

Abstract
Chemokine receptor CXCR4 and its sole ligand SDF-1 are key players in regulating cancer cell invasion and metastasis. Plerixafor (AMD3100) is a small-molecule CXCR4 antagonist that prevents binding of SDF-1 to CXCR4 and has potential in prevention of cancer metastasis. This study investigates the influence of biodegradability of a recently reported polymeric Plerixafor (PAMD) on CXCR4 antagonism, antimetastatic activity, and transfection efficacy of PAMD polyplexes with plasmid DNA. We show that PAMD exhibits CXCR4 antagonism and inhibition of cancer cell invasion in vitro regardless of its biodegradability. Biodegradable PAMD showed considerably enhanced transfection efficiency and decreased cytotoxicity when compared with the non-degradable PAMD. Despite similar CXCR4 antagonism in vitro, only biodegradable PAMD displayed antimetastatic activity in experimental lung metastasis model in vivo.
1. Introduction
Reducible polycations with disulfide bonds in the structure show great promise for delivery of therapeutic nucleic acids to treat a variety of diseases caused by genetic disorders including cancer [1-9]. Taking advantage of the difference in the redox potential of the reducing intracellular environment and the oxidizing nature of the extracellular space, polyplexes based on the reducible polycations often exhibit significantly enhanced transfection activity and improved toxicity profile [10-14]. Intracellular degradation of the reducible polycations not only contributes to efficient disassembly of the polyplexes, but also to better spatial selectivity of release of the nucleic acids in the cytoplasm and to decreased cytotoxicity. Furthermore, the interactions between the reducible polyplexes and cell surface thiols are also playing an important role in improving cellular uptake of reducible polyplexes [15].
It is known that among most patients that die of aggressive types of cancer, it is not the primary tumors, but their metastases at distant sites that are the main cause of death. Consistent with the seed-and-soil hypothesis of metastatic dissemination [16-19], the potential for and the sites of cancer metastasis are determined not only by the characteristics of the primary cancer cells (the ‘seed’), but also by the microenvironment in specific organs (the ‘soil’) that supports tumor cell adhesion and subsequent growth and proliferation [20]. It has been well-established that diverse network of chemokines and their receptors play a crucial role in the invasion and metastasis of cancer cells. Mounting clinical and pre-clinical evidence has highlighted the involvement of CXCR4 along with its ligand, stromal cell-derived factor-1 (SDF-1, also known as CXCL12) in this process [21]. CXCR4 overexpression is associated with poor survival and aggressive types of cancer [22-26]. Some reports also suggest that CXCR4 overexpression is associated with high risk of cancer recurrence and decreased survival rate [27].
CXCR4 is a highly conserved G-protein-coupled receptor that binds its only ligand SDF-1. The ligand binding initiates divergent signaling transduction pathways and downstream effector molecules that regulate cell adhesion, survival, proliferation, invasion and angiogenesis. CXCR4/SDF-1 axis triggers phosphatidylinositol-3-kinase (PI3K) pathway that further activates protein kinase AKT, which is the key effector in mediating cancer cell migration and survival [21, 28]. In addition, activated CXCR4 increases secretion of matrix metalloproteinases (MMPs), which leads to the degradation of extracellular matrix and facilitating of the invasion process [21, 29, 30]. CXCR4/SDF-1 axis also stimulates mitogen-activated protein kinase (MAPK) pathways including protein kinase Erk1/2 that phosphorylates transcription factor Elk-1 to promote cancer cell proliferation and survival [31]. Some reports suggest that CXCR4/SDF-1 signaling promotes angiogenesis in both primary and metastatic cancers [32-34]. All the above evidence suggests the critical role of CXCR4/SDF-1 axis in metastatic cancer, which makes it a potential therapeutic target. Numerous studies have shown that blocking CXCR4 activation with commercial antagonists like Plerixafor or knocking down CXCR4 expression using RNA interference inhibits metastasis and controls the growth of the primary tumors [35-37].
Plerixafor (AMD3100) is an FDA-approved small molecular antagonist of CXCR4 (Scheme 1). Plerixafor contains six secondary and two tertiary amines, which provide opportunity for easy chemical modification. Importantly, chemical modification of Plerixafor is possible also because not all of the eight amines are required for binding to the CXCR4 receptor and pharmacologic function [38, 39]. Furthermore, the presence of total of 8 protonizable amines provides the molecule with strong positive charge, which makes it a suitable building block for synthesis of cationic polymers applicable for delivery of nucleic acids. Based on this rationale, we have recently reported synthesis of reducible polymeric Plerixafor (rPAMD). The synthesized polymer retained the pharmacologic activity of the small-molecule drug and also successfully delivered plasmid DNA [40]. However, our study did not address whether the CXCR4 antagonism was attributed directly to the polymeric Plerixafor or to its low molecular weight degradation products. The goal of the present study was to test the hypothesis that polymeric Plerixafor is the active component, which binds to the CXCR4 receptor and inhibits invasion of cancer cells. To test the hypothesis, we have synthesized rPAMD and its non-degradable analog PAMD and conducted head-to-head comparison of their physicochemical properties, cytotoxicity, CXCR4 antagonism, cancer cell invasion inhibition, and antimetastatic activity in vivo.
Scheme 1.
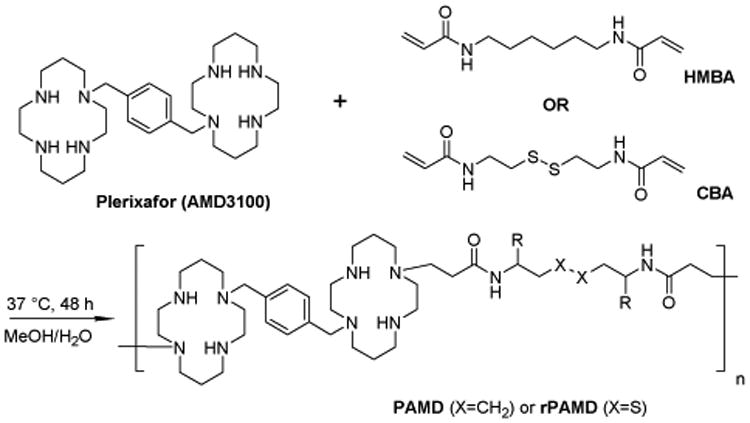
Synthesis of rPAMD and PAMD.
2. Materials and Methods
2.1. Materials
N,N′-cystaminebisacrylamide (CBA) and N,N′-hexamethylenebisacrylamide (HMBA) were obtained from Polysciences, Inc. (Warrington, PA). Salt form of AMD3100 (octahydrochloride) was purchased from Sigma-Aldrich (St. Louis, MO). Free-base form of AMD3100 was obtained from Ontario Chemicals, Inc. (Guelph, ON, Canada). Plasmid DNA, gWiz high-expression luciferase (gWiz-Luc) containing luciferase reporter gene was from Aldevron (Fargo, ND). Plasmid DNA containing blue fluorescent protein (BFP) reporter gene was a kind gift from Dr. Luker (University of Michigan). Cell culture inserts for 24-well plates (8.0 μm pores, Translucent PET Membrane, cat# 353097) and BD Matrigel™ Basement Membrane Matrix (cat# 356237) were purchased from BD Biosciences (Franklin Lakes, NJ). Human SDF-1 was from Shenandoah Biotechnology, Inc. (Warwick, PA). Dulbecco's Modified Eagle Medium (DMEM), Roswell Park Memorial Institute (RPMI) medium, Dulbecco's phosphate buffered saline (PBS), fetal bovine serum (FBS), L-Glutamine, and Penicillin-Streptomycin (Pen-Strep) solution were from Thermo Scientific (Waltham, MA). G418 sulfate and Minimum Essential Medium (MEM) were from Mediatech, Inc. (Manassas, VA). Diff-Quick staining kit was from IMEB Inc. (San Marcos, CA). XenoLight D-luciferin potassium salt was purchased from PerkinElmer (Waltham, MA). All other reagents and chemicals were obtained from Fisher Scientific or VWR International unless otherwise noted.
2.2. Synthesis and characterization of PAMD
Reducible polymeric Plerixafor (rPAMD) and non-reducible polymeric Plerixafor (PAMD) were synthesized by Michael polyaddition of 1:1 molar ratio of AMD3100 (free base) and either a reducible bisacrylamide CBA or non-reducible bisacrylamide HMBA (Scheme 1). In a typical reaction, CBA (104 mg, 0.4 mmol) or HMBA (90 mg, 0.4 mmol) and AMD3100 (200.8 mg, 0.4 mmol) were added into a glass vial containing methanol/water mixture (4 mL, 7/3 v/v). Polymerization was carried out under nitrogen atmosphere in dark at 37 °C for 72 h. Then, additional 20 mg of AMD3100 was added to the reaction mixture to consume any residual acrylamide groups, and the mixture was stirred for another 6 h. The reaction mixture was then added dropwise to excess of 1.25 M HCl in ethanol so that pH of the mixture was kept around 3. The resulting precipitated HCl salt of PAMD was isolated by centrifugation, washed twice with ethanol and dried in vacuum. The polymers were dissolved in water and dialyzed (MWCO 3.5 kDa) against water for 2 days before final freeze-drying.
The synthesized polymers were analyzed by 1H-NMR to confirm completion of the reaction from disappearance of the acrylamide signal of CBA and HMBA. The composition of the polymers was determined by elemental analysis (Atlantic Microlab). Removal of any potentially unreacted AMD3100 was confirmed by analyzing its content in PAMD and rPAMD using LC-MS/MS (AQUITY UPLC® TQD system, Waters, MA) equipped with AQUITY UPLC® BEH Shield RP18 column (2.1mm×100mm, 1.7 urn). A gradient of aqueous solution of (2 mM ammonium formate + 0.1% formic acid) and acetonitrile was used. AMD3100 was monitored at the parent/daughter ions (m/z) 503.61 → (m/z) 105.00.
Weight- and number-average molecular weights and polydispersity index (Mw/Mn) were determined by size exclusion chromatography using Viscotek GPCmax chromatography system equipped with a refractive index detector and a low- and right-angle light scattering detector (Malvern Instruments, UK). The columns used were single pore AquaGel™ columns (cat# PAA-202 and PAA-203) by PolyAnalytik (London, ON, Canada). Sodium acetate buffer (0.3 M, pH 5) was used as an eluent at flow rate of 0.3 mL/min.
2.3. Ethidium bromide (EtBr) exclusion assay
The ability of PAMD and rPAMD to condense plasmid DNA was determined by EtBr exclusion assay by measuring the changes in EtBr/DNA fluorescence. DNA solution at a concentration of 20 μg/mL in 10 mM HEPES buffer (pH 7.4) was mixed with EtBr (1 μg/mL) and fluorescence was measured and set to 100% using an excitation wavelength of 540 nm and an emission wavelength of 590 nm. Fluorescence readings were taken following a stepwise addition of a polycation solution, and the condensation curve for each polycation was constructed.
2.4. Preparation and physicochemical characterization of polyplexes
DNA solution in 10 mM HEPES (pH 7.4) was prepared to give a DNA concentration of 20 μg/mL in the final prepared polyplexes. Polyplexes were formed by adding predetermined volume of polymer to achieve the desired polycation/DNA (w/w) ratio and mixed by vigorous vortexing for 10 s. Polyplexes were further allowed to stand for 30 min prior to use. The determination of hydrodynamic diameters and zeta potentials of polyplexes was performed by dynamic light scattering. Results were expressed as mean ± standard deviation (SD) of 3-10 experimental runs.
2.5. Cell culture
Human hepatocellular carcinoma cell line HepG2 was purchased from ATCC (Manassas, VA) and maintained in MEM supplemented with 10% FBS. Human epithelial osteosarcoma U20S cells stably expressing human CXCR4 receptor fused to the N-terminus of enhanced green fluorescent protein (EGFP) were purchased form Fisher Scientific. The cells were cultured in DMEM supplemented with 2 mM L-Glutamine, 10% FBS, 1% Pen-Strep and 0.5 mg/mL G418. All the cells were maintained at 37 °C incubator with 5% CO2. Mouse melanoma cell line B16F10 was purchased from ATCC and maintained in DMEM supplemented with 10% FBS. B16F10 cells stably expression luciferase (B16F10.Luc) were purchased from PerkinElmer and cultured in RPMI supplemented with 10% FBS.
2.6. Cytotoxicity of polycations
Toxicity of the polycations was evaluated by MTS assay in U20S and HepG2 cells. The cells were plated in 96-well microplates at a density of 8,000 (U20S) or 20,000 (HepG2) cells/well. After 24 h, the medium was replaced by 150 μL of serial dilutions of a polymer in serum-containing medium and the cells were incubated for 24 h. Polymer solutions were aspirated and replaced by a mixture of 100 μL serum-free media and 20 μL of MTS reagent (CellTiter96® AQueous Non-Radioactive Cell Proliferation Assay, Promega). After 2 h incubation, the absorbance [A] was measured spectrophotometrically on Synergy 2 Microplate Reader (BioTek, VT) at λ = 490 nm. The relative cell viability (%) was calculated as [A]sample/[A]untreated × 100%. The IC50 were calculated as polymer concentration which inhibits growth of 50% of cells relative to untreated cells using a built-in curve fitting procedure in GraphPad Prism.
2.7. CXCR4 redistribution assay
U20S cells expressing EGFP-CXCR4 receptors were plated in black 96-well plates with optical bottom 18-24 h before the experiment at a seeding density of 8,000 cells per well. The cells were first washed twice with 100 μL assay buffer (DMEM supplemented with 2 mM L-Glutamine, 1% FBS, 1% Pen-Strep and 10 mM HEPES) and then incubated with rPAMD, PAMD, PEI and their DNA polyplexes in assay buffer containing 0.25% DMSO at 37 °C for 30 min. AMD3100 (300 nM) was used as the positive control. SDF-1 was then added to each well to make final concentration of 10 nM. Cells treated with SDF-1 alone were used as the negative control. After 1 h incubation at 37 °C, the cells were fixed with 4% formaldehyde at room temperature for 20 min and washed 4 times with PBS. The cell nuclei were stained with 1 μM Hoechst in PBS containing 0.5% Triton X-100. Images were taken by EVOS fl microscope at 20×. High-content analysis was applied to quantify the CXCR4 antagonistic activity based on the internalization of the EGFP-CXCR4 receptors from plasma membrane into the cells. ImageXpress Micro High Content Screening System (Molecular Devices) equipped with MetaXpress software (Transfluor module) were used for the imaging and analysis.
2.8. Cell invasion assay
The upper sides of the transwell inserts were coated with 40 μL Matrigel diluted 1:3 (v/v) with serum-free medium. The 24-well plates with coated inserts were then placed in 37 °C incubator for 2 h. CXCR4+ U20S cells were trypsinized and resuspended with different concentrations of Plerixafor, PAMD, or rPAMD in serum-free medium for 30 min before adding to the inserts at a final concentration of 10,000 cells in 300 μL medium per insert. 20 nM SDF-1 in serum-free medium as the chemo-attractant was then added to the corresponding wells in the companion plate. After 16 h, the non-invaded cells on the upper surface of the inserts were removed with a cotton swab. The invaded cells were then fixed and stained by dipping the inserts into staining Diff-Quick solution. The images were taken by EVOS ×l microscope. Five 40× imaging areas were randomly selected for each insert and each sample was conducted in triplicate.
2.9. Transfection of DNA polyplexes
All transfection experiments were conducted in 48-well plates with cells at logarithmic growth phase. Cells were seeded at a density of 40,000 cells/well 24 h prior to transfection. On the day of transfection, cells were incubated with 170 μL of the polyplexes at DNA concentration of 2.35 μg/mL in media with or without 10% FBS. After 4 h incubation, polyplexes were completely removed and the cells were cultured in medium with 10% FBS for 24 h prior to measuring luciferase expression. The medium was discarded and the cells were lysed in 100 μL of 0.5× cell culture lysis reagent buffer (Promega, Madison, WI) for 30 min. To measure the luciferase content, 100 μL of 0.5 mM luciferin solution was automatically injected into each well of 20 μL of cell lysate and the luminescence was integrated over 10 s using Synergy 2 Microplate Reader (BioTek, VT). Total cellular protein in the cell lysate was determined by the Bicinchoninic acid protein assay using calibration curve constructed with standard bovine serum albumin solutions (Pierce, Rockford, IL). Transfection activity was expressed as relative light units (RLU)/mg cellular protein ± SD of quadruplicate samples.
2.10. Simultaneous transfection and CXCR4 inhibition of rPAMD and PAMD polyplexes
U20S cells were plated in 48-well plates with optical bottom 24 h before the experiment at a seeding density of 20,000 cells per well. The cells were incubated with polyplexes containing BFP plasmid using the same method described in 2.9. The CXCR4 antagonism was evaluated in the same cells at 24 h after polyplex incubation by stimulating the cells with 10 nM SDF-1 for 1 h, followed by fixation and fluorescent imaging.
2.11. Intracellular distribution rPAMD and PAMD polyplexes
Plasmid DNA was labeled using Label IT®-TrackerTM CX-Rhodamine Kit (Mirus, Madison, WI) following the manufacturer's protocol and purified by precipitation. The polymers (rPAMD and PAMD) were labeled with a blue dye (AlexaFluor 350) using protocol recommended by the supplier (Life Technologies, Grand Island, NY) and purified by ultracentrifugation to remove unreacted dye. 120,000 CXCR4+ U20S cells were plated in glass-bottom dish (MatTek P35GC-0-14-C) 24 h before the experiment. The cells were incubated with rPAMD and PAMD polyplexes prepared at w/w 5 (2.35 μg/mL DNA) for 3 h before incubation with 10 nM SDF-1. The cells were incubated for another 1 h before a PBS wash, fixation and imaging by Leica TCS SP5 laser scanning confocal microscope.
2.12. Antimetastatic activity in vivo
All animal experiments followed a protocol approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Animals were placed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care upon arrival. Twenty 8-week old female C57BL/6 mice were randomly assigned into four groups (n=5) and one million B16F1 O.Luc cells was injected intravenously via the tail vein. The cells were treated before the injection for 15 min with AMD3100 (1 mM), PAMD (5 μg/mL) or rPAMD (5 μg/mL). Following injection of the B16F10.Luc cells, the animals were intravenously administrated with AMD3100 (1.25 mg/kg), PAMD (2.5 mg/kg), or rPAMD (0.5 mg/kg) on day 3, 5 and 7 for a total of three doses. On day 11, the animals were given 3 mg of D-luciferin in 100 μL PBS by intraperitoneal injection and humanely sacrificed 5 min after the luciferin injection. The lungs were harvested and washed with PBS before ex vivo bioluminescent imaging using Kodak FX In-Vivo Imaging System (10 min exposure). Tumor burden in the lungs was quantified from the bioluminescence intensity (RLU) per lung.
3. Results and Discussion
3.1. Synthesis and characterization of polymeric AMD3100
Our recent report described the synthesis and evaluation of CXCR4 antagonism and transfection activity of reducible polymeric Plerixafor (rPAMD). Here, we investigated how rPAMD biodegradability affects its pharmacologic and gene delivery properties. We synthesized non-degradable PAMD and degradable rPAMD using Michael polyaddition of AMD3100 with either non-reducible bisacrylamide HMBA or its reducible counterpart CBA (Scheme 1). AMD3100 contains 6 secondary amines that are reactive in the Michael polyaddition and can form insoluble crosslinked products. In order to avoid formation of insoluble product it was important to optimize the reaction stoichiometry, temperature and solvent. We have found that conducting the reaction at 37 °C in a mixture of methanol and water and using a molar ratio of 1:1 between AMD3100 and the acrylamides leads to soluble branched polymers with intermediate molecular weights. The used molar ratio of the reagents represents 3:1 excess of amines to acrylamide functional groups. Decreasing the ratio of functional groups below 3:1 resulted in crosslinked insoluble product. The polymers were purified by precipitation and extensive dialysis using a membrane with 3.5 kDa molecular weight cut-off. Complete consumption of potentially toxic unreacted acrylamide groups was confirmed from the lack of the methylene (CH2=) signal at 5.6 and 6.3 ppm in 1H-NMR. Size exclusion chromatography analysis of the molecular weight showed that PAMD had Mw = 12,800 (Mw/Mn = 1.23) and rPAMD had Mw = 13,900 (Mw/Mn = 1.26). The absence of any unreacted AMD3100 that could interfere with the analysis of the biological activity of the synthesized polymers was confirmed by LC-MS/MS, which found that the purity of rPAMD was >99.8% and the purity of PAMD was >99.9%.
3.2. Characterization of polyplexes
To test the DNA condensation capability of rPAMD and PAMD, EtBr exclusion assay was conducted (Fig. 1). Both rPAMD and PAMD were able to fully condense DNA and displayed typical sigmoidal condensation curves, which were comparable with control polycation PEI. Despite comparable molecular weight, PAMD exhibited better DNA condensation ability than rPAMD. PAMD fully condensed DNA at w/w > 1, while rPAMD required w/w > 2 to achieve the same outcome. Hydrodynamic size and zeta-potential of the PAMD and rPAMD polyplexes prepared at different w/w ratios were measured by light scattering (Fig. 2). The sizes of both rPAMD and PAMD polyplexes fit into a relatively narrow range of 56–70 nm. All the polyplexes exhibit positive surface charge ranging from 25 to 43 mV. No significant correlation between the formulation parameters (i.e., w/w ratio) and size or zeta-potential were observed.
Fig. 1.
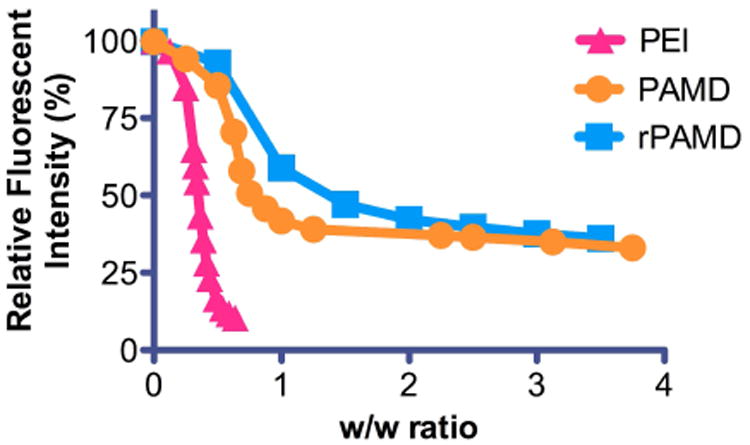
DNA condensation by rPAMD and PAMD. EtBr exclusion assay was conducted in 10 mM HEPES buffer (pH 7.4), and PEI was used a control.
Fig. 2.
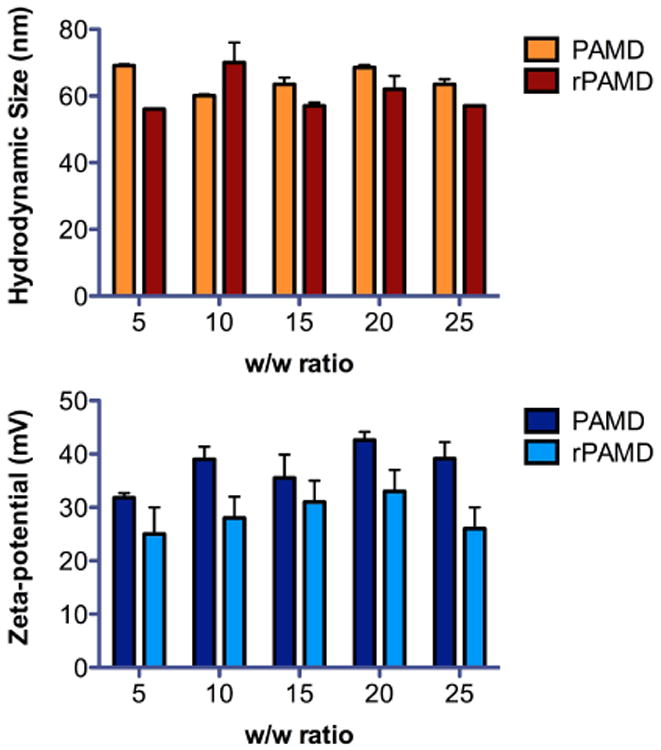
Physicochemical properties of rPAMD and PAMD polyplexes. Hydrodynamic size and zeta-potential of the DNA polyplexes prepared at different w/w ratios were measured by dynamic light scattering.
3.3. Cytotoxicity
Cytotoxicity of synthetic gene delivery vectors is one of the major hurdles that prevent their clinical advancement. It has been well documented that reducible polycations display considerably decreased cytotoxicity compared with non-reducible counterparts [12, 15, 41]. Here we evaluated the cytotoxicity of rPAMD and PAMD by MTS assay in human osteosarcoma U2OS cells and human liver hepatocellular carcinoma HepG2 cells. PEI was also tested as a control. The cell viability curves and the calculated IC50 values of each polymer in these two cell lines are summarized in Fig. 3. The results show that the HepG2 liver cells are more resistant to the adverse action of polycations than U2OS cells. As expected, biodegradability of rPAMD resulted in considerably lower toxicity in both cell lines when compared with PAMD. The IC50 of rPAMD was about 42-fold higher than PAMD in U2OS cells and about 9-fold higher in HepG2 cells. This finding confirms previous reports that suggest that the decrease in cytotoxicity of reducible polycations is strongly dependent on the used cell line and may be correlated with intracellular glutathione levels [41]. Comparison with the control PEI revealed that not only rPAMD but also the non-degradable PAMD had lower toxicity in both cell lines than PEI (3.2-times lower in U2OS and 5.1-times lower in HepG2). The lower toxicity of PAMD compared with PEI is due to a combination of lower molecular weight and lower charge density due to the unique chemical properties of the cyclam ring in PAMD. Unlike corresponding linear amines which can be fully protonated at neutral or slightly acidic pH, the geometric constrains of the cyclam ring result in only two of the four amines that can be protonated at neutral pH. The remaining two amines then become highly acidic with pKa = 2.4 and 1.6 [42].
Fig. 3.
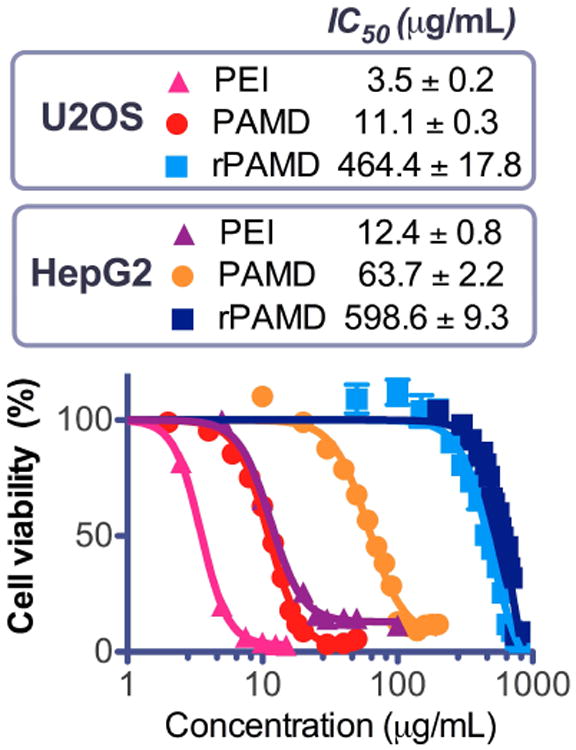
Cytotoxicity of PAMD and rPAMD in U20S and HepG2 cells. Cells were treated with increasing concentrations of the polymers for 24 h before measuring cell viability using MTS assay. Results shown as mean viability ± SD (n = 3).
3.4. CXCR4 antagonistic activity
We evaluated CXCR4 antagonism of the polycations and their DNA polyplexes using high content screening (HCS) based on the inhibition of SDF1-triggered endocytosis of EGFP-CXCR4 receptor. HCS is a phenotypic assay that, in this case, uses automatic image analysis to quantify the extent of EGFP-CXCR4 internalization into the cells. As shown in Fig. 4, the small-molecule CXCR4 antagonist AMD3100 (0.3 μM) inhibits CXCR4 internalization, as documented by the diffuse pattern of fluorescence. In contrast, untreated cells display punctate fluorescence indicative of EGFP-CXCR4 internalization into endosomes. PAMD and rPAMD, as well as their polyplexes, showed strong CXCR4 antagonism under conditions used in a typical transfection experiment. The CXCR4 antagonism of the polycations and their polyplexes was fully comparable to the activity of AMD3100. CXCR4 antagonism was quantified by calculating half-maximal inhibition concentrations (EC50) using the HCS analysis. The results showed that PAMD was about 1.9-times more potent CXCR4 antagonist than the reducible rPAMD (EC50 = 103 ± 13 μg/mL vs. 195 ± 26 μg/mL). Important conclusion from this experiment is that biodegradability, although beneficial for decreasing toxicity, is not required for CXCR4 inhibition by PAMD. Control PEI exhibited no ability to inhibit CXCR4 in any of the experiments.
Fig. 4.
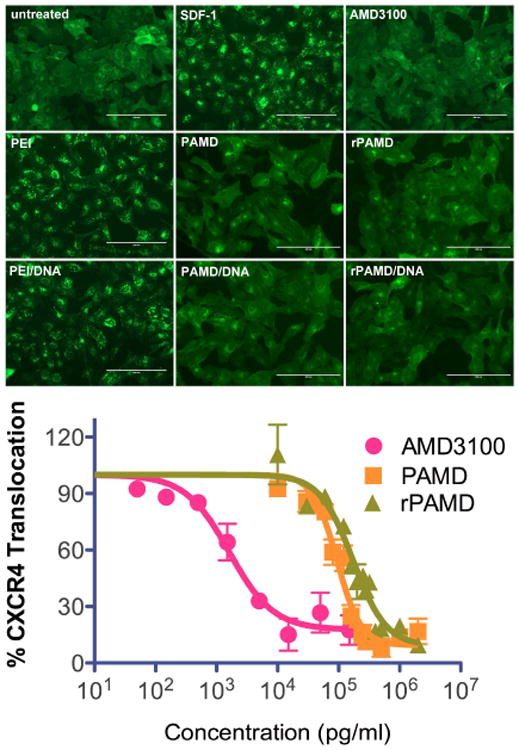
CXCR4 antagonism. U20S cells expressing EGFP-CXCR4 were treated with either polymer alone (2.5 μg/mL) or polyplexes (DNA dose 0.5 μg/mL, w/w 5) for 30 min before incubation with 20 nM SDF-1. AMD3100 (300 nM) was used as a positive and PEI as a negative control. Results shown as mean antagonism ± SD (n = 3).
3.5. Inhibition of cancer cell invasion
As discussed above, the CXCR4/SDF-1 axis is involved in migration of multiple types of cancer cells, and CXCR4 antagonists are known to inhibit invasion of those cancer cells. Binding of SDF-1 to CXCR4 receptor present on the cell membrane triggers activation of various signaling pathways leading to secretion of multiple MMPs that result in cancer cell invasion and metastasis. Here we determined if the CXCR4 antagonism of the studied polycations led also to inhibition of cancer cell invasion. We used a Boyden chamber method to evaluate the SDF1-induced invasion of the U20S cells (Fig. 5). At 2.5 μg/mL, both PAMD and rPAMD showed effective and comparable inhibition of cell invasion (73% and 71%, respectively). Then, polyplexes were prepared at polycation/DNA (w/w) ratio of 5 and their ability to inhibit cancer cell invasion was also assessed. Both PAMD and rPAMD polyplexes showed slightly improved, although not statistically significant, cell invasion inhibition when compared with free parent polycations at the same concentration. For comparison, the inhibition of cell invasion achieved with the positive control AMD3100 was 75%. None of the tested negative controls (PEI and PEI/DNA polyplexes) showed any significant inhibitory effect.
Fig. 5.
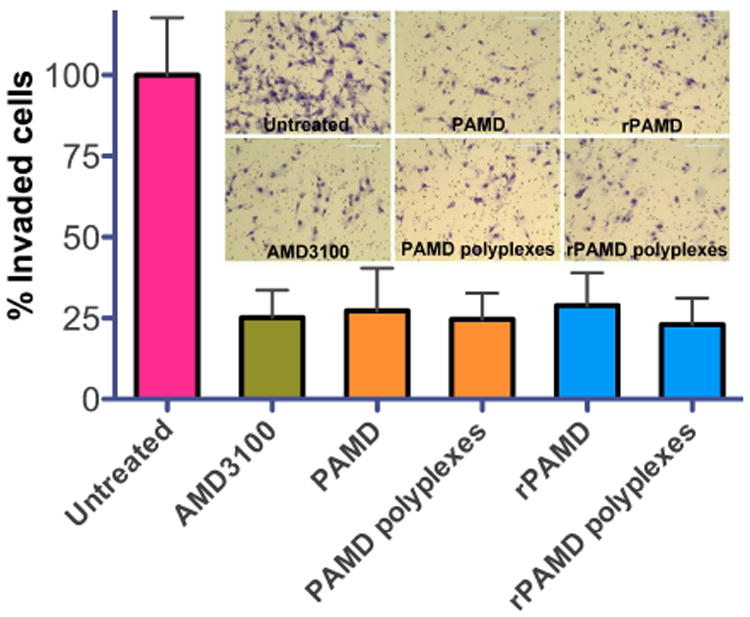
Inhibition of cancer cell invasion. U20S cells were treated with either polymer alone (2.5 μg/mL) or polyplexes (DNA dose 0.5 μg/mL, w/w 5) and allowed to invade through Matrigel upon stimulation with SDF-1 for 16 h. AMD3100 (300 nM) was used as positive control. Results shown as mean number of invaded cells ± SD (n = 3).
3.6. Antimetastatic activity of PAMD and rPAMD
To investigate if the ability of PAMD and rPAMD to inhibit cancer cell invasion in vitro translates into decreased metastasis in vivo, we used B16F10 mouse melanoma model known to metastasize readily to the lung via the CXCR4/SDF-1 axis [43-45]. Experimental lung metastasis was induced by tail vein injection of the melanoma cells and antimetastatic activity of the polymers was evaluated using bioluminescence imaging of the luciferase-expressing B16F10 cells in the lungs (Fig. 6). Treatment with AMD3100 resulted in decreased lung tumor burden compared with untreated control mice. The results with the synthesized polymers revealed that only the reducible rPAMD exhibited substantial antimetastatic activity. In contrast, PAMD had no significant effect on the growth of the melanoma tumors in the lung. Interestingly, the antimetastatic activity of rPAMD was higher than the activity of the commercial CXCR4 antagonist AMD3100. Histological analysis of the lungs showed that treatment with rPAMD resulted not only in smaller sizes of the tumor nodules in the lung but also in the lower number of the nodules. These in vivo results contrast with the CXCR4 antagonism and cancer cell inhibition data in vitro in which PAMD exhibited better or similar activity when compared with rPAMD (Figs. 4 and 5). These results also point to the unreliability of in vitro assays and importance of in vivo testing in development of non-viral gene delivery systems. We hypothesize that the observed differences between rPAMD and PAMD activity in vivo are due to different pharmacokinetics profiles of the two polycations.
Fig. 6.
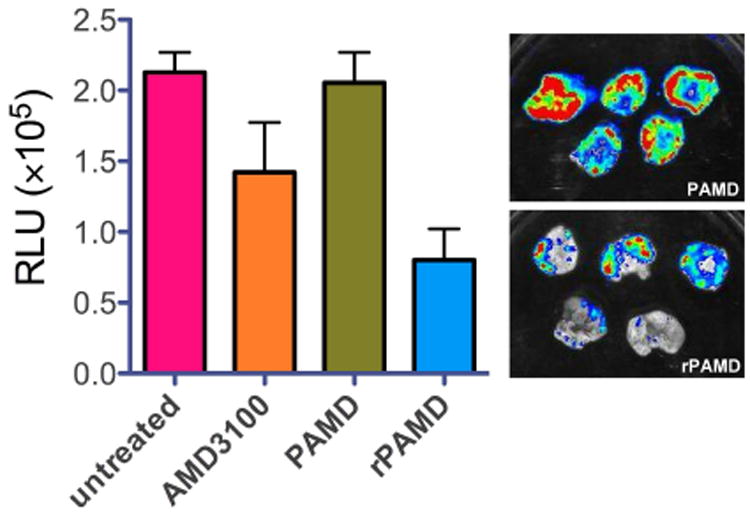
Antimetastatic activity in B16F10 lung metastatic model. B16F10 cells stably expressing luciferase were injected into C57BL/6 mice and the mice were treated by 3 intravenous doses of the polycation or AMD3100. Results shown as mean RLU/lung ± SD (n = 5).
3.7. Transfection
After establishing CXCR4 antagonism and related anti-metastatic activity, we have evaluated the ability of PAMD and rPAMD polyplexes to mediate transfection in the B16F10 cells used in the in vivo experiments. The polyplexes were formulated at varying polycation/DNA (w/w) ratios and PEI/DNA polyplexes (w/w = 1.2) were used as a control (Fig. 7). Due to the cytotoxicity of PAMD, only formulations prepared below w/w = 10 were included. All the transfection experiments were conducted in the presence of 10% FBS. Both CXCR4-inhibiting polycations were able to deliver DNA and mediate efficient transfection. However, polyplexes based on the reducible rPAMD showed considerably higher transfection activity than polyplexes based on the non-reducible PAMD polyplexes at all tested w/w ratios.
Fig. 7.
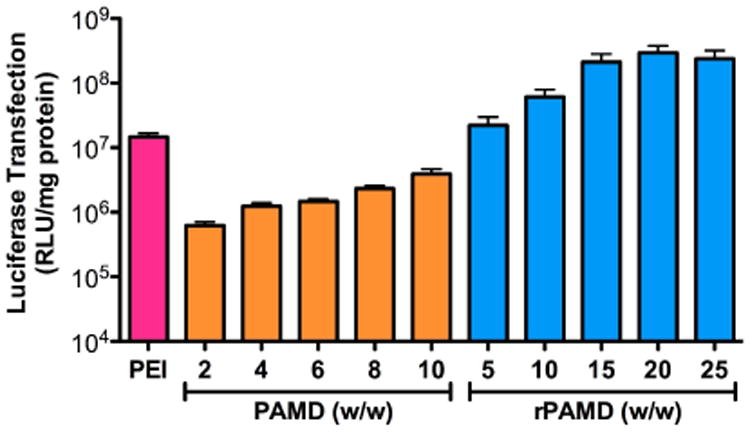
Transfection activity. Polyplexes were prepared using different polycation/DNA (w/w) ratios and used to transfect B16F10 cells in the presence of 10% FBS. Control PEI/DNA polyplexes were prepared at w/w 1.2. Results shown as mean RLU/mg protein ± SD (n = 3).
3.8. Simultaneous gene delivery and CXCR4 inhibition
The main goal of this study was to develop dual-function vectors that can simultaneously deliver genes and exhibit CXCR4 antagonism. In order to accomplish this goal, it was important to determine if both functions could be achieved in the same dose range and within a similar time-frame. We have used polyplexes prepared with plasmid DNA expressing BFP to permit concurrent microscopic evaluation of CXCR4 antagonism and transfection in the same cells (Fig. 8). Because of the superior in vivo activity of rPAMD, we have focused our attention on rPAMD/DNA polyplexes in these studies. As shown in Fig. 8, transfection activity of rPAMD polyplexes was significantly higher than transfection activity of control PEI polyplexes. This was documented by both the higher number of transfected (i.e., BFP-expressing) cells as well as higher average blue fluorescence intensity per cell. Using the same cells, we next analyzed CXCR4 antagonism by examining distribution of the green fluorescence of EGFP-CXCR4 receptors in the cell. The results show persistent CXCR4 antagonism of rPAMD/DNA recognized by the fact that, even 24 h after removal of the polyplexes from the cell culture medium, significant fraction of the CXCR4 receptors remained localized at the plasma membrane as indicated by the diffuse green fluorescence. In contrast, when treated with PEI/DNA polyplexes, no CXCR4 inhibition was observed as indicated by the punctate green fluorescence in the cells. Finally, examination of the overlay images revealed that CXCR4 inhibition does not preclude transfection of individual cells as confirmed by the fact that the individual BFP-expressing cells exhibited simultaneous inhibition of CXCR4. Overall, these results confirm that a single polyplex formulation can achieve simultaneous CXCR4 antagonism and gene transfection.
Fig. 8.
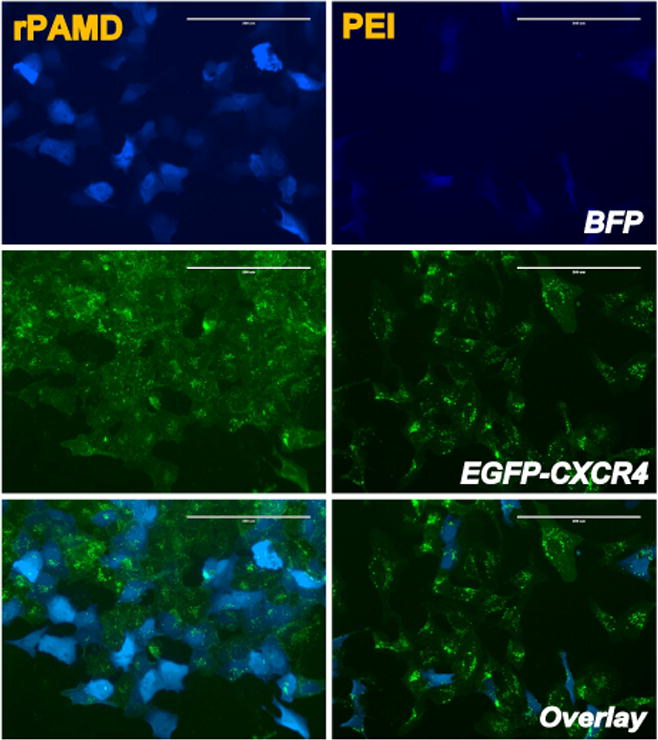
Simultaneous CXCR4 inhibition and transfection by rPAMD/DNA polyplexes. U2OS cells were transfected with polyplexes prepared with BFP plasmid at w/w 15. The cells were stimulated with 20 nM SDF-1 24 h after incubation with the polyplexes. Control PEI/DNA polyplexes were prepared at w/w 1.2.
3.9. Intracellular distribution of PAMD and rPAMD polyplexes
Understanding of the intracellular trafficking of polyplexes is vital for improving their transfection activity. Polyplexes have to be efficiently internalized into cells and routed into a proper intracellular compartment to achieve their therapeutic effect. The dual-function polyplexes developed in this study bind to CXCR4 receptor on cell membrane and inhibit its cellular internalization. It was therefore important to determine if any significant amount of polyplexes remains localized at the plasma membrane and is thus unavailable for transfection. We have used confocal microscopy to study intracellular trafficking of the polyplexes. In order to visualize both components of the polyplexes, we have fluorescently labeled the polycation with a blue dye (AlexaFluor-350) and DNA with a red dye (CX-Rhodamine). Using U2OS cells expressing the EGFP-CXCR4 receptor allowed us to compare distribution of the polycation, DNA, and CXCR4 receptors in a single experiment (Fig. 9). After 4 h incubation with the polyplexes, majority of PAMD and rPAMD polyplexes were internalized inside the cells, with no significant amount of DNA detected at the plasma membrane or associated with the green fluorescence of EGFP-CXCR4 receptors. Overlay images confirmed co-localization (purple) of the fluorescence signals of the DNA and the polycation, while no significant co-localization between any of the components of the polyplexes and the CXCR4 receptor was found. These results suggest that only negligible amounts of polycation or polyplexes are needed for the CXCR4 inhibition and that the small amounts of the polycations or polyplexes could not be visualized under the used experimental conditions. These results also suggest that, at this early time point, the DNA remained associated with polycations and reduction of the disulfide bonds in rPAMD did not occur to any significant extent. Interestingly, PAMD polyplexes showed a higher cell uptake compared with rPAMD polyplexes, indicating that inefficient intracellular trafficking and not cell uptake is the main cause of their low transfection activity observed in Fig. 7.
Fig. 9.
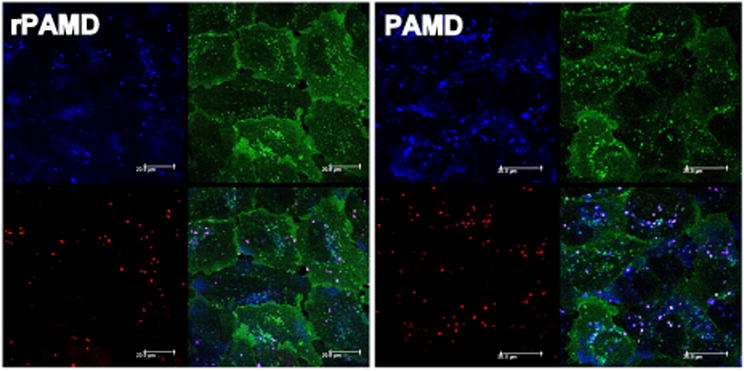
Intracellular distribution of PAMD and rPAMD polyplexes in U2OS cells expressing EGFP-CXCR4. DNA was fluorescently labeled with CX-Rhodamine (red), PAMD and rPAMD were labeled with Alexa Fluor 350 (blue) and the U2OS cells are overexpressing EGFP-tagged CXCR4 receptors (green). Cells were treated with the polyplexes for 3 h, followed by 1 h incubation with 20 nM SDF-1. The cells were fixed and imaged under confocal microscope.
4. Conclusions
We have synthesized two polycationic CXCR4 antagonists that differed in their biodegradability. Our results demonstrate that regardless of the biodegradability, both polycations are capable of inhibiting CXCR4-mediated cancer cell invasion while effectively delivering DNA and mediating transfection in vitro. The reducible polycation displayed better transfection efficiency and lower toxicity but less efficient CXCR4 antagonism than the non-reducible polycation in vitro. When tested in vivo, only the reducible polycation showed anti-metastatic activity in a lung metastasis melanoma model in mice. Future development of these dual-function systems will focus on improving the CXCR4 antagonism of rPAMD and optimization of in vivo transfection as well as identification of suitable therapeutic nucleic acids for the treatment and prevention of metastatic cancer.
Acknowledgments
This work was supported in part by NIH grants CA109711 and EB014570. We thank Steven Swaney (Center for Chemical Genomics, University of Michigan) for the help with the analysis of the CXCR4 redistribution assay and Mary Olive (Microscopy Core facility, Wayne State University) for help with confocal microscopy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Won YW, Lee M, Kim HA, Nam K, Bull DA, Kim SW. Synergistically combined gene delivery for enhanced VEGF secretion and antiapoptosis. Mol Pharm. 2013;10:3676–83. doi: 10.1021/mp400178m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao LY, Liu XY, Chen CJ, Wang JC, Feng Q, Yu MZ, et al. Core-shell type lipid/rPAA-Chol polymer hybrid nanoparticles for in vivo siRNA delivery. Biomaterials. 2014;35:2066–78. doi: 10.1016/j.biomaterials.2013.11.046. [DOI] [PubMed] [Google Scholar]
- 3.Kim SJ, Ise H, Kim E, Goto M, Akaike T, Chung BH. Imaging and therapy of liver fibrosis using bioreducible polyethylenimine/siRNA complexes conjugated with N-acetylglucosamine as a targeting moiety. Biomaterials. 2013;34:6504–14. doi: 10.1016/j.biomaterials.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Won YW, Kim KM, An SS, Lee M, Ha Y, Kim YH. Suicide gene therapy using reducible poly (oligo-D-arginine) for the treatment of spinal cord tumors. Biomaterials. 2011;32:9766–75. doi: 10.1016/j.biomaterials.2011.08.089. [DOI] [PubMed] [Google Scholar]
- 5.Kim SH, Jeong JH, Ou M, Yockman JW, Kim SW, Bull DA. Cardiomyocyte-targeted siRNA delivery by prostaglandin E(2)-Fas siRNA polyplexes formulated with reducible poly(amido amine) for preventing cardiomyocyte apoptosis. Biomaterials. 2008;29:4439–46. doi: 10.1016/j.biomaterials.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Wang Y, Zhu Y, Oupicky D. Recent advances in delivery of drug-nucleic acid combinations for cancer treatment. J Control Release. 2013;172:589–600. doi: 10.1016/j.jconrel.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SH, Jeong JH, Kim TI, Kim SW, Bull DA. VEGF siRNA delivery system using arginine-grafted bioreducible poly(disulfide amine) Mol Pharm. 2009;6:718–26. doi: 10.1021/mp800161e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parmar RG, Busuek M, Walsh ES, Leander KR, Howell BJ, Sepp-Lorenzino L, et al. Endosomolytic bioreducible poly(amido amine disulfide) polymer conjugates for the in vivo systemic delivery of siRNA therapeutics. Bioconjug Chem. 2013;24:640–7. doi: 10.1021/bc300600a. [DOI] [PubMed] [Google Scholar]
- 9.Yin Q, Gao Y, Zhang Z, Zhang P, Li Y. Bioreducible poly (β-amino esters)/shRNA complex nanoparticles for efficient RNA delivery. J Control Release. 2011;151:35–44. doi: 10.1016/j.jconrel.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Wu C, Oupicky D. Bioreducible hyperbranched poly(amido amine)s for gene delivery. Biomacromolecules. 2009;10:2921–7. doi: 10.1021/bm900724c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryu JK, Choi MJ, Kim TI, Jin HR, Kwon KD, Batbold D, et al. A guanidinylated bioreducible polymer as a novel gene carrier to the corpus cavernosum of mice with high-cholesterol diet-induced erectile dysfunction. Andrology. 2013;1:216–22. doi: 10.1111/j.2047-2927.2012.00057.x. [DOI] [PubMed] [Google Scholar]
- 12.Lin C, Zhong Z, Lok MC, Jiang X, Hennink WE, Feijen J, et al. Novel bioreducible poly(amido amine)s for highly efficient gene delivery. Bioconjug Chem. 2007;18:138–45. doi: 10.1021/bc060200l. [DOI] [PubMed] [Google Scholar]
- 13.Drake CR, Aissaoui A, Argyros O, Thanou M, Steinke JH, Miller AD. Examination of the effect of increasing the number of intra-disulfide amino functional groups on the performance of small molecule cyclic polyamine disulfide vectors. J Control Release. 2013;171:81–90. doi: 10.1016/j.jconrel.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Beloor J, Choi CS, Nam HY, Park M, Kim SH, Jackson A, et al. Arginine-engrafted biodegradable polymer for the systemic delivery of therapeutic siRNA. Biomaterials. 2012;33:1640–50. doi: 10.1016/j.biomaterials.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Manickam DS, Chen J, Oupicky D. Effect of cell membrane thiols and reduction-triggered disassembly on transfection activity of bioreducible polyplexes. Eur J Pharm Sci. 2012;46:173–80. doi: 10.1016/j.ejps.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–7. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 17.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 19.Paget S. The distribution of secondary growths in cancer of the breast. 1889 Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 20.Harvey JR, Mellor P, Eldaly H, Lennard TW, Kirby JA, Ali S. Inhibition of CXCR4-mediated breast cancer metastasis: a potential role for heparinoids? Clin Cancer Res. 2007;13:1562–70. doi: 10.1158/1078-0432.CCR-06-1987. [DOI] [PubMed] [Google Scholar]
- 21.Luker KE, Luker GD. Functions of CXCL12 and CXCR4 in breast cancer. Cancer Lett. 2006;238:30–41. doi: 10.1016/j.canlet.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 22.Hiller DJ, Meschonat C, Kim R, Li BD, Chu QD. Chemokine receptor CXCR4 level in primary tumors independently predicts outcome for patients with locally advanced breast cancer. Surgery. 2011;150:459–65. doi: 10.1016/j.surg.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Chu QD, Panu L, Holm NT, Li BD, Johnson LW, Zhang S. High chemokine receptor CXCR4 level in triple negative breast cancer specimens predicts poor clinical outcome. J Surg Res. 2010;159:689–95. doi: 10.1016/j.jss.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 24.Salvucci O, Bouchard A, Baccarelli A, Deschenes J, Sauter G, Simon R, et al. The role of CXCR4 receptor expression in breast cancer: a large tissue microarray study. Breast Cancer Res Treat. 2006;97:275–83. doi: 10.1007/s10549-005-9121-8. [DOI] [PubMed] [Google Scholar]
- 25.Andre F, Xia W, Conforti R, Wei Y, Boulet T, Tomasic G, et al. CXCR4 expression in early breast cancer and risk of distant recurrence. Oncologist. 2009;14:1182–8. doi: 10.1634/theoncologist.2009-0161. [DOI] [PubMed] [Google Scholar]
- 26.Kato M, Kitayama J, Kazama S, Nagawa H. Expression pattern of CXC chemokine receptor-4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. 2003;5:R144–50. doi: 10.1186/bcr627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chu QD, Holm NT, Madumere P, Johnson LW, Abreo F, Li BDL. Chemokine receptor CXCR4 overexpression predicts recurrence for hormone receptor-positive, node-negative breast cancer patients. Surgery. 2011;149:193–9. doi: 10.1016/j.surg.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 29.Hartmann TN, Burger JA, Glodek A, Fujii N, Burger M. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene. 2005;24:4462–71. doi: 10.1038/sj.onc.1208621. [DOI] [PubMed] [Google Scholar]
- 30.Fernandis AZ, Prasad A, Band H, Klosel R, Ganju RK. Regulation of CXCR4-mediated chemotaxis and chemoinvasion of breast cancer cells. Oncogene. 2004;23:157–67. doi: 10.1038/sj.onc.1206910. [DOI] [PubMed] [Google Scholar]
- 31.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 32.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 33.Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002;99:2703–11. doi: 10.1182/blood.v99.8.2703. [DOI] [PubMed] [Google Scholar]
- 34.Bachelder RE, Wendt MA, Mercurio AM. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res. 2002;62:7203–6. [PubMed] [Google Scholar]
- 35.Rhodes LV, Short SP, Neel NF, Salvo VA, Zhu Y, Elliott S, et al. Cytokine receptor CXCR4 mediates estrogen-independent tumorigenesis, metastasis, and resistance to endocrine therapy in human breast cancer. Cancer Res. 2011;71:603–13. doi: 10.1158/0008-5472.CAN-10-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lapteva N, Yang AG, Sanders DE, Strube RW, Chen SY. CXCR4 knockdown by small interfering RNA abrogates breast tumor growth in vivo. Cancer Gene Ther. 2005;12:84–9. doi: 10.1038/sj.cgt.7700770. [DOI] [PubMed] [Google Scholar]
- 37.Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005;65:967–71. [PMC free article] [PubMed] [Google Scholar]
- 38.Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002;527:255–62. doi: 10.1016/s0014-5793(02)03143-5. [DOI] [PubMed] [Google Scholar]
- 39.Bridger GJ, Skerlj RT, Hernandez-Abad PE, Bogucki DE, Wang Z, Zhou Y, et al. Synthesis and structure-activity relationships of azamacrocyclic C-X-C chemokine receptor 4 antagonists: analogues containing a single azamacrocyclic ring are potent inhibitors of T-cell tropic (X4) HIV-1 replication. J Med Chem. 2010;53:1250–60. doi: 10.1021/jm901530b. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Zhu Y, Hazeldine ST, Li C, Oupicky D. Dual-function CXCR4 antagonist polyplexes to deliver gene therapy and inhibit cancer cell invasion. Angew Chem Int Ed Engl. 2012;51:8740–3. doi: 10.1002/anie.201203463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu C, Li J, Zhu Y, Chen J, Oupický D. Opposing influence of intracellular and membrane thiols on the toxicity of reducible polycations. Biomaterials. 2013;34:8843–50. doi: 10.1016/j.biomaterials.2013.07.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hancock RD, Motekaitis RJ, Mashishi J, Cukrowski I, Reibenspies JH, Martell AE. The unusual protonation constants of cyclam. A potentiometric, crystallographic and molecular mechanics study. J Chem Soc, Perkin Trans 2. 1996;2:1925–9. [Google Scholar]
- 43.Murakami T, Maki W, Cardones AR, Fang H, Tun Kyi A, Nestle FO, et al. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62:7328–34. [PubMed] [Google Scholar]
- 44.Kim M, Koh YJ, Kim KE, Koh BI, Nam DH, Alitalo K, et al. CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res. 2010;70:10411–21. doi: 10.1158/0008-5472.CAN-10-2591. [DOI] [PubMed] [Google Scholar]
- 45.Takenaga M, Tamamura H, Hiramatsu K, Nakamura N, Yamaguchi Y, Kitagawa A, et al. A single treatment with microcapsules containing a CXCR4 antagonist suppresses pulmonary metastasis of murine melanoma. Biochem Biophys Res Commun. 2004;320:226–32. doi: 10.1016/j.bbrc.2004.05.155. [DOI] [PubMed] [Google Scholar]