

Abstract
Aims: Phosphoinositide 3-kinases (PI3Ks) relay growth factor signaling and mediate cytoprotection and cell growth. The cystine/glutamate antiporter system xc− imports cystine while exporting glutamate, thereby promoting glutathione synthesis while increasing extracellular cerebral glutamate. The aim of this study was to analyze the pathway through which growth factor and PI3K signaling induce the cystine/glutamate antiporter system xc− and to demonstrate its biological significance for neuroprotection, cell growth, and epilepsy. Results: PI3Ks induce system xc− through glycogen synthase kinase 3β (GSK-3β) inhibition, general control non-derepressible-2-mediated eukaryotic initiation factor 2α phosphorylation, and the subsequent translational up-regulation of activating transcription factor 4. This pathway is essential for PI3Ks to modulate oxidative stress resistance of nerve cells and insulin-induced growth in fibroblasts. Moreover, the pathway is active in human glioblastoma cells. In addition, it is induced in primary cortical neurons in response to robust neuronal activity and in hippocampi from patients with temporal lobe epilepsy. Innovation: Our findings further extend the concepts of how growth factors and PI3Ks induce neuroprotection and cell growth by adding a new branch to the signaling network downstream of GSK-3β, which, ultimately, leads to the induction of the cystine/glutamate antiporter system xc−. Importantly, the induction of this pathway by neuronal activity and in epileptic hippocampi points to a potential role in epilepsy. Conclusion: PI3K-regulated system xc− activity is not only involved in the stress resistance of neuronal cells and in cell growth by increasing the cysteine supply and glutathione synthesis, but also plays a role in the pathophysiology of tumor- and non-tumor-associated epilepsy by up-regulating extracellular cerebral glutamate. Antioxid. Redox Signal. 20: 2907–2922.
Introduction
Different kinds of intracellular stress are relayed through phosphorylation of the eukaryotic initiation factor 2α (eIF2α) by one of the following four eIF2α kinases: protein kinase R (PKR), heme-regulated eIF2α kinase (HRI), PKR-like endoplasmic reticulum kinase (PERK) and general control non-derepressible-2 (GCN2), and subsequent translational up-regulation of activating transcription factor 4 (ATF4) (61). The mechanism underlying the translational up-regulation of ATF4 is based on two upstream open reading frames (ORFs) within the 5′ untranslated region (5′UTR) of its mRNA, the second of which overlaps with the ATF4 ORF and inhibits ATF4 protein synthesis when eIF2α phosphorylation is low (22). The re-establishment of cellular homeostasis by ATF4-induced gene transcription is called the integrated stress response (ISR) (3).
Innovation.
Phosphoinositide 3-kinases (PI3Ks) as well as system xc− have been shown to induce cell growth (48) and neuroprotection (36, 58, 70, 72). In addition, both PI3Ks and system xc− are involved in tumor growth (62, 66). We show that PI3Ks induce system xc− through general control non-derepressible-2-mediated eukaryotic initiation factor 2α phosphorylation and activating transcription factor 4 translation. The pathway is important for the neuroprotective and growth-stimulatory effects of PI3K activation, is active in glioblastoma cells and, as it is induced by robust neuronal activity in neurons and in human epileptic hippocampi, it might be involved in the pathophysiology of epilepsy.
ATF4 activates the transcription of genes that are involved in amino-acid import, glutathione (GSH) biosynthesis, and resistance against oxidative stress (23), including xCT mRNA, which encodes the light chain of the amino-acid transporter, system xc− (62). System xc− imports cystine into cells while exporting glutamate in a 1:1 ratio (64). Intracellularly, cystine is reduced to cysteine, which is limiting for the synthesis of the important antioxidant GSH (51). Due to its high concentration, the ratio of reduced GSH to oxidized GSH (glutathione disulfide [GSSG]) determines the overall intracellular redox state (67).
We recently reported that the eIF2α/ATF4/xCT signaling module is an important determinant of the oxidative stress resistance of cells (39). However, in the brain, system xc− might represent a double-edged sword, as its activity increases extracellular glutamate (15) and can, therefore, positively regulate epileptic activity (15) and neurodegeneration (65).
Strong activation of the ISR reduces protein synthesis (31), whereas anabolic signaling through growth factors promotes protein synthesis and cell growth. One of the major downstream effectors of growth factors are phosphoinositide 3-kinases (PI3Ks) (48). On activation, PI3Ks phosphorylate membrane inositol lipids, thereby generating phosphoinositide 3,4,5-triphosphate (PIP3). This is followed by phosphoinositide-dependent kinase 1-induced phosphorylation and activation of Akt, which then phosphorylates and inhibits glycogen synthase kinase 3β (GSK-3β) (13). In addition to its role in cell proliferation, the PI3K/Akt/GSK-3β pathway has been repeatedly found to be neuroprotective (58, 72).
Here, we show that the PI3K/Akt/GSK-3β pathway is linked to the activation of the eIF2α/ATF4/xCT signaling module, a connection which is not only important in the pro-proliferative and neuroprotective effects of PI3K signaling but might also play a role in epilepsy.
Results
The PI3K inhibitor LY294002 sensitizes HT22 cells to oxidative glutamate toxicity through down-regulation of the eIF2α/ATF4/xCT signaling module
Recently, we demonstrated that eIF2α phosphorylation is a major determinant of oxidative stress resistance by regulating ATF4 protein levels and system xc− activity (39). While examining how the eIF2α/ATF4/xCT signaling module is connected to other neuroprotective pathways, we found that the broad-spectrum PI3K inhibitor LY294002 (LY) rapidly and dose dependently down-regulates eIF2α phosphorylation and ATF4 in hippocampal HT22 cells (Fig. 1A, Supplementary Fig. 1A). When transfected with a luciferase (Luci) reporter construct containing the ATF4 5′UTR and AUG of the ATF4 ORF fused to Luci in a pGL3 backbone (22), relative Luci activity was decreased by >50% in HT22 cells that were treated with LY as compared with control cells (Fig. 1B, upper panel). However, a 24 h treatment with LY did not down-regulate ATF4 mRNA (Fig. 1B, lower panel). These findings confirm that ATF4 is down-regulated by LY due to decreased translation. Next, we asked whether LY also reduces xCT expression and system xc− activity. A 24 h treatment with LY decreased xCT mRNA expression by ∼80% and system xc− activity by ∼50% (Fig. 1C, left panels). Consistent with these observations, we detected an ∼20% decrease in total GSH levels and an ∼1.7-fold decrease in the GSH/GSSG ratio on a 24 h treatment with LY (Fig. 1C, middle panels). System xc− expression determines the resistance of HT22 cells to oxidative glutamate toxicity (37, 40). Correspondingly, LY pre-treated HT22 cells were significantly more sensitive to oxidative glutamate toxicity when measured by the MTT assay (Fig. 1C, right panel) with trypan blue exclusion and visual inspection yielding similar results (Supplementary Fig. S2; Supplementary Data are available online at www.liebertpub.com/ars).
FIG. 1.

The PI3K inhibitor LY294002 sensitizes HT22 cells to oxidative glutamate toxicity through down-regulation of the eIF2α/ATF4/xCT signaling module. (A) Western blotting for phospho-Ser51 eIF2α (p-eIF2α) and ATF4 using cytosolic and nuclear extracts, respectively, of HT22 cells incubated with LY at the indicated concentrations or durations. Total eIF2α (eIF2α) and actin served as loading control for phospho-eIF2α and ATF4, respectively. [(B), upper panel] ATF4 translation assessed by the relative luciferase activity in HT22 cells co-transfected with ATF4 5′UTR luciferase (Luci) reporter and Gal plasmids treated with 10 μM LY or vehicle for 24 h. [(B), lower panel] ATF4 mRNA abundance in HT22 cells treated with 10 μM LY for 24 h. (C) xCT mRNA abundance (left upper panel), system xc− activity (left lower panel), total GSH (central panel, upper graph), GSH/GSSG ratio (central panel, lower graph), and survival in response to oxidative glutamate toxicity (right panel) of HT22 cells exposed to LY or vehicle for 24 h measured by the MTT assay. Similar results were obtained by visual inspection and trypan blue exclusion assays (see Supplementary Fig. S2). (D) Western blotting as in (A) using HT22R cells treated with either 10 μM LY or vehicle for 4 h (left panel). System xc− activity (central panel) and sensitivity against oxidative glutamate toxicity (right panel) in HT22R cells treated with 10 μM LY or vehicle for 24 h. The graphs represent the mean±SEM of three [(A), upper and lower left panels; (D), lower left panel and right panel], four [(B, C), left and right panels; (D), middle panel], four to five [(A), lower right panel], five [(D), upper left panel], and six [(C), middle panel] independent experiments. Statistical analysis was performed using one way ANOVA with Bonferroni's post test compared with vehicle treatment (A), one sample t-test [(B–D) left and middle panels], or two-way ANOVA with Bonferroni's post test [(C, D), right panels], *p<0.05, **p<0.01, and ***p<0.001. 5′UTR, 5′ untranslated region; ANOVA, analysis of variance; ATF4, activating transcription factor 4; eIF2α, eukaryotic initiation factor 2α; GSH, glutathione; PI3K, phosphoinositide 3-kinase; SEM, standard error of the mean.
To confirm that the observed LY-mediated translational down-regulation of ATF4 is essential for decreased system xc− activity and increased glutamate sensitivity, we used the HT22-derived line HT22R in which a deletion in the second ORF within the ATF4 5′UTR leads to the expression of a second ORF–ATF4 fusion protein (ATF4h) that is not regulated by eIF2α phosphorylation (40). In HT22R cells, LY suppressed eIF2α phosphorylation but ATF4h levels did not decrease (Fig. 1D, left panel). Moreover, LY treatment had no effect on system xc− activity, which depends on ATF4h in these cells (40), nor did it increase glutamate sensitivity (Fig. 1D, middle and right panel). Thus, translational down-regulation of ATF4 is a prerequisite for the LY-mediated reduction in system xc− activity and the exacerbation of oxidative glutamate toxicity.
PI3Kα regulates eIF2α phosphorylation and ATF4 levels through inhibition of GSK-3β in HT22 cells
LY inhibits several kinases that are related to PI3K (17). However, intracellular delivery of PIP3 (73), the PI3K product, and transient over-expression of the constitively active PI3K p110* (7) increased eIF2α phosphorylation and ATF4 expression (Fig. 2A, B), supporting the idea that the LY effect is mediated through PI3Ks. To analyze which PI3K isoform mediates the observed effect of LY, we focused on PI3Kα and β, which are expressed in all cells while PI3Kγ and δ play specific roles in the immune system (48). Two structurally unrelated PI3K inhibitors, PIK-90, which preferentially inhibits PI3Kα (IC50 PI3Kα 0.011 vs. PI3Kβ 0.35 μM, PI3Kγ 0.018 μM, and PI3Kδ 0.058 μM) (48), and TGX-221, which preferentially inhibits PI3Kβ (IC50 PI3Kα 0.784 vs. PI3Kβ 0.010 μM, PI3Kγ 3.24 μM, and PI3Kδ 0.065 μM) (28, 48), were tested. Treatment of HT22 cells with 0.03 μM PIK-90 for 2 h significantly reduced Akt phosphorylation (Fig. 2C, right panel), whereas 1 μM TGX-221 had little effect (Fig. 2C, left panel). A similar pattern was seen for the downstream targets GSK-3β, eIF2α, and ATF4 (Fig. 2C). PIK-90 also reduced Luci activity after transfection with the ATF4 5′UTR Luci reporter construct (Fig. 2D), verifying reduced ATF4 translation and decreased system xc− activity by ∼50% (Fig. 2E). Of note, quantitative polymerase chain reaction (qPCR) showed that HT22 express the p110δ subunit of PI3K but not the p110γ subunit (Fig. 2F and Supplementary Fig. S3). In combination with the IC50s of PIK-90 and TGX221 for the different p110 isoforms, this indicates that PI3Kα determines the activity of the PI3K pathway in these cells, as inhibition of PI3Kγ cannot explain the effect of PIK90 because p110γ is not expressed and both PI3Kβ and δ are sensitive to TGX221, which has no effect (Supplementary Table S1).
FIG. 2.

PI3Kα regulates eIF2α phosphorylation and ATF4 and xCT expression in hippocampal HT22 cells. (A) Relative eIF2α phosphorylation (p-eIF2α) and ATF4 expression in HT22 cells exposed to PIP3 at the indicated concentrations for 1 h. Total eIF2α and actin were used as Western blot loading controls, respectively. (B, C) Relative Akt, GSK-3β, and eIF2α phosphorylation as well as ATF4 expression assessed by Western blotting in HT22 cells (B) transfected with the constitutively active PI3K p110* or (C) treated with the indicated concentrations of TGX-221 or PIK-90 for 2 h. Cytosolic extracts were used for Western blotting for phospho-Ser473-Akt (p-Akt), Akt, phospho-Ser9-GSK-3β, GSK-3β, p-eIF2α, and eIF2α and nuclear extracts for ATF4 blotting with actin as a loading control. Vertical white lines indicate the juxtaposition of non-adjacent lanes from the same gel (same exposure). (D) Relative luciferase acitivty in HT22 cells transfected as in Figure 1B treated with 1 μM PIK-90 or vehicle (Ctrl) for 24 h. (E) System xc− activity in HT22 cells treated with 1 μM PIK-90 or vehicle for 15 h. (F) Expression of the tour PI3K catalytic subunits in HT22 cells. The graphs represent the mean±SEM of three [(A–B), C right panels (except eIF2α: N=4) F]; (E) and four (C), left panels (except eIF2α: N=3); (D) independent experiments. Statistical analysis was performed using one-way ANOVA with Bonferroni's post test compared with vehicle treatment (A, C) or one sample t-test compared with 1 (B, D) or 100 (E), *p<0.05, **p<0,01, and ***p<0.001. GSK-3β, glycogen synthase kinase 3β; PIP3, phosphoinositide 3,4,5-triphosphate.
The PI3K pathway leads to the inactivating phosphorylation of GSK-3β (13). Thus, GSK-3β inhibition should mimic the effect of PI3K activation on the eIF2α/ATF4/xCT signaling module. Indeed, 20 mM lithium chloride (LiCl), a GSK-3β inhibitor (19, 33), strongly induced eIF2α phosphorylation, ATF4 protein expression (Fig. 3A), and ATF4 5′UTR translational activity in the presence as well as the absence of LY; while the effect of LiCl on ATF4 mRNA levels was not significant (Fig. 3B). Moreover, LiCl increased xCT mRNA expression, system xc− activity, cellular GSH, and survival in response to glutamate (Fig. 3C–F). Moreover, siRNA-mediated GSK-3β knock-down (Fig. 3G) and the GSK-3β inhibitors (2′Z,3′E)-6-bromoindirubin-3′-oxime and CT99021 (Supplementary Fig. S4) yielded results similar to those with LiCl. To substantiate the hypothesis that the regulation of system xc− activity is crucial for the PI3K/GSK3β pathway to influence GSH levels and sensitivity to glutamate, we used mouse embryonic fibroblasts (MEFs) derived from xCT−/− mice which were stably transfected with xCT driven by a CMV/chicken β actin promoter (pCAGxCT MEFs, see Supplementary Materials and Methods) as previously described for HH514 Burkitt's Lymphoma cells (4). As expected, 10 μM LY significantly down-regulated system xc− activity in wild-type MEFs, an effect that was more than reversed by 20 mM LiCl, whereas no significant changes in system xc− activity by these two compounds were detected in pCAGxCT MEFs (Fig. 3H). Matching patterns were observed for GSH levels (Fig. 3I) and the sensitivity to glutamate (Fig. 3J), although LY did not exacerbate cell death in both cell lines but LiCl only protected in the wild-type MEFs.
FIG. 3.

PI3K-regulated phospho-eIF2α- and ATF4-dependent xCT and system xc− expression is mediated via GSK-3β inhibition. (A) Relative eIF2α phosphorylation (p-eIF2α) and ATF4 expression in HT22 cells treated with either 20 mM LiCl or NaCl in combination with either 10 μM LY or vehicle for 4 h. (B) Relative luciferase activity (upper panel) in HT22 cells co-transfected with the ATF4 5′UTR luciferase (Luci) reporter and a Gal control plasmid and relative ATF4 mRNA abundance (lower panel) in HT22 cells, both treated as in (A) but for 24 h. (C, D) xCT mRNA abundance (C) and system xc− activity (D) in HT22 cells treated as in (B). (E) Relative GSH levels in HT22 cells treated as in (B) before exposure to 1 mM glutamate for 6 h. (F) Survival in response to 1 mM glutamate of HT22 cells treated as in (B) before exposure to glutamate for 24 h. (G) Relative GSK-3β expression, eIF2α phosphorylation, and ATF4 expression in HT22 cells transfected with siRNA specific for GSK-3β or control siRNA (Ctrl). (H, I) Relative (H) system xc− activity and total GSH (I) in response to 24 h of 10 μM LY and additional 20 mM LiCl in WT and pCAGxCT MEFs. The basal relative system xc− activity and cellular GSH levels of pCAGxCT MEFs compared with wild-type MEFs were 15.8- and 4.1-fold higher, respectively. (J) The effect of 24 h pre-treatment with 10 μM LY or LY plus 20 mM LiCl on the sensitivity of WT (left graph) and pCAGxCT MEFs (right graph). The graphs show the mean±SEM of five to seven (A, upper panel), six (J, right graph), four to five (A, lower panel, I), four (B, upper panel/D, E, G, middle and lower panel/H, I), and three (B, lower panel, C, F, G, upper panel, J, left graph) experiments. For (F), the survival of cells treated with NaCl and vehicle in the presence of 1 mM glutamate of each experiment was normalized to the mean survival (12.9%) of all experiments under these conditions. Total eIF2α or actin served as Western blot loading controls. Statistical analysis was performed using two-way ANOVA with Bonferroni's post test with LiCl-treated cells compared with cells with NaCl treatment (A–F) or by comparing Veh/NaCl with Ly/NaCl and Ly/NaCl with Ly/LiCl (H, I) or one-sample t-test compared with 1 (G), *p<0.05, **p<0,01, and ***p<0.001. GSH, glutathione; LiCl, lithium chloride; MEF, mouse embryonic fibroblast; WT, wild type.
The eIF2α/ATF4 pathway is involved in the neuroprotective as well as the pro-proliferative action of insulin, possibly by regulating the cellular redox potential
Insulin signals through the PI3K/Akt/GSK-3β signaling pathway (13) and has been shown to be neuroprotective (26, 72). In HT22 cells, 100 nM insulin induced eIF2α phosphorylation and ATF4 levels within 2 h (Fig. 4A). Moreover, insulin robustly induced system xc− activity ∼2.5-fold on 24 h of exposure (Fig. 4B). These effects were largely reduced in the presence of LY. The increased xc− activity in response to insulin was accompanied by increased resistance against oxidative glutamate toxicity (Fig. 4C). Growth factors such as insulin also promote cell proliferation and protein synthesis (57). In response to insulin, increased GSH synthesis enabled by the induction of the eIFα/ATF4/xCT pathway (39) might lead to a more reduced cellular redox environment, which will, in turn, support cell proliferation (67). Indeed, the complete absence of the eIFα/ATF4/xCT signaling module in MEFs expressing a genetically engineered eIF2α mutant where the phosphorylation site is inactivated by changing Serine 51 to Alanine (eIF2αS51A) (68) reversed the growth stimulatory effect of insulin observed in wild-type MEFs (Fig. 4D). Correspondingly, siRNA-mediated knock-down of ATF4 (40) strongly decreased cell proliferation by ∼40%, an effect that was completely reversed by augmenting cellular GSH (Fig. 4E). In addition, knock-down of ATF4 lowered the GSH/GSSG ratio (Fig. 4F), a change indicative of a more oxidized intracellular redox environment (67).
FIG. 4.

The eIF2α/ATF4 pathway is involved in the insulin-induced LY-sensitive up-regulation of system xc− and protection against oxidative glutamate toxicity as well as in the pro-proliferative activity of insulin. (A) Relative eIF2α phosphorylation and ATF4 expression in HT22 cells grown in medium with 0.5% FCS and treated with 100 nM insulin with or without 10 μM LY for 4 h. Total eIF2α or actin served as loading controls. (B, C) System xc− activity and survival in response to 10 mM glutamate in HT22 cells grown and treated as in (A) but for 24 h. The survival of vehicle-treated cells was normalized to the mean survival of all experiments (23.8%) under these conditions. (D) Relative cell growth over 48 h in response to 200 ng/ml insulin in WT and eIF2αS51A MEFs. (E, F) HT22 cells were transfected with control siRNA (Ctrl siRNA) or siRNA against ATF4. Twenty-four hours after transfection, cells were re-plated and grown for 48 h. (E) Relative cell number in control and ATF4 siRNA-transfected cells in the presence or absence of 2 mM GSH or 1 mM GEE. (F) The GSH/GSSG ratio of control and ATF4 siRNA-transfected cells calculated as a measure of the cellular redox status. The graphs represent the mean±SEM of three (A, B, F), four (C), five (E), and three to five (D) independent experiments. Statistical analysis was performed by two-way ANOVA with Bonferroni's post test with insulin treatment compared with control in the presence or absence of LY (A–D) and ATF4 siRNA compared with control siRNA (E) or by one-sample t test (F), *p<0.05, **p<0.01, and ***p<0.001. FCS, fetal calf serum; GEE, glutathione ethyl ester; GSSG, glutathione disulfide.
GCN2-mediated eIF2α phosphorylation is essential for the regulation of ATF4 and system xc− activity via PI3K/GSK-3β signaling, while nuclear factor (erythroid-derived 2)-like 2 is not involved
PI3K has been reported to up-regulate the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (11, 30), which also up-regulates xCT mRNA expression and, thus, system xc− (63). However, LY did not down-regulate basal Nrf2 levels in HT22 cells (Fig. 5A). To further examine whether the PI3K/Akt/GSK-3β pathway depends on eIF2α phosphorylation or Nrf2 to regulate system xc− activity, we used MEFs derived from Nrf2 knock-out (−/−) mice (63) and the eIF2αS51A MEFs. Consistent with previous findings that both Nrf2 and eIF2α phosphorylation induces xCT expression (36, 39), basal system xc− activity was reduced to ∼41% and ∼8% of control activity in Nrf2−/− and eIF2αS51A MEFs, respectively (Fig. 5B, left panel). Both PIK-90 and LY suppressed and LiCl rescued the LY effect on system xc− activity equally in wild-type and Nrf2−/− MEFs (Fig. 5B, right panel). In contrast, in eIF2αS51A MEFs, no changes in system xc− activity were detected with any of these compounds.
FIG. 5.

GCN2-mediated eIF2α phosphorylation is essential for the regulation of ATF4 and system xc− activity via PI3K/GSK-3β signaling, while Nrf2 is not involved. (A) Nuclear Nrf2 and ATF4 levels in HT22 cells treated with 10 μM LY for 2 h with actin as a Western blot loading control. (B) System xc− activity in WT, Nrf2−/−, or eIF2αS51A MEFs treated with PIK-90 and LY with 20 mM NaCl or LY in combination with 20 mM LiCl for 24 h. (C) Nuclear ATF4 levels in WT, PKR−/−, PERK−/−, HRI−/−, and GCN2−/− MEFs treated with vehicle (veh), 10 μM LY (LY), 20 mM LiCl (Li), or a LiCl and LY for 4 h. Actin served as a loading control. (D) Phosphorylation of eIF2α in cytosolic extracts from GCN2−/− and GCN2+/+ MEFs treated as in (C) with total eIF2α as a loading control. (E) GCN2 phosphorylation as a surrogate marker for its activity. HT22 cells were treated as indicated in (C), and then, cell lysates were prepared and GCN2 was immunoprecipitated with a rabbit GCN2 antibody and protein A-Sepharose. The immunoprecipitates were blotted with either an antibody to phospho-GCN2 (p-GCN2) or an antibody to total GCN2 (GCN2). (F) Regulation of system xc− activity, total GSH, and resistance against oxidative glutamate toxicity by the PI3K/GSK-3β pathway in GCN2−/− compared with GCN2+/+ MEFs. After a 24 h treatment with PI3K inhibitors and LiCl as in (B) system xc− (left panel) and total GSH (middle panel) were measured. Relative system xc− activity and relative total GSH in GCN2−/− compared with GCN2+/+ was 79% and 109%, respectively. (right panel) GCN2−/− and GCN2+/+ MEFs were seeded onto 96-well plates and after 24 h, they were treated with indicated concentrations of glutamate for 24 h before survival was quantified by the MTT assay. Mean survival of GCN2−/− was 74.1% at 2.5 mM glutamate, while GCN2+/+ MEFs showed 70% viability at 50 mM (Supplementary Fig. S5). Relative effect on survival of LY and LiCl at these glutamate concentrations (GCN2+/+ MEFs 50 mM, GCN2−/− MEFs 2.5 mM). The graphs represent the mean±SEM of three [(F), left panel], four [(F), middle and right panel], five (A), or four to six independent experiments (B). The non-quantified Western blots show representative results from at least three independent experiments. Statistical analysis was performed by a one-sample t test (A), one-way ANOVA (B), or two-way ANOVA (F) with Bonferroni's post tests;*p<0.05, **p<0.01, and ***p<0.001. GCN2, general control non-derepressible-2; HRI, heme-regulated eIF2α kinase; Nrf2, nuclear factor (erythroid-derived 2)-like 2; PERK, PKR-like endoplasmic reticulum kinase; PKR, protein kinase R.
We next asked how the PI3K/Akt/GSK-3β and eIF2α/ATF4/xCT pathways were connected. LY down-regulated and LiCl increased ATF4 expression in MEFs from wild-type, PKR−/−, PERK−/−, and HRI−/− mice, whereas this effect was completely abolished in MEFs deficient in the eIF2α kinase GCN2 (Fig. 5C). Phosphorylation of eIF2α was reduced by LY and increased by LiCl in wild-type MEFs, but no response to either of these treatments was seen in GCN2−/− MEFs (Fig. 5D). Activation of GCN2 leads to its autophosphorylation (60). In HT22 cells, GCN2 phosphorylation was decreased by LY and increased by LiCl (Fig. 5E). Downstream, the suppression of system xc− activity by either PIK-90 or LY was largely reduced in GCN2−/− MEFs compared with GCN2+/+ MEFs and the opposing effect of LiCl was even reversed. A similar pattern was observed for total GSH in response to LY and LiCl (Fig. 5F). GCN2−/− cells were considerably more sensitive to glutamate. When glutamate concentrations were used that led to a similar ∼70% survival (50 mM for GCN2+/+, 2.5 mM for GCN2−/− MEFs, see Supplementary Fig. S5), the effect of LY on viability was significantly less in GCN2−/− MEFs compared with wild-type MEFs, and again, the protective effect of LiCl was missing and even reversed in GCN2−/− MEFs. Taken togeter, these data show that Nrf2 is dispensable, whereas eIF2α phosphorylation is essential for the regulation of system xc− by PI3K/Akt/GSK-3β signaling under the experimental conditions employed in the present study and connected to PI3K/Akt/GSK-3β signaling via GCN2.
PI3Ks regulate xCT expression and system xc− activity via eIF2α phosphorylation and ATF4 in glioblastoma cells
Glioblastomas are highly malignant brain tumors in which the PI3K pathway is frequently hyperactive (16), and both ATF4 (6) and xCT (65) have been found to be up-regulated. Increased system xc− activity in glioblastomas has been linked not only to invasive growth but also to tumor-induced epilepsy (9, 63–66, 72, 74). Indeed, as described for regular glioma cell lines (12, 53), pharmacological inhibition of system xc− activity by two structurally different inhibitors, sulfasalazine and (S)-4-carboxyphenylglycine, also reduced proliferation of serum-differentiated cells derived from glioblastoma-initiating cancer stem cell line G38 (Supplementary Fig. S6). In these cells and a similarly generated cell line, G35, (5), LY-mediated PI3K inhibition down-regulated eIF2α phosphorylation, ATF4 protein levels, and system xc− activity (Fig. 6), indicating that the PI3K pathway is involved in the activity of the eIFα/ATF4/xCT signaling module in glioblastoma cells in vitro.
FIG. 6.

The phosphorylation of eIF2α, ATF4 expression, and system xc− activity are regulated by PI3Ks in human cancer stem cell-derived glioblastoma cells. Relative eIF2α phosphorylation and nuclear ATF4 levels in glioblastoma cells derived from G35 (A) and G38 (B) glioblastoma-initiating cancer stem cells treated with 10 μM LY for 2 h compared with vehicle (Veh) (left panels). Total eIF2α and actin served as loading controls. Relative system xc− activity in G35 and G38 glioblastoma cells treated with 10 μM LY (LY) for 24 h compared with vehicle (Veh). Graphs represent the mean±SEM of three [(A), left lower and right, (B), left upper and right panel] to four [(A), left upper, (B) left lower panel] independent experiments. Statistical analysis was performed by one-sample t tests, *p<0.05, **p<0.01, and ***p<0.001.
High-frequency neuronal activity up-regulates system xc− activity via the PI3K/Akt/GSK-3β/eIF2α/ATF4 pathway in primary cortical neurons
In glioblastoma patients, not only was high tumor xCT expression found to be associated with epileptic seizures but also xCT up-regulation in the peritumoral tissue was associated with epilepsy (75), possibly indicating epilepsy-induced changes of the affected brain tissue. To test this hypothesis, we induced high-frequency discharges, as found in epilepsy, in rat cortical neurons using the GABAA receptor antagonist bicuculline (Bic) plus the K+ channel blocker 4-aminopyridine (Bic/4-AP) (24). Bic/4-AP was reported to activate the PI3K/Akt/GSK-3β pathway through synaptic N-methyl-D-aspartate receptors in this paradigm (2, 54). Bic/4-AP led to a robust and LY-sensitive increase in system xc− activity as well as xCT mRNA abundance (Fig. 7A, B). In addition, the GSK-3β inhibitor CT99021 increased xCT mRNA levels (Fig. 7C). The up-regulation of xCT mRNA did not depend on Nrf2, as the Bic/4-AP-mediated induction of xCT mRNA was fully preserved in Nrf2−/− neurons (Fig. 7D). In contrast, Bic/4-AP treatment significantly increased ATF4 protein levels in an LY-sensitive manner (Fig. 7E) and stimulated the translational activity of the 5′UTR of ATF4 (Fig. 7F), indicating increased ATF4 protein translation induced by eIF2α phosphorylation. Moreover, the Bic/4-AP-induced xCT promoter activity in neurons transfected with an xCT promoter-Luci reporter was suppressed when co-transfected with a dominant-negative ATF4 mutant (Fig. 7G).
FIG. 7.

In cortical neurons, high-frequency neuronal activity GSK-3β-dependently induces system xc− via ATF4. (A) System xc− activity in rat cortical neurons treated for 24 h with or without 50 μM Bic plus 250 μM 4-AP (Bic/4-AP) and 30 μM LY (LY). (B–D) xCT mRNA expression of neurons (B, D) stimulated as in (A) or (C) co-treated with 2 μM of the GSK-3β inhibitor CT99021 (CT) with and without LY was measured by qPCR normalized to the expression of the housekeeping gene GAPDH. (D) xCT mRNA abundance measured by qPCR in neurons from Nrf2 knock-out (Nrf2−/−) and WT mice. (E) Relative ATF4 protein levels in neurons on stimulation with Bic/4-AP assayed by Western blotting with actin as a loading control. Vertical white lines indicate the juxtaposition of non-adjacent lanes from the same gel (same exposure). (F) Bic/4-AP-induced ATF4 translation was assessed in cortical neurons that were transfected with the ATF4 5′UTR luciferase (Luci) reporter along with a control Renilla luciferase plasmid. (G) Cortical neurons were transfected with an xCT-promoter luciferase reporter construct, a control plasmid expressing Renilla luciferase, and either a β-globin control plasmid (Ctrl) or plasmids expressing dominant-negative ATF4 (ATF-DN) or ATF4 before stimulation with Bic/4-AP. Firefly luciferase activity was compared with Renilla luciferase activity. Graphs represent the mean±SEM of (A) six, (B, C, G) four, (D) three, (E) eight, and three to five (F) independent experiments. Statistical analysis was performed by one-way ANOVA compared with control or 0 h Bic/4-Ap (A, F, G) or two-way ANOVA compared with cells not treated with Bic/4-AP (B, D, E) or not treated with CT99021 (C) with Bonferroni's post test, *p<0.05, **p<0.01, and ***p<0.001. 4-AP, 4-aminopyridine; Bic, bicuculline; qPCR, quantitative polymerase chain reaction.
The PI3K/Akt/GSK-3β/eIF2α/ATF4/xCT pathway is activated in hippocampi from patients with temporal lobe epilepsy
Next, we asked whether the new pathway described here is up-regulated in human hippocampal tissue samples from patients with temporal lobe epilepsy (TLE) obtained during lobectomy. Hypothetically, this should be followed by an increase in extracellular glutamate that is known to decrease the epileptic threshold (15). Indeed, Akt and GSK-3β phosphorylation in epileptic tissue was strongly increased compared with autoptic control hippocampi (Fig. 8A). This was associated with a significant increase in eIF2α phosphorylation and ATF4 and xCT protein expression. Linear regression analysis testing the relationship of the four pairs of connected parts of the pathway across the whole group of samples showed positive results for all pairs (Fig. 8B). Importantly, neither Akt, GSK-3β, and eIF2α phosphorylation nor ATF4 and xCT expression were influenced by the postmortem interval in the control group (Fig. 8C).
FIG. 8.

The PI3K/GSK-3β/eIF2α/ATF4/xCT pathway is activated in human epileptic hippocampal tissue. Protein extracts from surgical specimens from human epileptic hippocampi (Epilepsy) and control hippocampi (Control) were tested for the relative phosphorylation of Akt (phospho-Akt), GSK-3β (phospho-GSK-3β), and eIF2α (phospho-eIF2α) as well as ATF4 and xCT expression by Western blotting. Either antibodies that recognize the proteins irrespective of their phosphorylation state (total) or actin were used as loading controls. (A) Representative blots are shown. Longer exposures than those shown were used for the quantification of some of the samples. Graphs show the quantitative results with epileptic (Epi) compared with control (Ctrl) tissue with the mean value of the control group normalized to 1. For eIF2α phosphorylation, one sample (Epi sample 2, p-eIF2α/eIF2α=55.8) was excluded, as this value was classified as an extreme outlier (see the “Materials and Methods” section). Statistical analysis was performed by one-tailed Student's t tests, ***p<0.001, **p<0.01, and *p<0.05. (B) Pair-wise linear regression of the four pairs of connected parts of the pathway across the whole group of samples (Epi sample 2 excluded). The goodness of fit is given in the graphs. The slope was significantly different from zero in all cases (GSK-3β/p-Akt, p<0.001; p-eIF2α/p-GSK-3β and ATF4/p-eIF2, p<0.01; xCT/ATF4, p<0.05). (C) Relationship of Akt, GSK-3β and elF2α phosphorylation and ATF4 and xCT expression with the postmortem interval in control hippocampi.
Discussion
The results of the present study tie together two apparently functionally disparate signaling pathways, namely, the PI3K/Akt/GSK-3β pathway and the eIF2α/ATF4/xCT signaling module (Fig. 9). Inhibition of the first pathway by two independent inhibitors, LY and PIK-90, down-regulated the second signaling module. Activation of the PI3K/Akt/GSK-3β pathway by insulin, over-expression of a constitutively active PI3K, or mimicking its activation by PIP3, three different GSK-3β inhibitors or GSK-3β knock-down yielded the opposite results. Moreover, in cells unable to phosphorylate eIF2α, the regulation of system xc− activity by manipulation of the PI3K/Akt/GSK-3β pathway was completely lost. We found evidence of this new connection in cells as diverse as hippocampal HT22 cells, MEFs, glioblastoma cells, and primary neurons and present data strongly supporting the hypothesis that this pathway is responsible for up-regulating xCT expression in the human epileptic hippocampus.
FIG. 9.
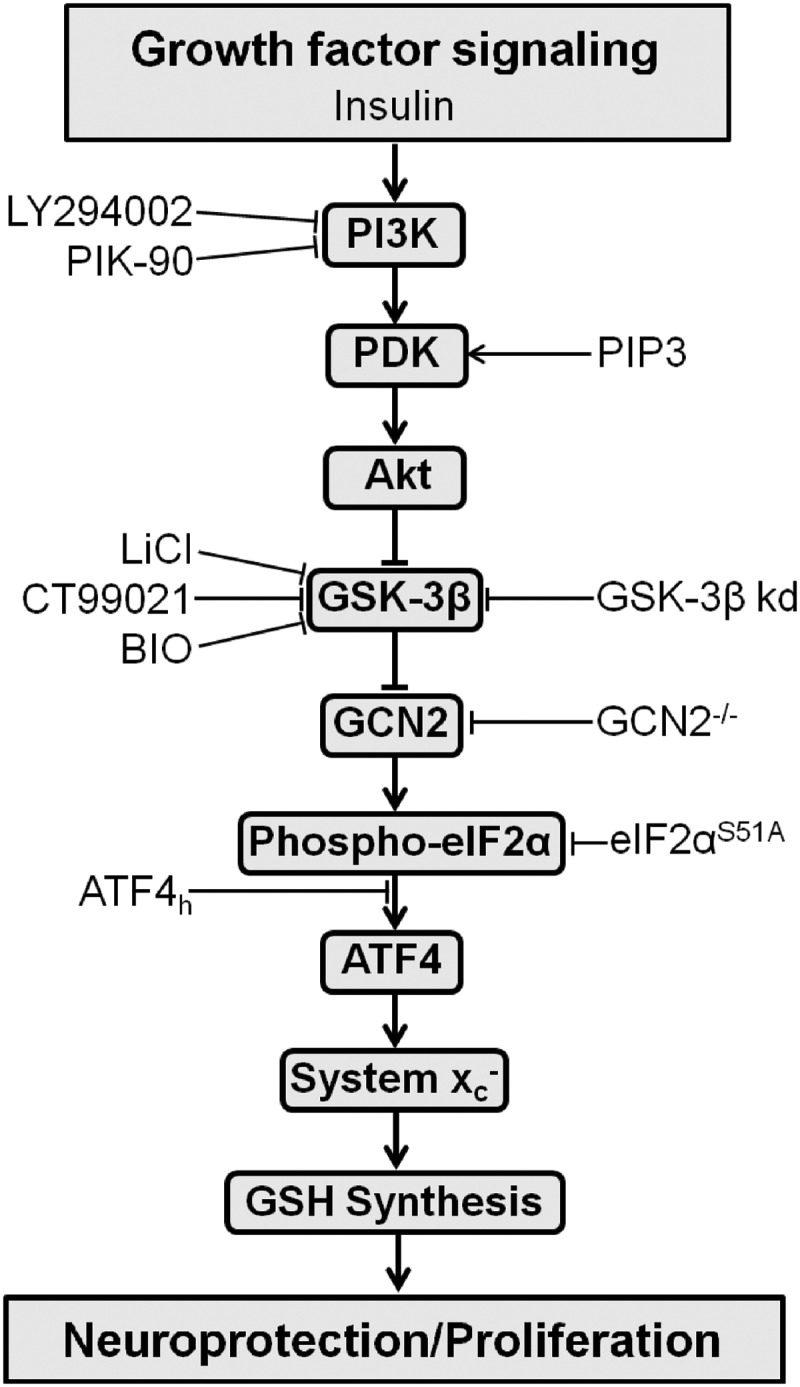
Summary of the new pathway and the experimental evidence supporting it. Activation is depicted as arrows, inhibition as ⊤, kd: siRNA-mediated knock down.
The link between the PI3K/Akt/GSK-3β pathway and the eIF2α/ATF4/xCT signaling module is the eIF2α kinase GCN2. This conclusion was drawn from three lines of evidence. First, in HT22 cells, manipulation of the PI3K/Akt/GSK-3β pathway changes GCN2 autophosphorylation, a surrogate marker for its activity (60), consistent with the observed changes in eIF2α phosphorylation and ATF4 protein levels. Second, the regulation of ATF4 through the PI3K/Akt/GSK-3β pathway is selectively lost in GCN2-deficient cells. Third, the regulation of system xc− activity in GCN2-deficient cells through the PI3K/Akt/GSK-3β pathway is largely reduced. Classically, GCN2 is activated not only by uncharged tRNAs when amino acids are in limited supply (32) but also by UV radiation (29). Thus, our findings describe a new role for this eIF2α kinase. However, how the PI3K/GSK-3β pathway regulates GCN2 activity remains to be determined.
To our knowledge, the observation that the eIF2α/ATF4/xCT signaling module is modulated by the PI3K pathway has not been previously recognized. However, that insulin can up-regulate ATF4 protein levels has been described (1, 27, 47). Similarly, macrophage colony-stimulating factor, which also binds to a receptor tyrosine kinase (8), induced LY-sensitive ATF4 up-regulation in osteoclasts (10). However, two of these reports concluded that the observed increase in ATF4 protein is mediated by reduced degradation (10, 27).
Downstream of eIF2α and ATF4, the new pathway connects PI3K signaling with the expression of the specific subunit of the cystine/glutamate antiporter system xc−, xCT, and, therefore, GSH metabolism. Insulin was reported to induce system xc− in human fibroblasts (43). Moreover, system xc− activity has been shown to be up-regulated by other factors that activate the PI3K/Akt/GSK-3β pathway, including erythropoietin (70), insulin-like growth factor 1 (55), and fibroblast growth factor 2 (42). Whether these observations are explained by the pathway described in the present study should be determined.
Of note, Nrf2, the other transcription factor that stimulates xCT expression (63), has been shown to be up-regulated though the activation of the PI3K/Akt/GSK-3β pathway (11, 35, 59) by inhibiting proteasomal degradation which is initiated by β-TrCP after GSK-3β-mediated Nrf2 phosphorylation. However, the impact of GSK-3β on Nrf2 may not be pronounced when Nrf2 degradation is directed by the “classical” pathway through Keap1 [see ref. (50)]. At present, it is unclear what dictates the relative importance of the two pathways. At least in HT22 cells, we found that basal Nrf2 protein levels are unaffected by PI3K inhibition. Moreover, our data showing that the regulation of system xc− activity by the PI3K/Akt/GSK-3β pathway as well as the up-regulation of the xCT mRNA in response to high-frequency discharges are preserved in Nrf2−/− MEFs and primary neurons, respectively, excluding Nrf2 as a major regulator of system xc− through the PI3K/Akt/GSK-3β pathway under the experimental conditions employed in our study.
The connection between the PI3K/Akt/GSK-3β pathway and the eIF2α/ATF4/xCT signaling module has functional consequences at least in vitro, because through this connection cellular GSH levels and the sensitivity of cells to oxidative glutamate toxicity are regulated. The transcriptional regulation of xCT by the PI3K/GCN2/eIF2α/ATF4 is essential for the pathway to influence cellular GSH and resistance against oxidative stress, as cells expressing xCT that were driven by β actin promoter did not show any difference in GSH or cell death in response to oxidative glutamate toxicity when this pathway was manipulated pharmacologically. This suggests that the PI3K/Akt/GSK-3β/GCN2/eIF2α/ATF4/xCT pathway might be relevant for the cytoprotective consequences of PI3K activation, including neuroprotection against diverse insults (58, 72) although direct evidence for neuroprotective action of xCT induction in vivo is lacking, and it plays a role in the malignant transformation and chemoresistance of tumor cells (21, 62). However, in addition to GSK-3β inhibition and subsequent neuroprotection via the pathway described here, Akt activation has been described to be neuroprotective by inducing BAD phosphorylation (69) and activation of the mammalian target of rapamycin (41). Due to its high concentration in cells, the GSH/GSSG redox couple determines the general redox environment of cells with a more reduced redox state that is linked to proliferation (67). ATF4 knock-down shifted the GSH/GSSG redox couple to a more oxidized state while inhibiting cell proliferation in a GSH-sensitive manner. Moreover, we show that insulin loses its growth-stimulating effect in fibroblasts lacking eIF2α phosphorylation. These findings strongly support the view that the eIF2α/ATF4/xCT signaling module is an essential constituent of the pro-proliferative machinery downstream of growth factor signaling by keeping the cellular redox potential in a reduced, growth-permissive state. This finding is surprising, as eIF2α phosphorylation has been viewed as a mechanism that reduces protein translation, and therefore growth, in response to cellular stress [reviewed in ref. (56)]. Thus, our data indicate that the formerly accepted role of eIF2α phosphorylation may be overly simplistic and applied only to very high levels of eIF2α phosphorylation.
Glioblastomas exhibit increased PI3K signaling (34), ATF4 (6) and xCT expression (65). The latter has been associated with invasive growth (44) and poor outcome (74) as well as with tumor-induced seizures that are possibly generated by system xc− mediated glutamate release (9, 75). Our data show that the connection between the PI3K/Akt/GSK-3β pathway and the eIF2α/ATF4/xCT signaling module is active in human glioblastoma cells which are derived from glioblastoma-initiating cancer stem cells. Thus, PI3K signaling might induce xCT expression via the eIF2α/ATF4 signaling module in glioblastomas and, therefore, tumor-associated seizures. Importantly, the eIF2α/ATF4/xCT branch of PI3K signaling that we describe might also explain other types of seizures. We show that this pathway is also induced by bursts of action potentials as found in epilepsy in primary neurons and in human epileptic hippocampi when compared with non-epileptic controls. Of note, since our control samples were derived from autopsy material and compared with biopsies obtained during lobectomy of the TLE patients, we cannot exclude changes in protein phosphorylation or expression due to the different tissue handling. However, since these data are highly consistent with the in vitro results and we found no association of the markers examined with the postmortem interval in control tissues, we assume that this potential bias is negligible. Thus, we hypothesize that activation of the PI3K pathway in response to epileptic discharges might result in pro-epileptic molecular changes via induction of eIF2α phosphorylation, ATF4 protein expression, and, finally, up-regulation of system xc− and, therefore, glutamate release.
Materials and Methods
For materials, see Supplementary Materials and Methods.
Human samples
Epileptic patients underwent resection of the hippocampus for medically intractable TLE and were neuropathologically diagnosed to have hippocampal sclerosis of Wyler grade 3 (18) (Supplementary Table S2). Control hippocampal tissue was obtained at autopsy from patients without a history of seizures. There were no significant differences in sex distribution or age between the two groups. Informed consent was obtained for the use of brain tissue and for access to medical records for research purposes. All samples were obtained and used in a manner that was compliant with the Declaration of Helsinki.
Cell culture
HT22 cells were grown in high-glucose Dulbecco's modified Eagle medium (DMEM) (Invitrogen) that was supplemented with 10% fetal calf serum (FCS; Hyclone) as described earlier (14). For viability assays, 2.5×103 HT22 cells, 5×103 GCN2+/+, 1×104 GCN2−/−, and 1×103 wild-type and pCAGxCT MEFs were plated in 96-well plates with LY, PIK-90, TGX-221, insulin, CT99021, or LiCl or vehicle or, in the case of LiCl, NaCl. In the case of HT22 cells treated with insulin and CT99021, the serum concentration was reduced to 0.5%, for wild type, and pCAGxCT MEFs medium cystine concentration had to be reduced to 50 μM to induce cell death in pCAGxCT MEFs. After 24 h of culture, the medium was exchanged with fresh medium without the respective compound, and the indicated concentrations of glutamate for 24 h before viability were quantified by the MTT assay as described earlier (38).
eIF2αS51A and wild-type control MEFs (68) were a kind gift from Randal J. Kaufman and Donalyn Scheuner (Sanford-Burnham Institute), Nrf2−/− MEFs from Hideyo Sato (Yamagata University), PKR−/−, PERK−,/− and GCN2−/− MEFs from Antonis Koromilas (McGill University), and HRI−/− MEFs from John Bartlett and Megan Sierant (Forsyth Institute). pCAG-xCT MEFs were generated as described in the “Supplementary Materials and Methods” section. MEFs were propagated in high-glucose DMEM (Invitrogen) with 10% FCS (Hyclone) that was additionally supplemented with essential and non-essential amino acids (Invitrogen). Cells were re-plated for no more that 10 passages by trypsinization when confluent. Glioblastoma-initiating cancer stem cells derived from surgical specimens were grown as described (5). The use of material for cell cultures was approved by the ethics committee, Medical Faculty, University of Ulm, and informed consent was obtained from the patients before surgery. G35 and G38 glioblastoma-initiating cancer stem cells were differentiated by culture in DMEM (GIBCO by Life Technologies) that was supplemented with 10% FCS (GIBCO), and stem cell-derived glioblastoma cells were passaged by trypsination for approximately 20 times.
Cortical neurons from E21 Sprague–Dawley rats were cultured as described earlier (2), and mouse cortical neurons from E17.5 Nrf2+/+ and Nrf2−/− mice were cultured as described (20). The Nrf2−/− mice were kindly provided by Masayuki Yamamoto of the University of Tsukuba (now University of Tohoku). In both cases, neurons were cultured in growth medium consisting of Neurobasal A that was supplemented with B27 (Invitrogen), 1% rat serum (Harlan SeraLab), and 1 mM glutamine (Sigma-Aldrich). To obtain astrocyte-free neuronal preparations (>98% NeuN-positive neurons and <0.2% GFAP-positive astrocytes), cultures were treated with the anti-mitotic cytosine-arabinoside immediately post-plating. Experiments were performed after 8–10 days in vitro (DIV). Before stimulation, neurons were transferred to a trophically deprived medium containing 10% Minimum Essential Medium (Invitrogen) and 90% Salt/Glucose/Glycine medium consisting of: 114 mM NaCl, 32.7 mM NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM glycine, 30 mM glucose, 0.5 mM sodium pyruvate, and 0.1% phenol red; osmolarity 325 mOsm/L (2) and allowed to equilibrate for at least 3 h. Bursts of action potentials were induced through stimulation with 50 μM Bic and 250 μM 4-AP for typically 24 h (24) unless stated otherwise.
Transfection and luciferase reporter assays
For luciferase (Luci) reporter assays, HT22 cells were transfected with a pSV-β-galactosidase (Gal) plasmid (Promega) and a vector containing the ATF4 5′UTR and AUG fused to Luci by the TK promoter in a pGL3 backbone (22) (a generous gift from David Ron, Metabolic Research Laboratories, University of Cambridge) as previously described (40). When using a pcDNA3.1-EGFP construct and 4′,6-diamidino-2-phenylindole to visualize nuclei, the transfection efficacy was 72.7±8.9% (not shown). Twenty-four hours after transfection, cells were treated with the indicated compounds for 24 h. Next, cells were lysed, and Luci and Gal enzyme activities were measured as previously described (39). For siRNA transfection, HT22 cells were plated in 60 mm dishes at 5×105 cells/dish and 20 pmol ATF4 siRNA (#sc-35113), GSK-3β siRNA (#sc-35525), or control siRNA (#sc-37007), all from Santa Cruz Biotechnology, were used along with Lipofectamine 2000 as previously described (40). Neurons were transfected at DIV 8 using Lipofectamine 2000 as described (2). For xCT-Luci assays, 0.1 μg pTK-Renilla (Promega), 0.2 μg pGL3-4.7 xCT promoter Luci reporter plasmid (63) (a kind gift from Hideyo Sato, Yamagata University), and 0.3 μg of a β-globin control plasmid [a gift from Richard Maurer, Oregon Health Sciences University (71)], or plasmids encoding ATF4 (39) or a dominant negative ATF4 mutant [a gift from Jawed Alam, Ochsner Medical Center (25)] were used. To assay ATF4 5′UTR-mediated translation, the ATF4 5′UTR Luci reporter was co-transfected with the pTK-Renilla plasmid (22). Twenty-four hours after transfection, neurons were stimulated as indicated. Luci assays were performed using the Dual Glo assay kit (Promega) with Firefly Luci reporter gene activity normalized to Renilla control.
Enzymatic measurement of GSH
To measure GSH, 1.65×105 HT22 cells were plated in 60 mm dishes and grown in the presence of 10 μM LY or vehicle for 24 h. Total GSH was measured as described (45). To measure the GSSG/GSH ratio, the GSH/GSSG-Glo Assay kit (Promega) was used.
Measurement of system xc− activity
3×104 HT22 cells were seeded into 24-well plates and treated with LY, LiCl or NaCl, or insulin for 24 h and PIK-90 for 15 h. System xc− activity after 24 h of PIK-90 exposure returned to normal levels (data not shown), either due to instability of the compound or due to up-regulation of PI3Ks other than PI3Kα. For insulin and CT99021, FCS concentration was reduced to 0.5%. 6×104 eIF2αS51A MEFs, Nrf2−/−, GCN2−/− MEFs, or control MEFs with or without 10 μM LY, 1 μM PIK-90, or vehicle (dimethylsulfoxide) with 20 mM LiCl or NaCl were grown for 24 h. For pCAGxCT MEFs and controls, the number was reduced to 3×104. In experiments including eIF2αS51Aand Nrf2−/− MEFs, extra amino acids and 1 mM GSH were added to the growth medium to prevent artifacts due to reduced amino-acid availability or GSH levels as a results of the genotype. G35 and G38 glioblastoma cells were seeded at a density of 1×104 cells per cm2 cells per well in 24-well plates that were coated with poly-l-lysine. Cells were washed thrice with sodium-free Hank's buffered salt solution (52). System xc− activity was measured as sodium-independent, homocysteic acid-inhibitable uptake of 3H-glutamate (Perkin Elmer NEN) as previously described (39). Primary cortical neurons stimulated in 90% Salt/Glucose/Glycine medium for 24 h were transferred to Na+-free Krebs' solution (containing 120 mM choline chloride, 25 mM Tris, 5 mM KCl, 1 mM K2HPO4, 1 mM MgCl2, 2 mM CaCl2, and 30 mM glucose). Then, 1 μCi/ml 3H-glutamate (Perkin Elmer NEN) was added to solution in the presence or absence of 1 mM cystine and cells were incubated for 30 min at 37°C. Cells were subsequently washed twice in ice-cold Na+-free Krebs' solution and lysed in K2HPO4 buffer containing 0.5% Triton. Radioactivity of lysates was measured using a scintillation counter, with a portion of the lysate used to determine protein concentration using the Bradford or bicinchoninic acid method (both from Pierce) for normalization purposes. System xc− activity was calculated as sodium-independent cystine-inhibitable 3H-glutamate uptake.
Subcellular fractionation and preparation of protein samples
Subcellular fractionation and the preparations of protein samples was performed as previously described (46, 49) with minor modifications; for details, see the “Supplementary Materials and Methods” section.
Western blotting
Western blotting was performed as previously reported (5, 39) and described in detail, including the primary antibodies and dilutions in the “Supplementary Materials and Methods” section. The specificity of the ATF4 and Nrf2 bands detected by both of these antibodies was extensively characterized previously (Supplementary Fig. 1B) [see ref. (40)]. For relative eIF2α and GSK-3β phosphorylation, parallel blots were performed. For all other antibodies, the same membrane was re-probed for actin or the antiserum reacting with the total protein produced in a different species from the phospho-specific antibody. Relative ATF4 and xCT expression was normalized to actin band density. For cellular extracts, each Western blot was repeated with at least three independent protein samples.
RNA preparation, reverse transcription, and qPCR
Total RNA from HT22 cells was isolated using the RNAeasy mini kit (Qiagen). qPCR that was used to quantify ATF4 and xCT mRNA expression was performed as previously described (40). For primers used for qPCR for the cDNAs encoding the four p110 isoforms, see the “Supplementary Materials and Methods” section. RNA from primary cortical neurons was prepared using the Roche isolation reagents, including a 15 min DNase I treatment to avoid genomic DNA contamination of samples (Roche). cDNA was synthesized using the Transcriptor One-Step RT-PCR Kit (Roche). qPCR was performed in an Mx3000P qPCR System (Stratagene; Agilent Technologies) using 2× FastStart Universal SYBR Green Master Mix (Roche) according to the manufacturer's instructions. Expression of the gene of interest was calculated using the efficiency corrected ΔΔCt method, normalizing to either GAPDH or 18S ribosomal RNA as housekeeping genes. For primers used for qPCR for cDNA derived from primary neurons, see the “Supplementary Materials and Methods” section.
Statistical analysis
Data from at least three independent experiments were normalized, pooled, and analyzed using Graph Pad Prism 4 software followed by appropriate statistical tests. For exclusion of outliers, the established definition of an extreme outlier was used as follows: values lower or higher than the 25% percentile (Q1) or 75% percentile (Q3) minus or plus three times the interquartile range (Q3–Q1), respectively.
Supplementary Material
Abbreviations Used
- 4-AP
4-aminopyridine
- 4-CPG
(S)-4-carboxyphenylglycine
- 5′UTR
5′ untranslated region
- ANOVA
analysis of variance
- ATF4
activating transcription factor 4
- Bic
bicuculline
- BIO
(2′Z,3′E)-6-bromoindirubin-3′-oxime
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- DIV
days in vitro
- DMSO
dimethylsulfoxide
- EDTA
ethylenediaminetetraaceticacid
- eIF2α
eukaryotic initiation factor 2α
- FCS
fetal calf serum
- GABA
γ-aminobutyric acid
- Gal
β-galactosidase
- GCN2
general control non-derepressible-2
- GEE
glutathione ethyl ester
- GFAP
glial fibrillary acidic protein
- GSH
glutathione
- GSK-3β
glycogen synthase kinase 3β
- GSSG
glutathione disulfide
- HCA
homocysteic acid
- HRI
heme-regulated eIF2α kinase
- ISR
integrated stress response
- LiCl
lithium chloride
- mBIO, MeBIO
methyl-BIO
- MEF
mouse embryonic fibroblast
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- ORF
open reading frame
- PBS
phosphate-buffered saline
- PERK
PKR-like endoplasmic reticulum kinase
- PI3K
phosphoinositide 3-kinase
- PIP3
phosphoinositide 3,4,5-triphosphate
- PKR
protein kinase R
- qPCR
quantitative polymerase chain reaction
- RT
room temperature
- SAS
sulfasalazine
- SDS
sodium dodecyl sulfate
- SEM
standard error of the mean
- TBS
Tris-buffered saline
- TLE
temporal lobe epilepsy
Acknowledgments
E.A. was funded by National Epilepsy Funds, NEF 09-05; G.E.H. by the Medical Research Council, the Wellcome Trust, and the Biotechnology and Biological Research Council; P.M. by the Alzheimer's Association and NIH; and P.J.M. by Diabetes UK. J.V.L. is supported by the agency for Innovation by Science and Technology (IWT/SB/101344). J.V.L., I.S., and A.M. acknowledge the Fund for Scientific Research Flanders (FWO, Belgium, grant G038412N) and the Queen Elizabeth Medical Foundation (G.S.K.E.) for financial support. The authors are grateful to Masayuki Yamamoto for providing the Nrf2−/− mice.
Author Disclosure Statement
The authors declare that they have no conflicts of interest to disclose.
References
- 1.Adams CM. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J Biol Chem 282: 16744–16753, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Al-Mubarak B, Soriano FX, and Hardingham GE. Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. Channels (Austin) 3: 233–238, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baird TD. and Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr 3: 307–321, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, Kolle P, Tschoep K, Issels RD, Daniel PT, Conrad M, and Bornkamm GW. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene 27: 1618–1628, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Berger R, Jennewein C, Marschall V, Karl S, Cristofanon S, Wagner L, Vellanki SH, Hehlgans S, Rodel F, Debatin KM, Ludolph AC, and Fulda S. NF-kappaB is required for Smac mimetic-mediated sensitization of glioblastoma cells for gamma-irradiation-induced apoptosis. Mol Cancer Ther 10: 1867–1875, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG, and Koumenis C. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 24: 3470–3481, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blume-Jensen P, Janknecht R, and Hunter T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr Biol 8: 779–782, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Bourette RP. and Rohrschneider LR. Early events in M-CSF receptor signaling. Growth Factors 17: 155–166, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, and Sontheimer H. Glutamate release by primary brain tumors induces epileptic activity. Nat Med 17: 1269–1274, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao H, Yu S, Yao Z, Galson DL, Jiang Y, Zhang X, Fan J, Lu B, Guan Y, Luo M, Lai Y, Zhu Y, Kurihara N, Patrene K, Roodman GD, and Xiao G. Activating transcription factor 4 regulates osteoclast differentiation in mice. J Clin Invest 120: 2755–2766, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, and Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32: 3765–3781, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, and Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci 25: 7101–7110, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cross DA, Alessi DR, Cohen P, Andjelkovich M, and Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785–789, 1995 [DOI] [PubMed] [Google Scholar]
- 14.Davis JB. and Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res 652: 169–173, 1994 [DOI] [PubMed] [Google Scholar]
- 15.De Bundel D, Schallier A, Loyens E, Fernando R, Miyashita H, Van Liefferinge J, Vermoesen K, Bannai S, Sato H, Michotte Y, Smolders I, and Massie A. Loss of system xc- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci 31: 5792–5803, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, and Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21: 2683–2710, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, Timms JF, and Waterfield MD. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J 404: 15–21, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorter JA, Zurolo E, Iyer A, Fluiter K, van Vliet EA, Baayen JC, and Aronica E. Induction of sodium channel Na(x) (SCN7A) expression in rat and human hippocampus in temporal lobe epilepsy. Epilepsia 51: 1791–1800, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Grimes CA. and Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 65: 391–426, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Gupta K, Patani R, Baxter P, Serio A, Story D, Tsujita T, Hayes JD, Pedersen RA, Hardingham GE, and Chandran S. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ 19: 779–787, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, McCubrey JA, Travali S, and Libra M. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance (review). Int J Oncol 40: 639–644, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, and Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, and Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Hardingham GE, Arnold FJ, and Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci 4: 261–267, 2001 [DOI] [PubMed] [Google Scholar]
- 25.He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, and Alam J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem 276: 20858–20865, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Hui L, Pei DS, Zhang QG, Guan QH, and Zhang GY. The neuroprotection of insulin on ischemic brain injury in rat hippocampus through negative regulation of JNK signaling pathway by PI3K/Akt activation. Brain Res 1052: 1–9, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Inageda K. Insulin modulates induction of glucose-regulated protein 78 during endoplasmic reticulum stress via augmentation of ATF4 expression in human neuroblastoma cells. FEBS Lett 584: 3649–3654, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM, Yuan Y, Sturgeon SA, Prabaharan H, Thompson PE, Smith GD, Shepherd PR, Daniele N, Kulkarni S, Abbott B, Saylik D, Jones C, Lu L, Giuliano S, Hughan SC, Angus JA, Robertson AD, and Salem HH. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med 11: 507–514, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Jiang HY. and Wek RC. GCN2 phosphorylation of eIF2alpha activates NF-kappaB in response to UV irradiation. Biochem J 385: 371–380, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson DA, Andrews GK, Xu W, and Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem 81: 1233–1241, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Kaufman RJ, Davies MV, Pathak VK, and Hershey JW. The phosphorylation state of eucaryotic initiation factor 2 alters translational efficiency of specific mRNAs. Mol Cell Biol 9: 946–958, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kilberg MS, Shan J, and Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab 20: 436–443, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein PS. and Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A 93: 8455–8459, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knobbe CB. and Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol 13: 507–518, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JM, Hanson JM, Chu WA, and Johnson JA. Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J Biol Chem 276: 20011–20016, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, Wiedau-Pazos M, Schubert D, Maher P, and Methner A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem 111: 332–343, 2009 [DOI] [PubMed] [Google Scholar]
- 37.Lewerenz J, Klein M, and Methner A. Cooperative action of glutamate transporters and cystine/glutamate antiporter system Xc- protects from oxidative glutamate toxicity. J Neurochem 98: 916–925, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Lewerenz J, Letz J, and Methner A. Activation of stimulatory heterotrimeric G proteins increases glutathione and protects neuronal cells against oxidative stress. J Neurochem 87: 522–531, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Lewerenz J. and Maher P. Basal levels of eIF2alpha phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem 284: 1106–1115, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewerenz J, Sato H, Albrecht P, Henke N, Noack R, Methner A, and Maher P. Mutation of ATF4 mediates resistance of neuronal cell lines against oxidative stress by inducing xCT expression. Cell Death Differ 19: 847–858, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L, Xu B, Zhu Y, Chen L, and Sokabe M. DHEA prevents Abeta25-Abeta35-impaired survival of newborn neurons in the dentate gyrus through a modulation of PI3K-Akt-mTOR signaling. Neuropharmacology 59: 323–333, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Liu X, Resch J, Rush T, and Lobner D. Functional upregulation of system xc-by fibroblast growth factor-2. Neuropharmacology 62: 901–906, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Longo N, Franchi-Gazzola R, Bussolati O, Dall'Asta V, Foa PP, Guidotti GG, and Gazzola GC. Effect of insulin on the activity of amino acid transport systems in cultured human fibroblasts. Biochim Biophys Acta 844: 216–223, 1985 [DOI] [PubMed] [Google Scholar]
- 44.Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, and Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res 67: 9463–9471, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maher P, Lewerenz J, Lozano C, and Torres JL. A novel approach to enhancing cellular glutathione levels. J Neurochem 107: 690–700, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maher PA. Nuclear translocation of fibroblast growth factor (FGF) receptors in response to FGF-2. J Cell Biol 134: 529–536, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malmberg SE. and Adams CM. Insulin signaling and the general amino acid control response: two distinct pathways to amino acid synthesis and uptake. J Biol Chem 283: 19229–19234, 2008 [DOI] [PubMed] [Google Scholar]
- 48.Marone R, Cmiljanovic V, Giese B, and Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 1784: 159–185, 2008 [DOI] [PubMed] [Google Scholar]
- 49.Massie A, Schallier A, Mertens B, Vermoesen K, Bannai S, Sato H, Smolders I, and Michotte Y. Time-dependent changes in striatal xCT protein expression in hemi-Parkinson rats. Neuroreport 19: 1589–1592, 2008 [DOI] [PubMed] [Google Scholar]
- 50.McMahon M, Lamont DJ, Beattie KA, and Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci U S A 107: 18838–18843, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meister A. Metabolism and function of glutathione. In: Glutathione: Chemical, Biochemical and Medical Aspects, edited by Dolphin D, Poulsen R, and Avramovic O. New York: John Wiley and Sons, 1989, pp. 367–474 [Google Scholar]
- 52.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, and Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2: 1547–1558, 1989 [DOI] [PubMed] [Google Scholar]
- 53.Ogunrinu TA. and Sontheimer H. Hypoxia increases the dependence of glioma cells on glutathione. J Biol Chem 285: 37716–37724, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, and Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 11: 476–487, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pauly K, Fritz K, Furey A, and Lobner D. Insulin-like growth factor 1 and transforming growth factor-beta stimulate cystine/glutamate exchange activity in dental pulp cells. J Endod 37: 943–947, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Proud CG. eIF2 and the control of cell physiology. Semin Cell Dev Biol 16: 3–12, 2005 [DOI] [PubMed] [Google Scholar]
- 57.Proud CG. Regulation of protein synthesis by insulin. Biochem Soc Trans 34: 213–216, 2006 [DOI] [PubMed] [Google Scholar]
- 58.Quesada A, Lee BY, and Micevych PE. PI3 kinase/Akt activation mediates estrogen and IGF-1 nigral DA neuronal neuroprotection against a unilateral rat model of Parkinson's disease. Dev Neurobiol 68: 632–644, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, and Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol 31: 1121–1133, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, and Hinnebusch AG. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol Cell Biol 18: 2282–2297, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rutkowski DT. and Kaufman RJ. All roads lead to ATF4. Dev Cell 4: 442–444, 2003 [DOI] [PubMed] [Google Scholar]
- 62.Samuels Y. and Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol 347: 21–41, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, and Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem 277: 44765–44771, 2002 [DOI] [PubMed] [Google Scholar]
- 64.Sato H, Nomura S, Maebara K, Sato K, Tamba M, and Bannai S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun 325: 109–116, 2004 [DOI] [PubMed] [Google Scholar]
- 65.Savaskan NE, Heckel A, Hahnen E, Engelhorn T, Doerfler A, Ganslandt O, Nimsky C, Buchfelder M, and Eyupoglu IY. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat Med 14: 629–632, 2008 [DOI] [PubMed] [Google Scholar]
- 66.Savaskan NE, Seufert S, Hauke J, Trankle C, Eyupoglu IY, and Hahnen E. Dissection of mitogenic and neurodegenerative actions of cystine and glutamate in malignant gliomas. Oncogene 30: 43–53, 2011 [DOI] [PubMed] [Google Scholar]
- 67.Schafer FQ. and Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30: 1191–1212, 2001 [DOI] [PubMed] [Google Scholar]
- 68.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, and Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7: 1165–1176, 2001 [DOI] [PubMed] [Google Scholar]
- 69.Shen J, Wu Y, Xu JY, Zhang J, Sinclair SH, Yanoff M, Xu G, Li W, and Xu GT. ERK- and Akt-dependent neuroprotection by erythropoietin (EPO) against glyoxal-AGEs via modulation of Bcl-xL, Bax, and BAD. Invest Ophthalmol Vis Sci 51: 35–46, 2010 [DOI] [PubMed] [Google Scholar]
- 70.Sims B, Clarke M, Njah W, Hopkins ES, and Sontheimer H. Erythropoietin-induced neuroprotection requires cystine glutamate exchanger activity. Brain Res 1321: 88–95, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun P, Enslen H, Myung PS, and Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev 8: 2527–2539, 1994 [DOI] [PubMed] [Google Scholar]
- 72.Sun X, Yao H, Douglas RM, Gu XQ, Wang J, and Haddad GG. Insulin/PI3K signaling protects dentate neurons from oxygen-glucose deprivation in organotypic slice cultures. J Neurochem 112: 377–388, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sweeney G, Garg RR, Ceddia RB, Li D, Ishiki M, Somwar R, Foster LJ, Neilsen PO, Prestwich GD, Rudich A, and Klip A. Intracellular delivery of phosphatidylinositol (3,4,5)-trisphosphate causes incorporation of glucose transporter 4 into the plasma membrane of muscle and fat cells without increasing glucose uptake. J Biol Chem 279: 32233–32242, 2004 [DOI] [PubMed] [Google Scholar]
- 74.Takeuchi S, Wada K, Toyooka T, Shinomiya N, Shimazaki H, Nakanishi K, Nagatani K, Otani N, Osada H, Uozumi Y, Matsuo H, and Nawashiro H. Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastoma. Neurosurgery 72: 33–41, 2012 [DOI] [PubMed] [Google Scholar]
- 75.Yuen TI, Morokoff AP, Bjorksten A, D'Abaco G, Paradiso L, Finch S, Wong D, Reid CA, Powell KL, Drummond KJ, Rosenthal MA, Kaye AH, and O'Brien TJ. Glutamate is associated with a higher risk of seizures in patients with gliomas. Neurology 79: 883–889, 2012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.