenzymes present in the synapse enzymatically release cysteine from glutathione after which cysteine is transported into neurons by the membrane bound EAAT3 transporter (excitatory amino acid transporter 3)[77-82]. H2O2 can react non-enzymatically with cysteine in the synaptic cleft to produce cystine[83]. This removes cysteine from the synapse and prevents its importation into the neuron resulting in oxidative stress by decreasing the synthesis of neuronal glutathione. The presence of thiols (i.e., cysteine) in the synaptic cleft suggests that this region can function as a sink for H2O2 resulting in disruption of synaptic transmission as a result of peroxidation of synaptic cellular membranes.
Thus, circulating H2O2 can permeate the brain during the initial hypermetabolic systemic inflammatory response syndrome (SIRS) phase of sepsis and disrupt brain function in the early stages of disease. Due to their limited capacity to detoxify H2O2, brain neurons are the first cells to be affected by H2O2 induced oxidative stress[84]. This is consistent with the observation that encephalopathy is often the first sign of sepsis.
LACTIC ACIDOSIS
Sepsis related lactic acidosis is generally attributed to tissue hypoxia. Although tissue hypoxia can result in lactic acidosis it is unsuitable as a general mechanism to explain the appearance of lactic acidosis in septic patients when tissue oxygenation can be normal or even increased[85].
Under normal circumstances pyruvate, the end product of glycolysis in the cytoplasm, is transported into mitochondria where it is oxidized by the Krebs cycle. Lactate synthesis increases when the rate of pyruvate formation in the cytoplasm exceeds its rate of oxidation by the mitochondria. The excess pyruvate in the cytoplasm is then converted to lactate by lactate dehydrogenase and released into the blood stream resulting in lactic acidosis.
Inhibition of Krebs cycle enzymes will decrease pyruvate oxidation resulting in lactic acidosis. This has been observed with inherited deficiency of alpha-ketoglutarate dehydrogenase resulting in severe congenital lactic acidosis[86]. Alpha-ketoglutarate dehydrogenase is also highly sensitive to oxidative inhibition by hydrogen peroxide[87]. Rising systemic concentrations of H2O2 in sepsis can account for the observed lactic acidosis with normal tissue oxygen perfusion. This has been termed cytopathic hypoxia. In this case the lactic acidosis is an epiphenomenon of a much more serious underlying metabolic abnormality and treatment of the acidosis does not resolve the inhibition of the Krebs cycle.
DISCUSSION
A hypermetabolic state can develop very quickely after a generalized septic or non-septic insult to the body. At the heart of the hypermetabolic state is a significantly increased bioenergetic response resulting mainly from enhanced ETC activity. The ETC is an assembly of intra-mitochondrial protein complexes that converts the energy of high-energy electrons into a form that is used to synthesize ATP, a high energy molecule that powers most energy requiring biosynthetic reactions and physiological functions. Thus, the high energy demands of body systems resulting from a generalized septic or non-septic insult are principally met by increased ATP production, which is manifested as a hypermetabolic state and recognized by the same parameters used to define a SIRS such as increased body temperature, heart rate, respiratory rate and increased white blood cell count.
A principle metabolic by-product of ETC activity is hydrogen peroxide; a highly toxic oxidizing agent. Hydrogen peroxide is produced when electrons spontaneously escape from the ETC and combine with available vicinal oxygen to generate superoxide that is enzymatically converted to H2O2 by superoxide dismutase. The increased amount of H2O2 generated during a hypermetabolic state can overwhelm the cell’s anti-oxidant enzymatic defenses resulting in net intracellular H2O2 accumulation. The excess H2O2 can oxidatively inhibit enzyme systems including those needed to neutralize H2O2 resulting in a positive bio-feedback loop and a vicious cycle of ever increasing intracellular H2O2[88]. Glutathione functions as a cofactor for GPx, which enzymatically neutralizes H2O2. GPx is inhibited by the rising concentrations of H2O2, which explains why exogenously supplied N-acetylcysteine has no effect on the course of septic shock since glutathione cannot be utilized by GPx to neutralize H2O2[88,89].
Hydrogen peroxide is biomembrane permeable and can diffuse into the bloodstream where it is distributed to all organs of the body generating a state of severe systemic oxidative stress. Studies have documented high levels of H2O2 in the blood and urine of septic patients[27,90]. This can result in the multi-organ failure and microangiopathic dysfunction characteristic of septic shock. Genetic variation in glutathione levels as well as age related decline has been reported[91-93]. This may compromise the ability to neutralize H2O2 and pre-dispose individuals to vasoplegic (i.e., septic) shock and multi-organ failure during acute hypermetabolic periods, especially in older individuals. Studies have shown that glutathione is essential for cell survival[94].
CONCLUSION
Taken together, the evidence suggests that septic shock is a primary radical induction process that has its origins early in the development of sepsis with the accumulation and generalized dispersal of cytotoxic levels of H2O2. This arises secondary to glutathione depletion as a result of a systemic inflammatory mediated hypermetabolic state. Studies have shown that systemic inflammation significantly reduces GSH levels, and GSH deficient animals subjected to shock develop hypotension, kidney and liver failure, increased organ bacteria and dramatic increases in mortality rates[95-97].
The near universal requirement of glutathione for cellular function and the pathological accumulation of H2O2 that ensues when glutathione is deficient can affect every organ in the body. Studies have shown that H2O2 can reproduce the clinico-pathological abnormalities observed in septic shock (Figure 2).
Figure 2.
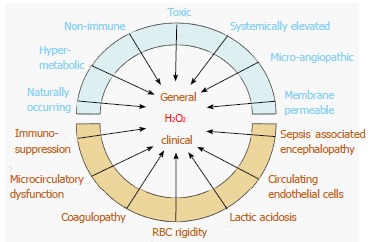
Pathologically elevated serum H2O2 levels can account for the general physiological, histological and clinical abnormalities observed in septic shock. Red blood cell glutathione accounts for a major portion of serum redox buffering capacity and is depleted in septic shock non-survivors vs. survivors. Brain neuron function is highly vulnerable to H2O2 oxidative stress and is manifested by electroencephalographic changes, which can appear before clinical encephalopathy is evident. Studies show that septic shock survivors upregulate serum antioxidant capacity (which decreases H2O2), while non-survivors are unable to do so. This suggests that elevated H2O2 is a necessary concomitant to the development of septic shock and recovery is preceded by decreasing H2O2. The individual clinical course, bookended by these extremes of H2O2, is influenced by parameters such as individual antioxidant capacity, susceptibility to oxidative stress, co-morbidities, age, general health and organ system involved.
Kept in check, the high membrane diffusability of H2O2 allows it to fulfill its physiological role as a cellular messenger but also creates the potential for a pathophysiological response during times of metabolic stress when reductive (anti-oxidant) mechanisms can become overwhelmed as a consequence of hyper-metabolic H2O2 production[98]. This is further exacerbated by nutritional deficits that may arise during the course of acute illness in addition to the effect of glutathione deficiency itself, which as master antioxidant of the cell, supplies reducing equivalents to maintain proteins in their reduced (and functional) state[14].
Footnotes
P- Reviewers: Carassiti M, Fink MP, Stover CM, Yao YM S- Editor: Song XX L- Editor: A E- Editor: Wu HL
References
- 1.Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med. 2011;3:88ps25. doi: 10.1126/scitranslmed.3002011. [DOI] [PubMed] [Google Scholar]
- 2.Marshall JC, Vincent JL, Guyatt G, Angus DC, Abraham E, Bernard G, Bombardier C, Calandra T, Jørgensen HS, Sylvester R, et al. Outcome measures for clinical research in sepsis: a report of the 2nd Cambridge Colloquium of the International Sepsis Forum. Crit Care Med. 2005;33:1708–1716. doi: 10.1097/01.ccm.0000174478.70338.03. [DOI] [PubMed] [Google Scholar]
- 3.Melamed A, Sorvillo FJ. The burden of sepsis-associated mortality in the United States from 1999 to 2005: an analysis of multiple-cause-of-death data. Crit Care. 2009;13:R28. doi: 10.1186/cc7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Cancer Society, Surveillance Research. Estimated New Cancer Cases and Deaths by Sex for All Sites, US, 2011. Available from: http://seer.cancer.gov/csr/1975_2008/results_single/sect_01_table.01.pdfAccessed 20/Jan/2014.
- 5.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208:2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suffredini AF, Munford RS. Novel therapies for septic shock over the past 4 decades. JAMA. 2011;306:194–199. doi: 10.1001/jama.2011.909. [DOI] [PubMed] [Google Scholar]
- 7.Bloch KC. Infectious Diseases. In: McPhee SJ, Hammer GD: Pathophysiology of Disease, editors. New York: McGraw-Hill; 2010. pp. 57–83. [Google Scholar]
- 8.Borgen L. Total parenteral nutrition in adults. Am J Nurs. 1978;78:224–228. [PubMed] [Google Scholar]
- 9.Depuydt M, Messens J, Collet JF. How proteins form disulfide bonds. Antioxid Redox Signal. 2011;15:49–66. doi: 10.1089/ars.2010.3575. [DOI] [PubMed] [Google Scholar]
- 10.Kelley EE, Khoo NK, Hundley NJ, Malik UZ, Freeman BA, Tarpey MM. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic Biol Med. 2010;48:493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schrader M, Fahimi HD. Mammalian peroxisomes and reactive oxygen species. Histochem Cell Biol. 2004;122:383–393. doi: 10.1007/s00418-004-0673-1. [DOI] [PubMed] [Google Scholar]
- 13.Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 15.Lyons J, Rauh-Pfeiffer A, Ming-Yu Y, Lu XM, Zurakowski D, Curley M, Collier S, Duggan C, Nurko S, Thompson J, et al. Cysteine metabolism and whole blood glutathione synthesis in septic pediatric patients. Crit Care Med. 2001;29:870–877. doi: 10.1097/00003246-200104000-00036. [DOI] [PubMed] [Google Scholar]
- 16.Biolo G, Antonione R, De Cicco M. Glutathione metabolism in sepsis. Crit Care Med. 2007;35:S591–S595. doi: 10.1097/01.CCM.0000278913.19123.13. [DOI] [PubMed] [Google Scholar]
- 17.Pacht ER, Timerman AP, Lykens MG, Merola AJ. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. 1991;100:1397–1403. doi: 10.1378/chest.100.5.1397. [DOI] [PubMed] [Google Scholar]
- 18.Hammarqvist F, Luo JL, Cotgreave IA, Andersson K, Wernerman J. Skeletal muscle glutathione is depleted in critically ill patients. Crit Care Med. 1997;25:78–84. doi: 10.1097/00003246-199701000-00016. [DOI] [PubMed] [Google Scholar]
- 19.Karapetsa M, Pitsika M, Goutzourelas N, Stagos D, Tousia Becker A, Zakynthinos E. Oxidative status in ICU patients with septic shock. Food Chem Toxicol. 2013;61:106–111. doi: 10.1016/j.fct.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 20.Sjövall F, Morota S, Hansson MJ, Friberg H, Gnaiger E, Elmér E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit Care. 2010;14:R214. doi: 10.1186/cc9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009;37:2539–2548. doi: 10.1093/nar/gkp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Malinouski M, Zhou Y, Belousov VV, Hatfield DL, Gladyshev VN. Hydrogen peroxide probes directed to different cellular compartments. PLoS One. 2011;6:e14564. doi: 10.1371/journal.pone.0014564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huet O, Cherreau C, Nicco C, Dupic L, Conti M, Borderie D, Pene F, Vicaut E, Benhamou D, Mira JP, et al. Pivotal role of glutathione depletion in plasma-induced endothelial oxidative stress during sepsis. Crit Care Med. 2008;36:2328–2334. doi: 10.1097/CCM.0b013e3181800387. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Gu Y, Shao Z, Luo J, Tan Z. Propofol protects against hydrogen peroxide-induced oxidative stress and cell dysfunction in human umbilical vein endothelial cells. Mol Cell Biochem. 2010;339:43–54. doi: 10.1007/s11010-009-0368-y. [DOI] [PubMed] [Google Scholar]
- 26.Li ZL, Liu JC, Hu J, Li XQ, Wang SW, Yi DH, Zhao MG. Protective effects of hyperoside against human umbilical vein endothelial cell damage induced by hydrogen peroxide. J Ethnopharmacol. 2012;139:388–394. doi: 10.1016/j.jep.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 27.Mathru M, Rooney MW, Dries DJ, Hirsch LJ, Barnes L, Tobin MJ. Urine hydrogen peroxide during adult respiratory distress syndrome in patients with and without sepsis. Chest. 1994;105:232–236. doi: 10.1378/chest.105.1.232. [DOI] [PubMed] [Google Scholar]
- 28.Cowley HC, Bacon PJ, Goode HF, Webster NR, Jones JG, Menon DK. Plasma antioxidant potential in severe sepsis: a comparison of survivors and nonsurvivors. Crit Care Med. 1996;24:1179–1183. doi: 10.1097/00003246-199607000-00019. [DOI] [PubMed] [Google Scholar]
- 29.Lehr HA, Bittinger F, Kirkpatrick CJ. Microcirculatory dysfunction in sepsis: a pathogenetic basis for therapy? J Pathol. 2000;190:373–386. doi: 10.1002/(SICI)1096-9896(200002)190:3<373::AID-PATH593>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Lupu C, Westmuckett AD, Peer G, Ivanciu L, Zhu H, Taylor FB, Lupu F. Tissue factor-dependent coagulation is preferentially up-regulated within arterial branching areas in a baboon model of Escherichia coli sepsis. Am J Pathol. 2005;167:1161–1172. doi: 10.1016/S0002-9440(10)61204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Birukova AA, Arce FT, Moldobaeva N, Dudek SM, Garcia JG, Lal R, Birukov KG. Endothelial permeability is controlled by spatially defined cytoskeletal mechanics: atomic force microscopy force mapping of pulmonary endothelial monolayer. Nanomedicine. 2009;5:30–41. doi: 10.1016/j.nano.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hastie LE, Patton WF, Hechtman HB, Shepro D. H2O2-induced filamin redistribution in endothelial cells is modulated by the cyclic AMP-dependent protein kinase pathway. J Cell Physiol. 1997;172:373–381. doi: 10.1002/(SICI)1097-4652(199709)172:3<373::AID-JCP11>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 33.López-Ongil S, Torrecillas G, Pérez-Sala D, González-Santiago L, Rodríguez-Puyol M, Rodríguez-Puyol D. Mechanisms involved in the contraction of endothelial cells by hydrogen peroxide. Free Radic Biol Med. 1999;26:501–510. doi: 10.1016/s0891-5849(98)00223-8. [DOI] [PubMed] [Google Scholar]
- 34.Kevil CG, Okayama N, Alexander JS. H(2)O(2)-mediated permeability II: importance of tyrosine phosphatase and kinase activity. Am J Physiol Cell Physiol. 2001;281:C1940–C1947. doi: 10.1152/ajpcell.2001.281.6.C1940. [DOI] [PubMed] [Google Scholar]
- 35.Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H(2)O(2)-mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol. 2000;279:C21–C30. doi: 10.1152/ajpcell.2000.279.1.C21. [DOI] [PubMed] [Google Scholar]
- 36.Pearse DB, Shimoda LA, Verin AD, Bogatcheva N, Moon C, Ronnett GV, Welsh LE, Becker PM. Effect of cGMP on lung microvascular endothelial barrier dysfunction following hydrogen peroxide. Endothelium. 2003;10:309–317. doi: 10.1080/10623320390272307. [DOI] [PubMed] [Google Scholar]
- 37.Valen G, Sondén A, Vaage J, Malm E, Kjellström BT. Hydrogen peroxide induces endothelial cell atypia and cytoskeleton depolymerization. Free Radic Biol Med. 1999;26:1480–1488. doi: 10.1016/s0891-5849(99)00009-x. [DOI] [PubMed] [Google Scholar]
- 38.Bradley JR, Thiru S, Pober JS. Hydrogen peroxide-induced endothelial retraction is accompanied by a loss of the normal spatial organization of endothelial cell adhesion molecules. Am J Pathol. 1995;147:627–641. [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Y, Davis HW. Hydrogen peroxide-induced cytoskeletal rearrangement in cultured pulmonary endothelial cells. J Cell Physiol. 1998;174:370–379. doi: 10.1002/(SICI)1097-4652(199803)174:3<370::AID-JCP11>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 40.Hirano S, Rees RS, Yancy SL, Welsh MJ, Remick DG, Yamada T, Hata J, Gilmont RR. Endothelial barrier dysfunction caused by LPS correlates with phosphorylation of HSP27 in vivo. Cell Biol Toxicol. 2004;20:1–14. doi: 10.1023/b:cbto.0000021019.50889.aa. [DOI] [PubMed] [Google Scholar]
- 41.Habib MP, Clements NC. Effects of low-dose hydrogen peroxide in the isolated perfused rat lung. Exp Lung Res. 1995;21:95–112. doi: 10.3109/01902149509031747. [DOI] [PubMed] [Google Scholar]
- 42.Gilmont RR, Dardano A, Young M, Engle JS, Adamson BS, Smith DJ, Rees RS. Effects of glutathione depletion on oxidant-induced endothelial cell injury. J Surg Res. 1998;80:62–68. doi: 10.1006/jsre.1998.5328. [DOI] [PubMed] [Google Scholar]
- 43.Shingu M, Yoshioka K, Nobunaga M, Yoshida K. Human vascular smooth muscle cells and endothelial cells lack catalase activity and are susceptible to hydrogen peroxide. Inflammation. 1985;9:309–320. doi: 10.1007/BF00916279. [DOI] [PubMed] [Google Scholar]
- 44.Lee HS, Namkoong K, Kim DH, Kim KJ, Cheong YH, Kim SS, Lee WB, Kim KY. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc Res. 2004;68:231–238. doi: 10.1016/j.mvr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 46.Arnalich F, Garcia-Palomero E, López J, Jiménez M, Madero R, Renart J, Vázquez JJ, Montiel C. Predictive value of nuclear factor kappaB activity and plasma cytokine levels in patients with sepsis. Infect Immun. 2000;68:1942–1945. doi: 10.1128/iai.68.4.1942-1945.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Böhrer H, Qiu F, Zimmermann T, Zhang Y, Jllmer T, Männel D, Böttiger BW, Stern DM, Waldherr R, Saeger HD, et al. Role of NFkappaB in the mortality of sepsis. J Clin Invest. 1997;100:972–985. doi: 10.1172/JCI119648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hotta N, Ichiyama T, Shiraishi M, Takekawa T, Matsubara T, Furukawa S. Nuclear factor-kappaB activation in peripheral blood mononuclear cells in children with sepsis. Crit Care Med. 2007;35:2395–2401. doi: 10.1097/01.ccm.0000284502.38701.e6. [DOI] [PubMed] [Google Scholar]
- 49.Schreck R, Albermann K, Baeuerle PA. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review) Free Radic Res Commun. 1992;17:221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- 50.Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J Biol Chem. 2003;278:24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- 51.van den Berg R, Haenen GR, van den Berg H, Bast A. Transcription factor NF-kappaB as a potential biomarker for oxidative stress. Br J Nutr. 2001;86 Suppl 1:S121–S127. doi: 10.1079/bjn2001340. [DOI] [PubMed] [Google Scholar]
- 52.Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klyubin IV, Kirpichnikova KM, Gamaley IA. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur J Cell Biol. 1996;70:347–351. [PubMed] [Google Scholar]
- 54.Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. 2006;80:1281–1288. doi: 10.1189/jlb.0506346. [DOI] [PubMed] [Google Scholar]
- 55.Wang L, Bastarache JA, Ware LB. The coagulation cascade in sepsis. Curr Pharm Des. 2008;14:1860–1869. doi: 10.2174/138161208784980581. [DOI] [PubMed] [Google Scholar]
- 56.Zeerleder S, Hack CE, Wuillemin WA. Disseminated intravascular coagulation in sepsis. Chest. 2005;128:2864–2875. doi: 10.1378/chest.128.4.2864. [DOI] [PubMed] [Google Scholar]
- 57.Crawley JT, Lane DA. The haemostatic role of tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2008;28:233–242. doi: 10.1161/ATVBAHA.107.141606. [DOI] [PubMed] [Google Scholar]
- 58.Ambrosio G, Tritto I, Golino P. Reactive oxygen metabolites and arterial thrombosis. Cardiovasc Res. 1997;34:445–452. doi: 10.1016/s0008-6363(97)00101-6. [DOI] [PubMed] [Google Scholar]
- 59.Wetter DA, Davis MD. Ulceration of the arm attributed to a spider bite and treated with intravenous hydrogen peroxide: a cautionary tale. Arch Dermatol. 2006;142:1658–1659. doi: 10.1001/archderm.142.12.1658. [DOI] [PubMed] [Google Scholar]
- 60.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol. 2011;6:19–48. doi: 10.1146/annurev-pathol-011110-130327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerella C, Coppola S, Maresca V, De Nicola M, Radogna F, Ghibelli L. Multiple mechanisms for hydrogen peroxide-induced apoptosis. Ann N Y Acad Sci. 2009;1171:559–563. doi: 10.1111/j.1749-6632.2009.04901.x. [DOI] [PubMed] [Google Scholar]
- 62.Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW, Peehl DM, Knox SJ. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002;9:252–263. doi: 10.1038/sj.cdd.4400959. [DOI] [PubMed] [Google Scholar]
- 63.Kinscherf R, Fischbach T, Mihm S, Roth S, Hohenhaus-Sievert E, Weiss C, Edler L, Bärtsch P, Dröge W. Effect of glutathione depletion and oral N-acetyl-cysteine treatment on CD4+ and CD8+ cells. FASEB J. 1994;8:448–451. [PubMed] [Google Scholar]
- 64.Snyder LM, Fortier NL, Trainor J, Jacobs J, Leb L, Lubin B, Chiu D, Shohet S, Mohandas N. Effect of hydrogen peroxide exposure on normal human erythrocyte deformability, morphology, surface characteristics, and spectrin-hemoglobin cross-linking. J Clin Invest. 1985;76:1971–1977. doi: 10.1172/JCI112196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Machiedo GW, Powell RJ, Rush BF, Swislocki NI, Dikdan G. The incidence of decreased red blood cell deformability in sepsis and the association with oxygen free radical damage and multiple-system organ failure. Arch Surg. 1989;124:1386–1389. doi: 10.1001/archsurg.1989.01410120032007. [DOI] [PubMed] [Google Scholar]
- 66.Erdbruegger U, Dhaygude A, Haubitz M, Woywodt A. Circulating endothelial cells: markers and mediators of vascular damage. Curr Stem Cell Res Ther. 2010;5:294–302. doi: 10.2174/157488810793351721. [DOI] [PubMed] [Google Scholar]
- 67.Wu H, Chen H, Hu PC. Circulating endothelial cells and endothelial progenitors as surrogate biomarkers in vascular dysfunction. Clin Lab. 2007;53:285–295. [PubMed] [Google Scholar]
- 68.Mutunga M, Fulton B, Bullock R, Batchelor A, Gascoigne A, Gillespie JI, Baudouin SV. Circulating endothelial cells in patients with septic shock. Am J Respir Crit Care Med. 2001;163:195–200. doi: 10.1164/ajrccm.163.1.9912036. [DOI] [PubMed] [Google Scholar]
- 69.Schlichting DE, Waxman AB, O’Brien LA, Wang T, Naum CC, Rubeiz GJ, Um SL, Williams M, Yan SC. Circulating endothelial and endothelial progenitor cells in patients with severe sepsis. Microvasc Res. 2011;81:216–221. doi: 10.1016/j.mvr.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 70.Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8:557–566. doi: 10.1038/nrneurol.2012.183. [DOI] [PubMed] [Google Scholar]
- 71.Lamar CD, Hurley RA, Taber KH. Sepsis-associated encephalopathy: review of the neuropsychiatric manifestations and cognitive outcome. J Neuropsychiatry Clin Neurosci. 2011;23:237–241. doi: 10.1176/jnp.23.3.jnp237. [DOI] [PubMed] [Google Scholar]
- 72.Siami S, Polito A, Sharshar T. Sepsis-associated Encephalopathy. In Vincent JL Yearbook of Intensive Care and Emergency Medicine. Berlin-Heidelberg: Springer; 2009. pp. 809–816. [Google Scholar]
- 73.Wilson JX, Young GB. Progress in clinical neurosciences: sepsis-associated encephalopathy: evolving concepts. Can J Neurol Sci. 2003;30:98–105. doi: 10.1017/s031716710005335x. [DOI] [PubMed] [Google Scholar]
- 74.Ringer TM, Axer H, Romeike BF, Zinke J, Brunkhorst F, Witte OW, Günther A. Neurological Sequelae of Sepsis: I) Septic Encephalopathy. Open Crit Care Med J. 2011;4:2–7. [Google Scholar]
- 75.Desagher S, Glowinski J, Premont J. Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci. 1996;16:2553–2562. doi: 10.1523/JNEUROSCI.16-08-02553.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dringen R, Pawlowski PG, Hirrlinger J. Peroxide detoxification by brain cells. J Neurosci Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- 77.Aoyama K, Watabe M, Nakaki T. Regulation of neuronal glutathione synthesis. J Pharmacol Sci. 2008;108:227–238. doi: 10.1254/jphs.08r01cr. [DOI] [PubMed] [Google Scholar]
- 78.Deth R, Muratore C. The redox/methylation hypothesis of autism. In: Chauhan A, Chauhan V, Brown WT : Autism: Oxidative Stress, Inflammation and immune abnormalities, editors. Florida: Florida CRC Press; 2010. pp. 113–130. [Google Scholar]
- 79.Cannon G, Caravati EM, Filloux FM. Hydrogen peroxide neurotoxicity in childhood: case report with unique magnetic resonance imaging features. J Child Neurol. 2003;18:805–808. doi: 10.1177/08830738030180111501. [DOI] [PubMed] [Google Scholar]
- 80.Cakir T, Alsan S, Saybasili H, Akin A, Ulgen KO. Reconstruction and flux analysis of coupling between metabolic pathways of astrocytes and neurons: application to cerebral hypoxia. Theor Biol Med Model. 2007;4:e48. doi: 10.1186/1742-4682-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- 82.Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 83.Luo D, Smith SW, Anderson BD. Kinetics and mechanism of the reaction of cysteine and hydrogen peroxide in aqueous solution. J Pharm Sci. 2005;94:304–316. doi: 10.1002/jps.20253. [DOI] [PubMed] [Google Scholar]
- 84.Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 85.Bellomo R, Ronco C. The pathogenesis of lactic acidosis in sepsis. Curr Opin Crit Care. 1999;5:452–457. [Google Scholar]
- 86.Bonnefont JP, Chretien D, Rustin P, Robinson B, Vassault A, Aupetit J, Charpentier C, Rabier D, Saudubray JM, Munnich A. Alpha-ketoglutarate dehydrogenase deficiency presenting as congenital lactic acidosis. J Pediatr. 1992;121:255–258. doi: 10.1016/s0022-3476(05)81199-0. [DOI] [PubMed] [Google Scholar]
- 87.Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cho CS, Lee S, Lee GT, Woo HA, Choi EJ, Rhee SG. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid Redox Signal. 2010;12:1235–1246. doi: 10.1089/ars.2009.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kinnula VL, Everitt JI, Mangum JB, Chang LY, Crapo JD. Antioxidant defense mechanisms in cultured pleural mesothelial cells. Am J Respir Cell Mol Biol. 1992;7:95–103. doi: 10.1165/ajrcmb/7.1.95. [DOI] [PubMed] [Google Scholar]
- 90.Pherwani AV, Puri VC, V Malhotra V. Estimation of hydrogen peroxide levels in the blood and urine of normal infants and infants with sepsis. Bombay Hosp J. 1999;41:8. [Google Scholar]
- 91.van ‘t Erve TJ, Wagner BA, Ryckman KK, Raife TJ, Buettner GR. The concentration of glutathione in human erythrocytes is a heritable trait. Free Radic Biol Med. 2013;65:742–749. doi: 10.1016/j.freeradbiomed.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Caprari P, Caforio MP, Cianciulli P, Maffi D, Pasquino MT, Tarzia A, Amadori S, Salvati AM. 6-Phosphogluconate dehydrogenase deficiency in an Italian family. Ann Hematol. 2001;80:41–44. doi: 10.1007/s002770000233. [DOI] [PubMed] [Google Scholar]
- 93.Lang CA, Naryshkin S, Schneider DL, Mills BJ, Lindeman RD. Low blood glutathione levels in healthy aging adults. J Lab Clin Med. 1992;120:720–725. [PubMed] [Google Scholar]
- 94.Hatem E, Berthonaud V, Dardalhon M, Lagniel G, Baudouin-Cornu P, Huang ME, Labarre J, Chédin S. Glutathione is essential to preserve nuclear function and cell survival under oxidative stress. Free Radic Biol Med. 2014;67:103–114. doi: 10.1016/j.freeradbiomed.2013.10.807. [DOI] [PubMed] [Google Scholar]
- 95.Keller GA, Barke R, Harty JT, Humphrey E, Simmons RL. Decreased hepatic glutathione levels in septic shock. Predisposition of hepatocytes to oxidative stress: an experimental approach. Arch Surg. 1985;120:941–945. doi: 10.1001/archsurg.1985.01390320065013. [DOI] [PubMed] [Google Scholar]
- 96.Ikegami K, Lalonde C, Young YK, Picard L, Demling R. Comparison of plasma reduced glutathione and oxidized glutathione with lung and liver tissue oxidant and antioxidant activity during acute inflammation. Shock. 1994;1:307–312. doi: 10.1097/00024382-199404000-00010. [DOI] [PubMed] [Google Scholar]
- 97.Robinson MK, Rounds JD, Hong RW, Jacobs DO, Wilmore DW. Glutathione deficiency increases organ dysfunction after hemorrhagic shock. Surgery. 1992;112:140–147; discussion 148-149. [PubMed] [Google Scholar]
- 98.Holmquist L, Stuchbury G, Steele M, Münch G. Hydrogen peroxide is a true first messenger. J Neural Transm Suppl. 2007;(72):39–41. doi: 10.1007/978-3-211-73574-9_6. [DOI] [PubMed] [Google Scholar]