

Abstract
Progesterone is an ovarian steroid hormone that is essential for normal breast development during puberty and in preparation for lactation and breastfeeding. The actions of progesterone are primarily mediated by its high-affinity receptors, which include the classical progesterone receptor (PR)-A and -B isoforms, located in diverse tissues, including the brain, where progesterone controls reproductive behavior, and the breast and reproductive organs. Progestins are frequently prescribed for contraception or during postmenopausal hormone replacement therapy, in which progestins are combined with estrogen as a means to block estrogen-induced endometrial growth. The role of estrogen as a potent breast mitogen is undisputed, and inhibitors of the estrogen receptor and estrogen-producing enzymes (aromatases) are effective first-line cancer therapies. However, PR action in breast cancer is grossly understudied and remains controversial. Herein, we review existing evidence and discuss the challenges to defining a role for progesterone in breast cancer.
Keywords: breast cancer, estrogen receptor, growth factor, hormone replacement therapy, progesterone, progesterone receptor, protein kinase, steroid hormone
Several factors contribute to the challenge of demonstrating a clear role for progesterone in breast cancer. Progesterone is difficult to study in isolation from other hormones (e.g., growth factors and prolactin) that also contribute to breast cancer biology. Progesterone receptor (PR) isoforms are primarily expressed in response to estrogen receptor (ER)-α-mediated transcriptional events, but can also occur independently from ER [1]. A subset of mammary epithelial cells (MECs) in the breast that express both PR-A and PR-B also express ERs, and estrogen is usually required in order to induce the expression of PR in these ER+ cells. For this reason, it has been difficult to separate the effects of progesterone alone from estrogen, itself a potent breast mitogen. Indeed, PR isoforms are grossly understudied relative to ER in both normal and cancerous breast cells.
Studies in ER and PR knockout mice have revealed that the concerted actions of estrogen and progesterone are required for normal mammary gland development [2,3]; estrogen/ER promotes the early growth of milk ducts that invade the mammary fat pad emanating from the nipple, while estrogen/ER and progesterone/PR isoforms are responsible for the development of the terminal end-buds (TEBs) or acini located at the ends of ducts that will become the milk-producing structures in the lactating mammary gland. Additional required hormones, EGF and IGF-1, promote the proliferation of terminal end-buds during normal breast development, and augment ductal outgrowth and side branching induced by estrogen plus progesterone [4,5]. In fact, PR isoform expression can not be induced in response to estrogen unless EGF is present [6], suggesting the existence of important cross talk between EGF receptors (EGFR) and/or family members (erbB2) and both steroid hormone receptors.
Another limitation to understanding progesterone/PR action in the context of breast cancer (i.e., uncontrolled proliferation) is that normal proliferating breast epithelial cells are steroid hormone receptor negative [7]. In the normal adult mammary gland, ER and PR are found in a minority luminal epithelial cell population of nondividing cells that lie adjacent to proliferating cells (Figures 1 & 2). These steroid receptor positive (ER+/PR+) cells represent only approximately 7–10% of the epithelial cell population. The most current information suggests that ER+/PR+ cells are capable of proliferating, but are ‘held’ in a nonproliferative or growth-inhibited state by inhibitory molecules, such as TGF-β or high levels of p27, the endogenous inhibitor of cell-cycle-dependent protein kinases (CDKs). Communication between the breast epithelial and stromal compartments mediates the proliferation of neighboring cells by expression and secretion of locally active proproliferative molecules such as Wnts, IGF-II [7], or stroma-derived HGF [8]. Recent evidence suggests that ER+/PR+ cells may serve as feeder cells by providing growth-promoting substances (e.g., Wnts) to nearby progenitor or stem cell populations [9].
Figure 1. Mammary gland architecture.
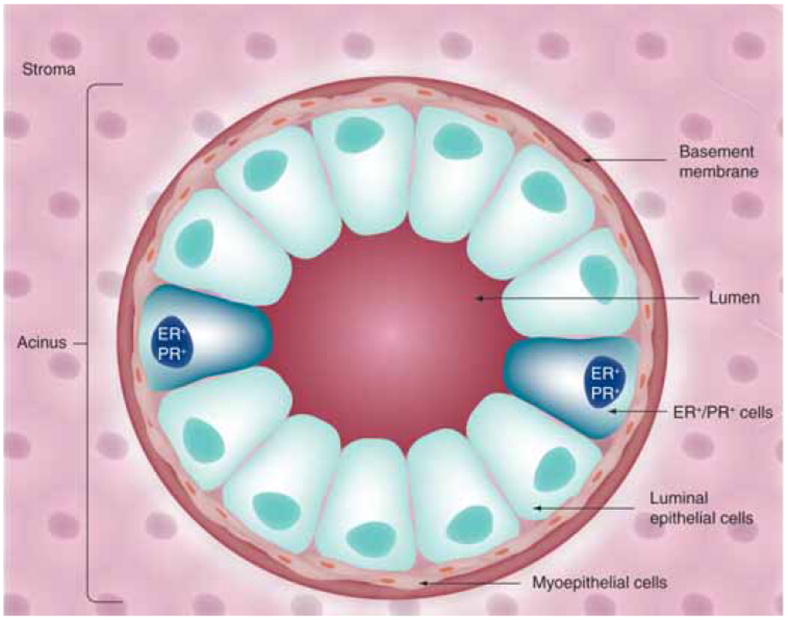
Acini, located at the ends of ducts in the mature mammary gland, are the functional units of the lactating mammary gland. Luminal epithelial cells (apical) are arranged as polar cells in contact with myoepithelial cells (basal). Epithelial cell populations are separated from the stromal environment by a basement membrane.
Figure 2. Paracrine signaling in the mammary gland.
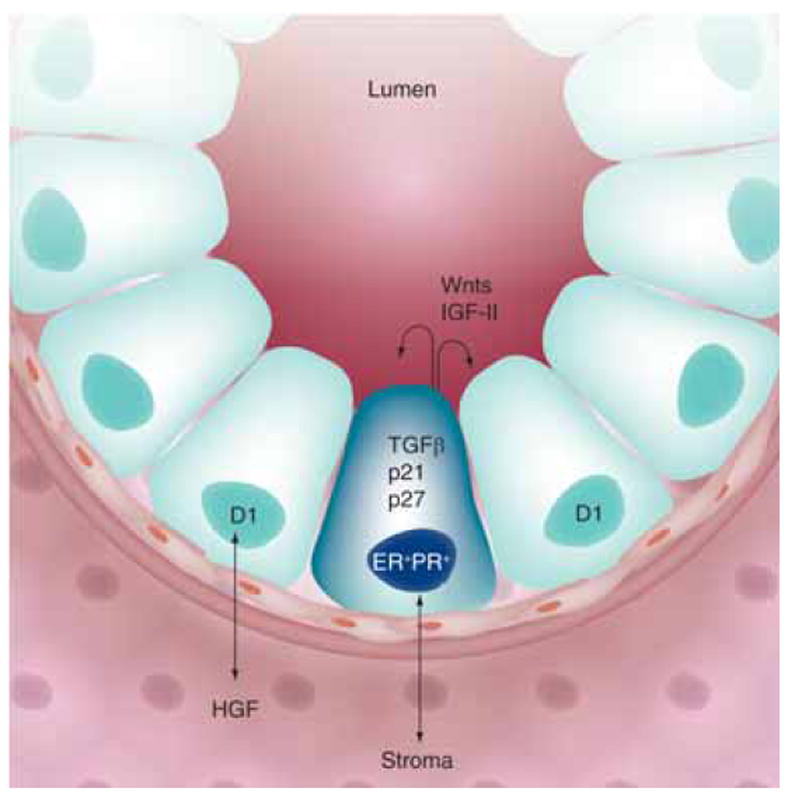
A minority population of steroid hormone receptor positive (ER+/PR+) cells occur adjacent to proliferating cells in the normal mammary gland. Communication between the epithelial and stromal compartments mediates proliferative paracrine signaling. Early events during breast cancer development may mediate switching from paracrine to autocrine mechanisms of proliferation.
In contrast to the normal breast, where proliferating cells are clearly devoid of steroid hormone receptors, the majority of breast cancers (~70%) express ER and PR at the time of diagnosis. Although hormone receptor-positive tumors are often slower growing relative to receptor-negative tumors [10], ER+/PR+ breast epithelial cells may undergo an early switch to autocrine or paracrine signaling mechanisms, whereby negative controls on proliferation are somehow lifted. Another setting where PR-containing cells divide is in the pregnant mammary gland, where PR-B colocalizes with cyclin D1 in BrdU-stained (dividing) cells, [11]. Thus, pathways involved in normal mammary gland growth and development may inappropriately ‘re-assert’ themselves during breast cancer progression. Experimental data in model organisms (primates, mice and rats) and humans suggest a proproliferative role for progestins [12-14]. Herein, we review the evidence regarding progesterone/PR action in breast cancer models, and relate these findings to the potential for future development of PR antagonists for clinical use as part of combined breast cancer therapies.
PR structure & function: classical & membrane-associated rapid signaling
PR isoforms are classically defined as ligand-activated transcription factors and members of a larger family of related steroid hormone receptors (including ER, androgen receptor [AR], glucocorticoid receptor [GR] and mineralocorticoid receptor) that regulate gene expression by binding directly or indirectly to specific sites in DNA (Figure 3). PRs are activated upon binding to the naturally occurring ovarian steroid hormone, progesterone, or via binding to synthetic ligands (progestins). Three PR isoforms (Figure 3) are the distinct protein products of a single gene located on chromosome 11 at q22–23 that undergoes transcription via the use of two alternate ‘distal’ and ‘proximal’ promoter regions [15]. The use of two internal translational start sites within common mRNAs results in the creation of three protein isoforms that consist of the full-length PR-B (116 kDa), N-terminally-truncated PR-A (94 kDa), and PR-C-isoforms (60 kDa). PR+ cells usually coexpress PR-A and PR-B isoforms; these receptors have different transcriptional activities within the same promoter context, but can also recognize entirely different gene promoters [16,17]. PR-B is required for normal mammary gland development [18], while PR-A is essential for uterine development and reproductive function [19]. PR-C is devoid of classical transcriptional activity, but when expressed, can enhance PR activity in breast cancer cells [20] or function as a dominant inhibitor of PR-B in the uterus [21].
Figure 3. Progesterone receptor structure.
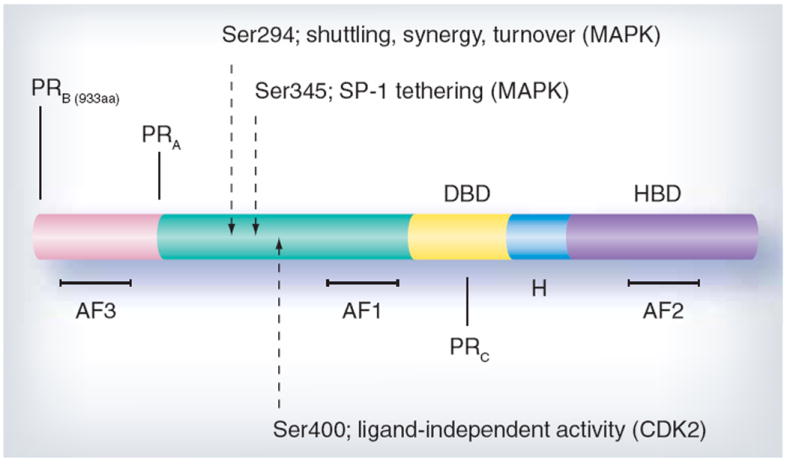
Progesterone receptor (PR)-B and -A are ligand-activated transcription factors containing a hormone-binding domain (HBD), hinge region (H), DNA-binding domain (DBD) and amino terminus. Activation functions (AFs) are the sites of coregulator interaction required for transcription. Serines 294, 345 and 400 are examples of regulatory sites that are phosphorlyated in response to progestins and/or mitogenic signaling pathways that modify PR function, in part by direct phosphorylation of PR or its coregulatory proteins.
In the absence of progesterone, PRs are complexed with chaperone molecules including heat shock proteins (HSPs); these interactions allow correct protein folding and assembly of stable PR molecules that are competent to bind hormone [22]. HSPs also function to connect PR to protein trafficking systems. After binding to progesterone, the receptors undergo conformational changes, dimerization and HSP dissociation (Figure 3). Activated receptors are withheld in the nucleus and associate with numerous coregulatory molecules, including histone acetyltransferases (CBP/p300, p160/SRC family steroid receptor coactivators 1–3), chromatin remodeling machines (SWI/SNF), and TRAP/DRIP complexes that recruit RNA polymerase II. Receptor/co-activator complexes bind directly to specific progesterone response elements (PREs) and PRE-like sequences in the promoter regions of target genes, such as c-myc [23], fatty acid synthetase [24] and MMTV [25]. Treatment with progestin also results in an upregulation of genes without classical PREs in their proximal promoter regions, such as epidermal growth factor receptor [26], c-fos [27], p21 [28], IRS-2 [29] and cyclin D1 [30]. Without canonical PREs, PR regulation of these genes can occur through PR tethering to other DNA-binding transcription factors, such as specificity protein 1 [28], activating protein 1 [31] or signal transducers and activators of transcription (Stats) [32,33].
The genomic or classical actions of steroid hormone treatment are delayed by several minutes to hours (i.e., following transcription and translation of target genes). Recently, however, rapidly occurring (within a few minutes) extra-nuclear or nongenomic effects of cell membrane-localized steroid hormone receptors have become appreciated as a major facet of steroid hormone receptor action. For example, progestin treatment of breast cancer cells causes a rapid and transient (2–10 min) activation of cytoplasmic protein kinases, including mitogen-activated protein kinase (MAPK), PI3K and p60-Src kinase [34-36]. Similar activities have been reported for membrane-associated ER and AR [37]. The mechanism of these effects involves direct binding of steroid hormone receptors to protein–protein interaction domains of signaling molecules located in or near the plasma membrane, in close proximity to growth factor receptors and their immediate effectors. Human PR contains a proline-rich (PXXP) motif that mediates direct binding to the Src-homology 3 (SH3) domains of signaling molecules in the p60-Src kinase family in a ligand-dependent manner [34]. In vitro experiments demonstrate that in the presence of progestin, purified PR-A and PR-B activate the c-Src-related protein kinase, Hck; PR-B but not PR-A activates c-Src and MAPKs in vivo. Mutation of the PXXP sequence in PR-B prevents c-Src/PR interaction and blocks progestin-induced activation of c-Src (or Hck) and p42/p44 MAPKs. Furthermore, mutation of the PR-B DNA-binding domain (DBD) abolished PR transcriptional activity without affecting progestin-induced c-Src or MAPK activation. Thus, nongenomic MAPK activation by progestin/PR-B/c-Src complexes probably occurs by way of a c-Src-dependent mechanism involving Ras activation of the MAPK module (Figure 4). ER, as well as other signaling and adaptor molecules, is suspected to reside in similar cytoplasmic signaling complexes, possibly in association with PR and c-Src [37].
Figure 4. Progesterone receptor function.
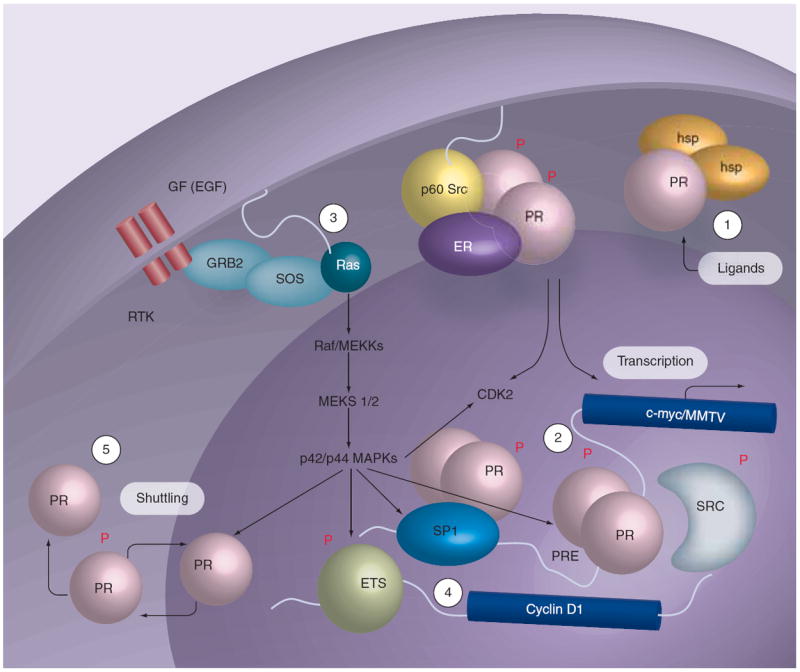
Phosphorylation (P) of specific sites in PR couples multiple receptor functions, including transcriptional synergy in the presence of steroid hormones and growth factors predicted to activate MAPK and/or CDK2, and nuclear import or export (shuttling) in response to MAPK activation. Rapid ligand-dependent PR downregulation occurs by the ubiquitin–proteasome pathway (degradation). 1. Ligand-binding mediates dissociation of heat-shock proteins and nuclear accumulation of PR. 2. Nuclear PR mediates gene regulation via the classical pathway; phosphorylated PR may recruit regulatory molecules that are phosphoproteins, and function in one or more interconnected processes (transcription, localization and turnover). 3. PR and growth factors activate MAPKs independently via a c-Src kinase-dependent pathway, and this may result in positive regulation of PR action via ‘feedback’ regulation (i.e., direct phosphorylation of liganded PR or co-activators), occurring in both the absence and presence of steroid hormone ligands and on PRE-containing or other PR-regulated gene promoters. 4. Activation of MAPKs by PR provides for regulation of gene targets whose promoters do not contain PREs and are otherwise independent of PR-transcriptional activities but utilize PR-activated MAPKs, such as regulation of the cyclin D1 promoter by ETS factors. 5. MAPK regulation of PR has been shown to mediate nuclear accumulation/shuttling and nuclear export that is coupled to regulation of PR transcriptional events.
ER: Estrogen receptor; PR: Progesterone receptor.
In studies using human breast or prostate cancer-cell lines, the rapid actions of AR, PR and/or ER have been shown to contribute to the regulation of cell proliferation in response to their respective hormone ligands [38-40]. While its role in human physiology (i.e., whole organisms) is less clear, steroid hormone receptor-mediated activation of cytoplasmic signaling molecules could theoretically serve to potentiate the nuclear functions of these receptors (Figures 3 & 4). For example, amplification of PR nuclear functions might occur through rapid, direct phosphorylation of PR proteins and/or their coregulators in response to activation of PR-induced cytoplasmic pathways that coincide with ligand binding. Thus, appropriately phosphorylated and activated receptor complexes are directed for efficient regulation of selected target genes. Clearly, such a positive feedback loop would explain the dramatic influence of activated signaling pathways on PR nuclear function. Indeed, several progestin-dependent functions of PR are also MAPK or c-Src-dependent, including upregulation of cyclins D1 and E, CDK2 activation, S-phase entry and anchorage-independent cell growth in soft agar [26,41,42]. C-Src and MAPK-dependent direct phosphorylation of PR is required for PR tethering to SP1 transcription factors on the p21 and EGFR promoters [43]. Kinases can also confer hyperactivity and ligand-independence to phosphorylated PR-B [42,44,45]. Although more studies are needed, it is becoming clear that activation of cytoplasmic protein kinases is an integral feature of steroid hormone receptor action in the nucleus (i.e., phosphorylation events are required for gene regulation leading to changes in cell biology).
How might the extranuclear actions of steroid hormone receptors, including PR, contribute to deregulated breast cancer cell growth and/or increased breast cancer risk? Perhaps by linking steroid hormone action to the regulation of MAPK-regulated genes (i.e., the end point of MAPK signaling is the regulation of transcription factor substrates). In support of this concept, the extranuclear actions of liganded ER are thought to induce a state of ‘adaptive hypersensitivity’ during endocrine therapy in which growth factor signaling pathways are co-opted by upregulated ER [46]. In this model of ER-dependent MAPK activation, liganded ER associated with the cell membrane interacts with the adapter protein Shc and induces its phosphorylation, leading to recruitment of adaptor molecules and activation of Ras and the Raf-1/MEK/MAPK module. MAPK then regulates genes via phosphorylation of ETS factors and/or AP1 components (i.e., independently of ER transcriptional activity). ER activation of MAPK may explain why many tumors respond well to aromatase inhibitors, yet fail to respond to selective ER modulators (SERMs) designed to inhibit ER transcriptional activity in the nucleus, but not ER-dependent MAPK activation in the cytoplasm. Breast cancers often exhibit heightened c-Src and MAPK activities relative to normal breast tissue [47,48]. Steroid hormone receptors including ER, AR, and PR may contribute to the constitutive signaling of these mitogenic kinases via their membrane-associated activities, thereby circumventing endocrine-based therapies (i.e., antiestrogen).
Progesterone action in animal models
Studies in rodents demonstrate that PR-A and PR-B are differentially expressed during pregnancy-induced mammary gland development, with PR-A predominantly expressed during ductal sidebranching, while PR-B expression coincided with the formation of alveoli [49,50]. PR-B but not PR-A was expressed in proliferating cells. Some but not all proliferating cells in both compartments were PR-B+, suggesting that progesterone can induce proliferation through either direct or paracrine mechanisms. By contrast, cells in adult virgin glands were PR and cyclin D1 positive, but failed to proliferate, possibly as a result of high levels of the cyclin-dependent protein kinase inhibitors, p21 and p27 [50]. During the menstrual cycle, cells in the mammary gland undergo sequential waves of proliferation and apoptosis. Notably, in primates (macaques and humans) increased terminal duct lobular unit (TDLU) cell proliferation coincides with peaking serum progesterone levels that occur during the luteal phase [13,51-53], again suggesting a paracrine mechanism for this hormone in adult tissues. Upregulation of local IGF-1 has been suggested as a cooperating factor in this regard [54]. In animal models of postmenopausal hormone-replacement therapy, either parous or nulliparous early and late postmenopausal mice were subjected to estrogen alone (E) or estrogen plus progestin (E+P). In this study, E+P produced a greater proliferative response relative to E regardless of parity or treatment time. E+P acted directly on the mammary gland, rather than via systemic effects [55]. Similar results were reported in surgically postmenopausal macaques [56] and in postmenopausal humans [57]. Although breast cancer development was not modeled in the above animal studies, the results (i.e., proliferation) are in good agreement with human clinical data [58,59], which revealed increased tumor number and size in women taking E+P, while E alone did not significantly alter breast cancer risk or tumor size (discussed below).
Progesterone may act primarily via proto-oncogenes and growth factors to affect breast cell proliferation and breast cancer etiology. As the majority of early breast cancer lesions express ER and PR and these receptors remain high in 60% of advanced disease, early events may include a switch in the ability of quiescent ER+/PR+ cells to respond directly to steroid hormones and proliferate. Notably, deregulation of the cell cycle appears to be a hallmark of breast cancer. At least 40% of breast cancers overexpress cyclin D1, while up to 30% have lost p27 or p21 and/or contain elevated CDK2 activity [60,61]. Loss or mutation of p53 is also a frequent occurrence [62]. Numerous in vitro studies have demonstrated linkage of PR action to cell-cycle control [42,63-65]. Namely, PR interacts directly with CDK2 and cyclins A and E [64,65]. PR activity is highest in the DNA synthesis (S) phase of the cell cycle, when CDK2 activity is also very high [63,64]. Furthermore, PR transcriptional activity becomes ligand-independent and CDK2-driven upon loss of p27 [42]. Progestins, acting through PR-B-dependent transcriptional events, induce cyclin D1 expression and cell cycle re-entry in antiestrogen-arrested breast cancer cells [66]. This suggests that in the breast, progesterone/PR action is tightly coupled to cell-cycle regulation. In breast cancer, the mitogenic potential of activated PR may manifest particularly during loss of checkpoint control and/or elevation of selected kinase activities. TGF-α transgenic mice develop ER+/PR+ proliferative hyperplasias (early lesions) that progress to ER+/PR- tumors [67]. Recent in vitro studies demonstrate that knockdown of BRCA-1 enhances progestin-induced PR activity, while progestin-induced proliferation is increased in genetically engineered mice that lack BRCA-1 in the mammary gland [68]. In related studies using mice lacking mammary gland expression of both BRCA-1 and p53, the development of aggressive mammary tumors in virgin mice was completely blocked by antiprogestins [69]. It will be important to define how negative regulation of cell proliferation in ER+/PR+ normal MECs is somehow lifted to allow progression of early lesions to full-blown cancer, and if early events include loss or alteration of DNA damage and repair pathways and/or checkpoint control in PR+ cells.
Progesterone action in human breast cancer cells
The biochemistry of PR action is well understood, having been largely defined using PR+ human breast cancer cell line models, or PR-null cells into which wild-type or modified PR has been re-expressed. Numerous studies have focused on PR interactions with other regulatory proteins, changes in PR subcellular localization, or post-translational modifications (i.e., phosphorylation, ubiquitinylation or sumoylation) or other conditions that affect PR transcriptional activities, usually measured on artificial gene promoters (reporter genes) that contain one or more tandem progestone responsive elements (PRE) sites [70]. Growth factors, including EGF or heregulin, promote transcriptional synergy with progestins on PR-target genes [45,71,72]. Phosphorylation events primarily serve to augment PR action in a promoter-selective manner [73]. Despite this depth of basic understanding, gene regulation and the associated changes in cell biology in response to PR activation remain elusive. Only a handful of endogenous progesterone-responsive genes have been described in moderate detail [23,29,74]. The majority of genes regulated in response to progesterone lack PR-binding consensus sequences or PREs, and the presence of one or more PREs or PRE half-sites does not predict progesterone-responsive regulation [17]. Numerous genes are regulated upon PR expression, but independently of progesterone [16,75]. Furthermore, many genes are downregulated in response to progesterone/PR-dependent transcriptional repression, largely by unknown mechanisms [16,17]. In most cases, the regulation of particular genes in response to progesterone/PR is only loosely tied (by correlation) to changes in cell biology. For example, many PR-regulated genes have been associated with aspects of tumor progression towards aggressive tumor phenotype. In addition, variation of the PR-A:PR-B ratio is a frequent occurrence in breast tumors relative to normal tissue [76], and is predicted to dramatically alter the genetic program [16,77].
Confounding the role of progesterone in breast cancer is that progesterone has biphasic effects on the proliferation of breast cancer cell lines grown in vitro (cells grown in plastic culture dishes supplemented with progestin-containing media). Cultured cell lines generally undergo an initial burst of proliferation characterized by increased S-phase entry that peaks at 18 h of progesterone treatment [30,78,79]. Cell-cycle progression is driven by upregulation of G1/S and G2 cyclins, p21 and elevated CDK2 activity. This is followed 24–48 h (1–3 cell cycles) later by a period of cell-growth inhibition in which p27 is upregulated and the cells ultimately arrest at the G1/S boundary. Thus, the response of cultured breast cancer cells to progesterone is both proliferative and inhibitory, in sharp contrast to the clear mitogenic effects of estrogens in the same cell-line models. For this reason, it has been suggested that progesterone acts primarily as a priming agent, with growth-promoting activity dependent upon cellular context [80]. For example, progestins upregulate many of the components of growth factor-initiated signaling pathways, including IRS-2 [29], and EGFR family members and their ligands; progestin-treated breast cancer cells are much more responsive to EGF-induced proliferation than are progestin-naive cells [30]. Thus, progesterone may act primarily by sensitizing breast cancer cells to growth factor and cytokine signals [81].
Notably, progesterone treatment of PR+ cells growing in culture has also been implicated in prosurvival (resistance to chemotherapy-induced apoptosis) [82], and tumor cell differentiation (from luminal to myoepithelial phenotype) without effects on tumor growth [83]; this transition is associated with poor prognosis in breast cancer. Similarly, epithelial to mesenchymal transition (EMT) is an early transitional event that precedes tumor cell invasion and metastasis, which may occur independently of changes in proliferation. During EMT, stationary epithelial cells become more like fibroblast cells and acquire the ability to migrate and invade locally. Interestingly, many of the genes regulated in response to progesterone include those encoding molecules involved in signal transduction and cell adhesion to extracellular matrix (ECM) or other basement membrane components [16,17]. IRS-2, a PR-B-specific target gene, is a mediator of increased cell motility [84]. The effects of progestins have recently been evaluated in soft agar, a form of 3D culture that allows breast cancer cells to grow as anchorage-independent colonies more similar to the organization of acini found in vivo, instead of cell monolayers. Progestins are clearly mitogenic in this system, wherein PR-B induces transcriptional upregulation of Wnt-1, leading to sustained MAPK activity, upregulation of cyclin D1 protein levels, and the formation of abundant large colonies [41]. This suggests that the mitogenic actions of progesterone may require the establishment of cell polarity, a property that is not supported in 2D culture systems. In addition to gaining further insight into the role of extensive cross talk between progesterone/PR and the components of signaling pathways, it will be important to more clearly define specific actions of progesterone/PR that are relevant to more advanced breast cancer cell biology (i.e., tumor progression to metastasis, including EMT). Perhaps this should include study conditions that mimic or preserve breast epithelial cell architecture in which PR+ luminal epithelial cells are in direct contact with basement membrane components (Figure 1). In support of this notion, normal MECs respond differently to estrogen and progesterone when cultured in the presence of different ECM components (i.e., collagen type I, fibronectin and laminin) [8]; ECM proteins increase the expression of EGF and IGF receptors, but E+P inhibits EGF/IGF-induced MEC proliferation in an ECM-dependent manner [85]. Clearly, ER and PR interactions with ECM are very complex [8]. Deregulation of ECM protein expression and/or integrin signaling (i.e., early events in cancer progression) is thus predicted to dramatically alter hormone responsiveness.
Clinical findings & future perspective
A direct role for PR in breast cancer is perhaps best illustrated by the clinical findings of the Women’s Health Initiative (WHI) and Million Women Study, demonstrating that women taking a progestin in combination with estrogen (EPT) as part of hormone replacement therapy, experienced a greater breast cancer risk relative to estrogen alone; tumors were larger and of higher grade [58,59]. The Million Women Study also found that women were more likely to die of breast cancer if they were taking EPT at the time of diagnosis. When the findings of the WHI were announced in mid-2002, the number of prescriptions for EPT dramatically decreased. Population-based studies have shown a decline in breast cancer incidence immediately after these data were made public [86,87]. While it is impossible to prove that the decreased use of EPT caused the decline in breast cancer incidence, this is certainly a plausible explanation. It is possible that discontinuation of EPT suppressed the appearance of breast cancers that would have become evident with continued hormone stimulation. Following the incidence trends will be necessary to determine the magnitude of the effect of EPT in changing breast cancer incidence.
Many aspects of PR action originally discovered in animal or cell-line models of breast cancer have not been well-established in humans. Convincing proof of the role of PR in breast cancer requires demonstration that perturbation of PR function results in important clinical outcomes. For example, we know that high-dose synthetic progestin administration (megestrol and medroxyprogesterone) has activity in advanced breast cancer [88] in a manner analogous to the clinical activity of the synthetic estrogen (diethylstilbesterol) in advanced breast cancer [89]. High-dose estrogen therapy for breast cancer has largely been replaced by selective estrogen receptor modulators (SERMs; tamoxifen, torimifene and fulvestrant) or ligand-deprivation strategies in which aromatase inhibitors are used to prevent the conversion of steroid hormone precursors into estradiol. If progesterone receptor modulators (PRMs) could be shown to alter the growth of PR-expressing breast cancer cells in human clinical trials, then this would clearly validate the findings made in vitro and in animal model systems.
Several PRMs have been described. Unfortunately, the clinically tested PRMs are not selective and have cross reactivity with GR [90]. Despite this concern, two small trials in breast cancer using these drugs have been reported. Mifepristone has been examined in women with advanced breast cancer and a low response rate was observed. In addition, the side effects of lethargy, nausea and anorexia were noted [91]. Mifepristone has also been tested in meningioma and given for longer periods of time with less toxicity [92]. In this study, glucocorticoids were given during the first 2 weeks of therapy and it is possible that this coadministration of steroids to avoid GR inhibition reduced the side effects. Onapristone, another PRM, has also been tested in breast cancer. This drug was associated with a substantial response rate and sustained stable disease (clinical benefit of 63%), but caused hepatotoxicity that necessitated discontinuation of the trial [93].
In sum, while the bulk of preclinical data suggest an important role for PR function in modulating breast cancer biology, validation of these findings are dependent on a clinical strategy to disrupt PR function in human breast cancers. It will then be important to decipher the contribution of nuclear and membrane PR activities, and target them appropriately with selective PRMs, in addition to targeting the relevant kinases (c-Src, MAPKs and CDK2) required for steroid hormone receptor action. We predict that PR activities will one day be routinely targeted as part of combination therapies aimed at blocking both ER-α and PR-B, along with the associated essential protein kinases.
Executive summary.
Background
Progesterone is difficult to study in isolation from other hormones and growth factors that similarly influence mammary gland biology and breast cancer (namely, estrogen, prolactin, EGF, IGF and HGF).
Progesterone receptor structure & function
Progesterone receptors (PRs) are classically defined as ligand-activated transcription factors, but also function at or near the plasma membrane to directly activate protein kinase pathways (namely, c-Src and the MAP kinase module consisting of Raf-1, MEK1/2 and ERK1/2). This is commonly called ‘rapid’ signaling.
The function of rapid signaling is unknown, but may provide positive regulation of PR-containing transcriptional complexes by direct phosphorylation of PR or coregulatory molecules. Phosphorylation also provides a mechanism for promoter selectivity.
Progesterone action in animal models
PR-B is required for normal mammary gland development; PR+ cells in the mammary gland may interact primarily with stromal components to mediate proliferative signaling of nearby or neighboring PR-null cells via the action of locally acting growth factors (namely IGF-II, Wnts and/or stromal HGF).
Studies in mice, rats, monkeys and humans support the concept that progesterone/PR contributes to proliferative signaling in the normal adult and neoplastic mammary gland.
Progesterone action in human breast cancer cells
Both the rapid actions and the transcriptional activity of PR-B contribute to breast cancer cell proliferation in response to progesterone.
Progesterone/PR act in concert with mitogenic protein kinases and cell-cycle mediators to induce regulation of PR target genes and proliferative responses.
Progesterone acts to sensitize breast cancer cells to the actions of growth factors by upregulation of target genes that include the components of signal transduction pathways (namely, EGFR and EGFR ligands, IRS-2, cyclins D and E, p21).
Understudied areas of progesterone/PR action include the regulation of breast cancer prosurvival and early events in breast cancer cell invasion and metastasis, including preneoplastic transitions during tumor progression (i.e., not necessarily related to altered proliferation).
Clinical findings & future perspective
Selective progesterone receptor modulators (PRMs) have significant activity against breast cancer in clinical trials, but have not been well tolerated by most patients, most likely owing to their high affinity and cross-reactivity to glucocorticoid receptors.
New selective PRMs must be developed before PR may be directly targeted. However, protein kinases that are critical for PR action may also provide effective therapeutic targets; c-Src, CDK2 and/or MAP kinase inhibitors should be considered as part of combination therapies that could be used to complement the use of anitestrogens and antiprogestins.
Acknowledgments
This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received, or pending or royalties.
Footnotes
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
No writing assistance was utilized in the production of this manuscript.
Bibliography
- 1.Hewitt SC, Korach KS. Progesterone action and responses in the αERKO mouse. Steroids. 2000;65(10–11):551–557. doi: 10.1016/s0039-128x(00)00113-6. [DOI] [PubMed] [Google Scholar]
- 2.Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-α expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci USA. 2007;104(37):14718–14723. doi: 10.1073/pnas.0706933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mulac-Jericevic B, Conneely OM. Reproductive tissue selective actions of progesterone receptors. Reproduction. 2004;128(2):139–146. doi: 10.1530/rep.1.00189. [DOI] [PubMed] [Google Scholar]
- 4.Haslam SZ, Counterman LJ, Nummy KA. Effects of epidermal growth factor, estrogen, and progestin on DNA synthesis in mammary cells in vivo are determined by the developmental state of the gland. J Cell Physiol. 1993;155(1):72–78. doi: 10.1002/jcp.1041550110. [DOI] [PubMed] [Google Scholar]
- 5.Ruan W, Monaco ME, Kleinberg DL. Progesterone stimulates mammary gland ductal morphogenesis by synergizing with and enhancing insulin-like growth factor-I action. Endocrinology. 2005;146(3):1170–1178. doi: 10.1210/en.2004-1360. [DOI] [PubMed] [Google Scholar]
- 6.Ankrapp DP, Bennett JM, Haslam SZ. Role of epidermal growth factor in the acquisition of ovarian steroid hormone responsiveness in the normal mouse mammary gland. J Cell Physiol. 1998;174(2):251–260. doi: 10.1002/(SICI)1097-4652(199802)174:2<251::AID-JCP12>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 7.Robinson GW, Hennighausen L, Johnson PF. Side-branching in the mammary gland: the progesterone–Wnt connection. Genes Dev. 2000;14(8):889–894. [PubMed] [Google Scholar]
- 8.Haslam SZ, Woodward TL. Host microenvironment in breast cancer development: Epithelial-cell-stromal-cell interactions and steroid hormone action in normal and cancerous mammary gland. Breast Cancer Res. 2003;5(4):208–215. doi: 10.1186/bcr615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Rosen JM. Stem/progenitor cells in mouse mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia. 2005;10(1):17–24. doi: 10.1007/s10911-005-2537-2. [DOI] [PubMed] [Google Scholar]
- 10.Wenger CR, Clark GM. S-phase fraction and breast cancer – a decade of experience. Breast Cancer Res Treat. 1998;51(3):255–265. doi: 10.1023/a:1006188512927. [DOI] [PubMed] [Google Scholar]
- 11.Aupperlee MD, Smith KT, Kariagina A, Haslam SZ. Progesterone receptor isoforms A and B: temporal and spatial differences in expression during murine mammary gland development. Endocrinology. 2005;146(8):3577–3588. doi: 10.1210/en.2005-0346. [DOI] [PubMed] [Google Scholar]
- 12.Chlebowski RT, Hendrix SL, Langer RD, et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women’s Health Initiative Randomized Trial. JAMA. 2003;289(24):3243–3253. doi: 10.1001/jama.289.24.3243. [DOI] [PubMed] [Google Scholar]
- 13.Graham JD, Clarke CL. Physiological action of progesterone in target tissues. Endocr Rev. 1997;18(4):502–519. doi: 10.1210/edrv.18.4.0308. [DOI] [PubMed] [Google Scholar]
- 14.Haslam SZ, Osuch JR, Raafat AM, Hofseth LJ. Postmenopausal hormone replacement therapy: effects on normal mammary gland in humans and in a mouse postmenopausal model. J Mammary Gland Biol Neoplasia. 2002;7(1):93–105. doi: 10.1023/a:1015726608146. [DOI] [PubMed] [Google Scholar]
- 15.Kraus WL, Montano MM, Katzenellenbogen BS. Cloning of the rat progesterone receptor gene 5′-region and identification of two functionally distinct promoters. Mol Endocrinol. 1993;7(12):1603–1616. doi: 10.1210/mend.7.12.8145766. [DOI] [PubMed] [Google Scholar]
- 16.Jacobsen BM, Richer JK, Schittone SA, Horwitz KB. New human breast cancer cells to study progesterone receptor isoform ratio effects and ligand-independent gene regulation. J Biol Chem. 2002;277(31):27793–27800. doi: 10.1074/jbc.M202584200. [DOI] [PubMed] [Google Scholar]
- 17.Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277(7):5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- 18.Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci USA. 2003;100(17):9744–9749. doi: 10.1073/pnas.1732707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulac-Jericevic B, Mullinax RA, DeMayo FJ, Lydon JP, Conneely OM. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science. 2000;289(5485):1751–1754. doi: 10.1126/science.289.5485.1751. [DOI] [PubMed] [Google Scholar]
- 20.Wei LL, Norris BM, Baker CJ. An N-terminally truncated third progesterone receptor protein, PR(C), forms heterodimers with PR(B) but interferes in PR(B)-DNA binding. J Steroid Biochem Mol Biol. 1997;62(4):287–297. doi: 10.1016/s0960-0760(97)00044-7. [DOI] [PubMed] [Google Scholar]
- 21.Condon JC, Hardy DB, Kovaric K, Mendelson CR. Up-regulation of the progesterone receptor (PR)-C isoform in laboring myometrium by activation of nuclear factor-kappaB may contribute to the onset of labor through inhibition of PR function. Mol Endocrinol. 2006;20(4):764–775. doi: 10.1210/me.2005-0242. [DOI] [PubMed] [Google Scholar]
- 22.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228(2):111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 23.Moore MR, Zhou JL, Blankenship KA, Strobl JS, Edwards DP, Gentry RN. A sequence in the 5′ flanking region confers progestin responsiveness on the human c-myc gene. J Steroid Biochem Mol Biol. 1997;62(4):243–252. doi: 10.1016/s0960-0760(97)00036-8. [DOI] [PubMed] [Google Scholar]
- 24.Chalbos D, Chambon M, Ailhaud G, Rochefort H. Fatty acid synthetase and its mRNA are induced by progestins in breast cancer cells. J Biol Chem. 1987;262(21):9923–9926. [PubMed] [Google Scholar]
- 25.Krusekopf S, Chauchereau A, Milgrom E, Henderson D, Cato AC. Co-operation of progestational steroids with epidermal growth factor in activation of gene expression in mammary tumor cells. J Steroid Biochem Mol Biol. 1991;40(1–3):239–245. doi: 10.1016/0960-0760(91)90188-b. [DOI] [PubMed] [Google Scholar]
- 26.Lange CA, Richer JK, Shen T, Horwitz KB. Convergence of progesterone and epidermal growth factor signaling in breast cancer. Potentiation of mitogen-activated protein kinase pathways. J Biol Chem. 1998;273(47):31308–31316. doi: 10.1074/jbc.273.47.31308. [DOI] [PubMed] [Google Scholar]
- 27.Richer JK, Lange CA, Wierman AM, et al. Progesterone receptor variants found in breast cells repress transcription by wild-type receptors. Breast Cancer Res Treat. 1998;48(3):231–241. doi: 10.1023/a:1005941117247. [DOI] [PubMed] [Google Scholar]
- 28.Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin-dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273(17):10696–10701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 29.Cui X, Lazard Z, Zhang P, Hopp TA, Lee AV. Progesterone crosstalks with insulin-like growth factor signaling in breast cancer cells via induction of insulin receptor substrate-2. Oncogene. 2003;22(44):6937–6941. doi: 10.1038/sj.onc.1206803. [DOI] [PubMed] [Google Scholar]
- 30.Groshong SD, Owen GI, Grimison B, et al. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1) Mol Endocrinol. 1997;11(11):1593–1607. doi: 10.1210/mend.11.11.0006. [DOI] [PubMed] [Google Scholar]
- 31.Tseng L, Tang M, Wang Z, Mazella J. Progesterone receptor (hPR) upregulates the fibronectin promoter activity in human decidual fibroblasts. DNA Cell Biol. 2003;22(10):633–640. doi: 10.1089/104454903770238102. [DOI] [PubMed] [Google Scholar]
- 32.Proietti C, Salatino M, Rosemblit C, et al. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 (Stat3) via a Jak- and Src-dependent mechanism in breast cancer cells. Mol Cell Biol. 2005;25(12):4826–4840. doi: 10.1128/MCB.25.12.4826-4840.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richer JK, Lange CA, Manning NG, Owen G, Powell R, Horwitz KB. Convergence of progesterone with growth factor and cytokine signaling in breast cancer. Progesterone receptors regulate signal transducers and activators of transcription expression and activity. J Biol Chem. 1998;273(47):31317–31326. doi: 10.1074/jbc.273.47.31317. [DOI] [PubMed] [Google Scholar]
- 34.Boonyaratanakornkit V, Scott MP, Ribon V, et al. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol Cell. 2001;8(2):269–280. doi: 10.1016/s1097-2765(01)00304-5. [DOI] [PubMed] [Google Scholar]
- 35.Migliaccio A, Piccolo D, Castoria G, et al. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 1998;17(7):2008–2018. doi: 10.1093/emboj/17.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saitoh M, Ohmichi M, Takahashi K, et al. Medroxyprogesterone acetate induces cell proliferation through up-regulation of cyclin D1 expression via phosphatidylinositol 3-kinase/Akt/nuclear factor-κB cascade in human breast cancer cells. Endocrinology. 2005;146(11):4917–4925. doi: 10.1210/en.2004-1535. [DOI] [PubMed] [Google Scholar]
- 37.Shupnik MA. Crosstalk between steroid receptors and the c-Src-receptor tyrosine kinase pathways: implications for cell proliferation. Oncogene. 2004;23(48):7979–7989. doi: 10.1038/sj.onc.1208076. [DOI] [PubMed] [Google Scholar]
- 38.Migliaccio A, Castoria G, Di Domenico M, Ballare C, Beato M, Auricchio F. The progesterone receptor/estradiol receptor association and the progestin-triggered S-phase entry. Ernst Schering Res Found Workshop; 2005. pp. 39–54. [DOI] [PubMed] [Google Scholar]
- 39.Migliaccio A, Castoria G, Di Domenico M, et al. Steroid-induced androgen receptor-oestradiol receptor β-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19(20):5406–5417. doi: 10.1093/emboj/19.20.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skildum A, Faivre E, Lange CA. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol. 2005;19(2):327–339. doi: 10.1210/me.2004-0306. [DOI] [PubMed] [Google Scholar]
- 41.Faivre E, Lange C. Progesterone receptors upregulate Wnt-1 to induce EGFR transactivation and c-Src-dependent sustained activation of Erk1/2 MAP kinase in breast cancer cells. Mol Cell Biol. 2006;27(2):466–480. doi: 10.1128/MCB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pierson-Mullany LK, Lange CA. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol. 2004;24(24):10542–10557. doi: 10.1128/MCB.24.24.10542-10557.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faivre E, Daniel AR, Hillard C, Lange CA. Progesterone receptor rapid signaling mediates Ser345 phosphorylation and tethering to Sp1 transcription factors. Mol Endocrinol. 2008 doi: 10.1210/me.2007-0437. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Labriola L, Salatino M, Proietti CJ, et al. Heregulin induces transcriptional activation of the progesterone receptor by a mechanism that requires functional ErbB-2 and mitogen-activated protein kinase activation in breast cancer cells. Mol Cell Biol. 2003;23(3):1095–1111. doi: 10.1128/MCB.23.3.1095-1111.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiu M, Lange CA. MAP kinases couple multiple functions of human progesterone receptors: degradation, transcriptional synergy, and nuclear association. J Steroid Biochem Mol Biol. 2003;85:147–157. doi: 10.1016/s0960-0760(03)00221-8. [DOI] [PubMed] [Google Scholar]
- 46.Santen R, Jeng MH, Wang JP, et al. Adaptive hypersensitivity to estradiol: potential mechanism for secondary hormonal responses in breast cancer patients. J Steroid Biochem Mol Biol. 2001;79(1–5):115–125. doi: 10.1016/s0960-0760(01)00151-0. [DOI] [PubMed] [Google Scholar]
- 47.Gee JM, Robertson JF, Ellis IO, Nicholson RI. Phosphorylation of ERK1/2 mitogen-activated protein kinase is associated with poor response to anti-hormonal therapy and decreased patient survival in clinical breast cancer. Int J Cancer. 2001;95(4):247–254. doi: 10.1002/1097-0215(20010720)95:4<247::aid-ijc1042>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 48.Wilson GR, Cramer A, Welman A, et al. Activated c-SRC in ductal carcinoma in situ correlates with high tumour grade, high proliferation and HER2 positivity. Br J Cancer. 2006;95(10):1410–1414. doi: 10.1038/sj.bjc.6603444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aupperlee MD, Haslam SZ. Differential hormonal regulation and function of progesterone receptor isoforms in normal adult mouse mammary gland. Endocrinology. 2007;148(5):2290–2300. doi: 10.1210/en.2006-1721. [DOI] [PubMed] [Google Scholar]
- 50.Kariagina A, Aupperlee MD, Haslam SZ. Progesterone receptor isoforms and proliferation in the rat mammary gland during development. Endocrinology. 2007;148(6):2723–2736. doi: 10.1210/en.2006-1493. [DOI] [PubMed] [Google Scholar]
- 51.Navarrete MA, Maier CM, Falzoni R, et al. Assessment of the proliferative, apoptotic and cellular renovation indices of the human mammary epithelium during the follicular and luteal phases of the menstrual cycle. Breast Cancer Res. 2005;7(3):R306–R313. doi: 10.1186/bcr994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soderqvist G, Isaksson E, von Schoultz B, Carlstrom K, Tani E, Skoog L. Proliferation of breast epithelial cells in healthy women during the menstrual cycle. Am J Obstet Gynecol. 1997;176(1 Pt 1):123–128. doi: 10.1016/s0002-9378(97)80024-5. [DOI] [PubMed] [Google Scholar]
- 53.Stute P, Wood CE, Kaplan JR, Cline JM. Cyclic changes in the mammary gland of cynomolgus macaques. Fertil Steril. 2004;82(Suppl. 3):1160–1170. doi: 10.1016/j.fertnstert.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 54.Dabrosin C. Increase of free insulin-like growth factor-1 in normal human breast in vivo late in the menstrual cycle. Breast Cancer Res Treat. 2003;80(2):193–198. doi: 10.1023/A:1024575103524. [DOI] [PubMed] [Google Scholar]
- 55.Haslam SZ. Experimental mouse model of hormonal therapy effects on the postmenopausal mammary gland. Breast Dis. 2005;24:71–78. doi: 10.3233/bd-2006-24106. [DOI] [PubMed] [Google Scholar]
- 56.Soderqvist G. Effects of sex steroids on proliferation in normal mammary tissue. Ann Med. 1998;30(6):511–524. [PubMed] [Google Scholar]
- 57.Hofseth LJ, Raafat AM, Osuch JR, Pathak DR, Slomski CA, Haslam SZ. Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in the normal postmenopausal breast. J Clin Endocrinol Metab. 1999;84(12):4559–4565. doi: 10.1210/jcem.84.12.6194. [DOI] [PubMed] [Google Scholar]
- 58.Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291(14):1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 59.Beral V. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362(9382):419–427. doi: 10.1016/s0140-6736(03)14065-2. [DOI] [PubMed] [Google Scholar]
- 60.Alkarain A, Jordan R, Slingerland J. p27 deregulation in breast cancer: prognostic significance and implications for therapy. J Mammary Gland Biol Neoplasia. 2004;9(1):67–80. doi: 10.1023/B:JOMG.0000023589.00994.5e. [DOI] [PubMed] [Google Scholar]
- 61.Steeg PS, Zhou Q. Cyclins and breast cancer. Breast Cancer Res Treat. 1998;52(1–3):17–28. doi: 10.1023/a:1006102916060. [DOI] [PubMed] [Google Scholar]
- 62.Kumar S, Walia V, Ray M, Elble RC. p53 in breast cancer: mutation and countermeasures. Front Biosci. 2007;12:4168–4178. doi: 10.2741/2378. [DOI] [PubMed] [Google Scholar]
- 63.Narayanan R, Adigun AA, Edwards DP, Weigel NL. Cyclin-dependent kinase activity is required for progesterone receptor function: novel role for cyclin A/Cdk2 as a progesterone receptor coactivator. Mol Cell Biol. 2005;25(1):264–277. doi: 10.1128/MCB.25.1.264-277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Narayanan R, Edwards DP, Weigel NL. Human progesterone receptor displays cell cycle-dependent changes in transcriptional activity. Mol Cell Biol. 2005;25(8):2885–2898. doi: 10.1128/MCB.25.8.2885-2898.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pierson-Mullany LK, Skildum A, Faivre E, Lange CA. Cross-talk between growth factor and progesterone receptor signaling pathways: implications for breast cancer cell growth. Breast Dis. 2003;18:21–31. doi: 10.3233/bd-2003-18104. [DOI] [PubMed] [Google Scholar]
- 66.McGowan EM, Russell AJ, Boonyaratanakornkit V, et al. Progestins reinitiate cell cycle progression in antiestrogen-arrested breast cancer cells through the B-isoform of progesterone receptor. Cancer Res. 2007;67(18):8942–8951. doi: 10.1158/0008-5472.CAN-07-1255. [DOI] [PubMed] [Google Scholar]
- 67.Rose-Hellekant TA, Sandgren EP. Transforming growth factor α- and c-myc-induced mammary carcinogenesis in transgenic mice. Oncogene. 2000;19(8):1092–1096. doi: 10.1038/sj.onc.1203350. [DOI] [PubMed] [Google Scholar]
- 68.Ma Y, Katiyar P, Jones LP, et al. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol. 2006;20(1):14–34. doi: 10.1210/me.2004-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314(5804):1467–1470. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- 70.Lange CA, Gioeli D, Hammes SR, Marker PC. Integration of rapid signaling events with steroid hormone receptor action in breast and prostate cancer. Annu Rev Physiol. 2007;69:171–199. doi: 10.1146/annurev.physiol.69.031905.160319. [DOI] [PubMed] [Google Scholar]
- 71.Daniel AR, Qiu M, Faivre EJ, Ostrander JH, Skildum A, Lange CA. Linkage of progestin and epidermal growth factor signaling: phosphorylation of progesterone receptors mediates transcriptional hypersensitivity and increased ligand-independent breast cancer cell growth. Steroids. 2007;72(2):188–201. doi: 10.1016/j.steroids.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shen T, Horwitz KB, Lange CA. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol Cell Biol. 2001;21(18):6122–6131. doi: 10.1128/MCB.21.18.6122-6131.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daniel AR, Faivre EJ, Lange CA. Phosphorylation-dependent antagonism of sumoylation de-represses progesterone receptor action in breast cancer cells. Mol Endocrinol. 2007;21(12):2890–906. doi: 10.1210/me.2007-0248. [DOI] [PubMed] [Google Scholar]
- 74.McGowan EM, Clarke CL. Effect of overexpression of progesterone receptor A on endogenous progestin-sensitive endpoints in breast cancer cells. Mol Endocrinol. 1999;13(10):1657–1671. doi: 10.1210/mend.13.10.0356. [DOI] [PubMed] [Google Scholar]
- 75.Jacobsen BM, Schittone SA, Richer JK, Horwitz KB. Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer: PR isoform-specific gene regulation and tumor biology. Mol Endocrinol. 2005;19(3):574–587. doi: 10.1210/me.2004-0287. [DOI] [PubMed] [Google Scholar]
- 76.Graham JD, Yeates C, Balleine RL, et al. Characterization of progesterone receptor A and B expression in human breast cancer. Cancer Res. 1995;55(21):5063–5068. [PubMed] [Google Scholar]
- 77.Jacobsen BM, Richer JK, Sartorius CA, Horwitz KB. Expression profiling of human breast cancers and gene regulation by progesterone receptors. J Mammary Gland Biol Neoplasia. 2003;8(3):257–268. doi: 10.1023/b:jomg.0000010028.48159.84. [DOI] [PubMed] [Google Scholar]
- 78.Hissom JR, Moore MR. Progestin effects on growth in the human breast cancer cell line T-47D – possible therapeutic implications. Biochem Biophys Res Commun. 1987;145(2):706–711. doi: 10.1016/0006-291x(87)91022-9. [DOI] [PubMed] [Google Scholar]
- 79.Musgrove EA, Lee CS, Sutherland RL. Progestins both stimulate and inhibit breast cancer cell cycle progression while increasing expression of transforming growth factor alpha, epidermal growth factor receptor, c-fos, and c-myc genes. Mol Cell Biol. 1991;11(10):5032–5043. doi: 10.1128/mcb.11.10.5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lange CA, Richer JK, Horwitz KB. Hypothesis: progesterone primes breast cancer cells for cross-talk with proliferative or antiproliferative signals. Mol Endocrinol. 1999;13(6):829–836. doi: 10.1210/mend.13.6.0290. [DOI] [PubMed] [Google Scholar]
- 81.Lange CA. Making sense of cross-talk between steroid hormone receptors and intracellular signaling pathways: who will have the last word? Mol Endocrinol. 2004;18(2):269–278. doi: 10.1210/me.2003-0331. [DOI] [PubMed] [Google Scholar]
- 82.Moore MR, Spence JB, Kiningham KK, Dillon JL. Progestin inhibition of cell death in human breast cancer cell lines. J Steroid Biochem Mol Biol. 2006;98(4–5):218–227. doi: 10.1016/j.jsbmb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 83.Sartorius CA, Harvell DM, Shen T, Horwitz KB. Progestins initiate a luminal to myoepithelial switch in estrogen-dependent human breast tumors without altering growth. Cancer Res. 2005;65(21):9779–9788. doi: 10.1158/0008-5472.CAN-05-0505. [DOI] [PubMed] [Google Scholar]
- 84.Byron SA, Horwitz KB, Richer JK, Lange CA, Zhang X, Yee D. Insulin receptor substrates mediate distinct biological responses to insulin-like growth factor receptor activation in breast cancer cells. Br J Cancer. 2006;95(9):1220–1228. doi: 10.1038/sj.bjc.6603354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Woodward TL, Lu H, Haslam SZ. Laminin inhibits estrogen action in human breast cancer cells. Endocrinology. 2000;141(8):2814–2821. doi: 10.1210/endo.141.8.7607. [DOI] [PubMed] [Google Scholar]
- 86.Ravdin PM, Cronin KA, Howlader N, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med. 2007;356(16):1670–1674. doi: 10.1056/NEJMsr070105. [DOI] [PubMed] [Google Scholar]
- 87.Robbins AS, Clarke CA. Regional changes in hormone therapy use and breast cancer incidence in California from 2001 to 2004. J Clin Oncol. 2007;25(23):3437–3439. doi: 10.1200/JCO.2007.11.4132. [DOI] [PubMed] [Google Scholar]
- 88.Pasqualini JR, Paris J, Sitruk-Ware R, Chetrite G, Botella J. Progestins and breast cancer. J Steroid Biochem Mol Biol. 1998;65(1–6):225–235. doi: 10.1016/s0960-0760(98)00028-4. [DOI] [PubMed] [Google Scholar]
- 89.Kennedy BJ. Hormonal therapies in breast cancer. Semin Oncol. 1974;1(2):119–130. [PubMed] [Google Scholar]
- 90.Allan GF, Palmer E, Musto A, Lai MT, Clancy J, Palmer S. Molecular properties and preclinical pharmacology of JNJ-1250132, a steroidal progesterone receptor modulator that inhibits binding of the receptor to DNA in vitro. Steroids. 2006;71(7):578–584. doi: 10.1016/j.steroids.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 91.Perrault D, Eisenhauer EA, Pritchard KI, et al. Phase II study of the progesterone antagonist mifepristone in patients with untreated metastatic breast carcinoma: a National Cancer Institute of Canada Clinical Trials Group study. J Clin Oncol. 1996;14(10):2709–2712. doi: 10.1200/JCO.1996.14.10.2709. [DOI] [PubMed] [Google Scholar]
- 92.Grunberg SM, Weiss MH, Russell CA, et al. Long-term administration of mifepristone (RU486): clinical tolerance during extended treatment of meningioma. Cancer Invest. 2006;24(8):727–733. doi: 10.1080/07357900601062339. [DOI] [PubMed] [Google Scholar]
- 93.Robertson JF, Willsher PC, Winterbottom L, Blamey RW, Thorpe S. Onapristone, a progesterone receptor antagonist, as first-line therapy in primary breast cancer. Eur J Cancer. 1999;35(2):214–218. doi: 10.1016/s0959-8049(98)00388-8. [DOI] [PubMed] [Google Scholar]