

Abstract
New analytical methods are needed for the successful outcome of experiments aimed at characterizing mechanisms of microtubule dynamics and at understanding the effects of drugs on microtubules. The identification of tubulin isotypes and of regions of the microtubule involved in drug interactions has been advanced by proteomic methodologies. The diversity of tubulin sequences and posttranslational modifications (PTMs) can generate a complex mixture of heterodimers with unique molecular dynamics driving specific functions. Mass spectrometry (MS)-based approaches have been developed, and in combination with chromatographic and/or electrophoretic separation of tubulin polypeptides or peptides, they have contributed to our understanding of tubulin proteomics. We present protocols that we have used for the analysis of tubulin isotypes and PTMs present in tubulin isolated from cells in culture or tissues and for the identification of tubulin regions altered by microtubule-stabilizing agents. Tubulin proteomics complements structural and computer modeling information for a high-resolution view of microtubule dynamics and its alteration by drugs. These methodologies will help in providing insights into tubulin isotype-specific functions and in the design of drugs targeting either all tubulin heterodimers indiscriminately or only those containing specific isotypes.
I. Introduction
In most laboratories, the tubulin that is used in in vitro assays is isolated from mammalian brains. This tubulin is usually obtained by cycles of polymerization/depolymerization of microtubules, and when necessary, microtubule-associated proteins (MAPs) are removed either by ion-exchange chromatography or by polymerization of microtubules in high-salt buffers (Andreu, 2007; Castoldi and Popov, 2003; Gaskin and Roychowdhury, 1986; Hamel and Lin, 1981, Hamel and Lin, 1984; Lee, 1982; Murphy and Hiebsch, 1979; Shelanski et al., 1973; Williams and Lee, 1982). The yield and purity of these preparations are assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie blue staining, and the functionality of isolated tubulin is checked for its ability to polymerize using turbidimetry- or pelleting-based assays (Gaskin et al., 1975).
Brain tubulin is composed of several a-tubulin (α1A, α1B) and β-tubulin (βI, βII, βIII, βIVa) isotypes and is highly posttranslationally modified, notably by polyglutamylation, glycylation, phosphorylation, acetylation, detyrosination, and/or loss of the last two amino-acid residues (Luduena, 1998; Verhey and Gaertig, 2007). Even though most cell lines studied may express several tubulin isotypes (α1B, α1C, α4A; βI, βIVb, βIII, βV, βII), the bulk of the tubulin pool is not posttranslationally modified (Verdier-Pinard et al., 2009). The ratio between β-tubulin isotypes is very different in cell lines than in preparations of brain tubulin. For instance, βI-tubulin is the major β-tubulin isotype in cell lines (Verdier-Pinard et al., 2009), whereas it is the minor one in brain where βII tubulin is the most abundant (Banerjee et al., 1992).
It is not proven one way or another that results from in vitro experiments with purified tubulin or tubulin after reconstitution with associated proteins would be systematically different, if one uses brain tubulin versus tubulin from cells in culture. Nevertheless, some differences have been noted over the years indicating that tubulin isotype composition may determine microtubule dynamics, protein association to the microtubule lattice, and drug-binding parameters (Banerjee et al., 1990; Derry et al., 1997; Lu and Luduena, 1994; Newton et al., 2002; Panda et al., 1994). Therefore, it is useful to analyze the content in tubulin preparations from nonneuronal tissues and cells in culture (Bellocq et al., 1992; Farrell, 1982; Fourest-Lieuvin, 2006; Kilmartin, 1981; MacRae and Gull, 1990; Macrae and Luduena, 1984; Maekawa and Sakai, 1978; Morejohn and Fosket, 1982; Murphy, 1991; Rudiger and Weber, 1993; Verdier-Pinard et al., 2009; Weatherbee et al., 1980). This analysis may help investigators in refining the interpretation of the data from their in vitro assays.
Working with tubulin from nonneuronal origin can be advantageous because its lower complexity facilitates the analysis of data [see the example of hydrogen-/deuterium-exchange mass spectrometry (HDX-MS) in the chapter] or aids in the discovery of microtubule-interacting drugs that are more active on tumor tissues and induce less neurological side effects. Immunoaffinity purification of tubulin fractions enriched in a particular isotype has been carried out for brain tubulin (Derry et al., 1997; Lu and Luduena, 1994; Paturle et al., 1989). In this case, or in any other tubulin fractionation process prior to in vitro assays, tubulin proteomics can assess the quality of these fractions. Ultimately, the MS-based methods for tubulin analysis presented in this chapter are also valuable in the characterization of antitubulin antibody specificity (Verdier-Pinard et al., 2009). Such validated antibodies allow straightforward and high-throughput analysis of tubulin-containing samples. Nevertheless, MS-based relative and absolute quantification are accurate on a much larger dynamic range than antibody-based detection systems. Furthermore, analysis of tubulin by MS may reveal novel forms of tubulin, for which no validated antibodies are available, as well as provide detailed characterization of tubulin posttranslational modifications (PTMs), including the exact number of glutamate residues added in a polyglutamylation modification, which can not be directly determined with antibodies.
We present protocols that we have implemented for tubulin purification from cell lines and tissues, for MS-based analysis of isolated tubulin, and for deciphering the binding sites of microtubule-stabilizing drugs and how these drugs affect tubulin molecular dynamics.
II. Methods
A. Taxol-Based Isolation of Tubulin from Cell or Tissue Extracts
One way to purify tubulin from nonneuronal sources is to take advantage of the microtubule-promoting and microtubule-stabilizing properties of Taxol. By lowering the critical tubulin concentration for assembly, this method allows the isolation of tubulin from 1 mg of total protein. Typically, Taxol [reducing the critical concentration of pure tubulin by 10-fold from 1–2 mg/ml to 0.1–0.2 mg/ml in the presence of guanosine triphosphate (GTP)] combined with the presence of MAPs induces tubulin assembly below 0.1 mg/ml. This approach was first implemented by Vallee et al. (Vallee, 1982) and has been applied since on a wide array of biological material. We adapted the method to cancer cell lines in culture and we also described a modified version that gave better results on murine tissues (Miller et al., 2008; Verdier-Pinard et al., 2005; Verdier-Pinard et al., 2003a,b).
1. Materials
Cell lines were maintained in the recommended culture medium supplemented with 10% heat-inactivated foetal bovine serum in the presence of 5% CO2. All animal studies were conducted under protocols approved by the Animal Care and Use Committee of Albert Einstein College of Medicine in accordance with National Institute of Health guidelines. For all surgical procedures, animals were anesthetized using isoflurane gas and were killed with a lethal dose of CO2 (10–15 psi). Taxol was provided by the Drug Development Branch of the National Cancer Institute (Bethesda, MD), dissolved in dimethylsulfoxide (DMSO), and kept as a 1 mM stock solution at −20°C. Protease inhibitor cocktail was from Roche Applied Science (Indianapolis, IN). All other chemicals were purchased from Sigma-Aldrich (St Louis, MO). We used a TLA-100.3 rotor from Beckman Coulter (Brea, CA) for all ultracentrifugation with teflon adaptors for 1.5-ml microfuge allomer tubes, but other fixed-angle rotors and thick-wall polycarbonate tubes can be used.
2. Protocols
a. Protocol 1
Depending on the cell line, 7–10 100-mm petri dishes at 80% confluence or 250 ml of exponentially growing cells in suspension yield enough tubulin material for each of the different analytical techniques described. Adherent cells are washed once with 10 ml of phosphate-buffered saline (PBS) per dish, scraped in 1 ml of PBS per dish, and pooled in a 15-ml conical tube marked with 0.1 ml graduation below 1 ml to facilitate the evaluation of the volume of packed cells after centrifugation at 1200×g.
PBS is completely removed and pellets of 0.3–0.4 ml of packed cells are typically obtained. Cells are resuspended by adding 1.5-fold the volume of packed cells (450-600 µl) of MME buffer.
At this stage, the cell suspensions can be frozen in liquid nitrogen in cryotubes or kept in the 15-ml conical tubes, flash frozen in liquid nitrogen, and stored at −70°C. Note that tubulin is sensitive to denaturation and liquid nitrogen is preferred for long period of storage.
If frozen, cell suspensions are rapidly thawed and kept on ice. If in cryotubes, cell suspensions are transferred to 15-ml conical tubes. A protease inhibitor cocktail from a 10-fold stock solution in MME buffer (75–100 µl) and 1 mM dithiothreitol (DTT) are added.
The 15-ml tube is placed in a beaker containing ice and water. The cell suspension is sonicated with a microtip probe (Branson, Danbury, CT), (duty cycle: 20, output cycle: 3) seven times for 30 s with 30 s rest intervals on melted ice.
The cell lysate is transferred to centrifuge tubes and spun at 120,000×g (Beckman TL100 centrifuge for 1 h at 4°C. Note that a whitish layer of lipids may appear at the surface of the supernatant at the end of centrifugation and should not be transferred; usually, tilting the tube while slowly pipetting out the supernatant deposits most of this layer on the wall of the centrifuge tube. The cytosolic supernatant (SI) is transferred to 1.5-ml tubes, and the DNA and cell debris pellets (PI) are discarded.
Cytosolic supernatants are incubated for 20 min at 37°C in the presence of 10 µM Taxol and 1 mM GTP.
Contamination of the microtubule pellet by non-copelleting components is prevented by cautiously layering the reaction mixture on a 0.1-ml cushion containing 5% sucrose (can be increased to 20%), 10 µM Taxol, and 1 mMGTP Samples are centrifuged at 80,000×g (Beckman TL100) for 30 min at 37°C.
After sequential removal of the supernatant and sucrose cushion, microtubule pellets (PII) are washed with 0.1 ml of warm MME buffer and resuspended in 0.1 ml of MME buffer containing 0.35 MNaCl and 10 µM Taxol on ice. Note that this last washing step with salt is optional, because tubulin is separated by either electrophoresis or liquid chromatography from most of the other proteins associated with the Taxol-stabilized microtubules.
After centrifugation at 80,000×g for 30 min at 37°C, microtubule pellets (PIII) are frozen on dry ice and kept at −70°C until their use.
We applied our method to mouse and rat tissue extracts, and depending on organ origin, we obtained variable results in terms of tubulin yield and separation on gels. Consequently, we implemented a modified protocol that significantly improved tubulin isolation from nonneuronal tissues.
b. Protocol 2
Rat or mouse tissues are washed with cold PBS and flash frozen in liquid nitrogen and kept at −70°C until use. The frozen tissue is crushed in a mortar and pestle in the presence of liquid nitrogen until it is reduced to a powder.
One volume of tissue powder is resuspended in 1.5 volume of MES/glutamate buffer [0.1 M 2-(N-morpholino)ethanesulfonic acid (pH 6.8), 0.5 mM MgCl2, 1 mM EGTA, and 1 M glutamate]. Protease inhibitor cocktail from a 10-fold stock solution in MES/glutamate buffer and 1 mM DTT are added.
The suspension was sonicated (Ultrasonics, duty cycle: 20, output cycle: 3) five times for 10 s with 30 s rest intervals on melted ice.
Sample is spun at 30,000×g (Beckman TL100) at 4°C for 15 min to remove cell debris. The supernatant is then centrifuged at 120,000×g at 4°C for 1 h.
The clarified supernatant is transferred to new tubes and 20 µM Taxol and 1 mMGTP are added prior to a 30-min incubation at 37°C.
Sample is layered on a 100 µl cushion containing 20% sucrose in MES/glutamate buffer and 20 µM Taxol, and spun at 30,000 × g for 30 min at 37°C.
The Taxol-microtubule pellet is resuspended in 30 µl of MES/glutamate buffer containing 0.35 MNaCl and 20 µM Taxol, and incubated for 10 min at 37°C.
Microtubules are pelleted by centrifugation at 30,000×g for 30 min at 37°C, flash frozen in liquid nitrogen, and stored at −80°C.
3. Discussion
The Taxol-based purification of tubulin can be monitored by SDS-PAGE and Western blot analyses using a pan α- or β-tubulin antibody and pure bovine brain tubulin as a standard. It could be argued that this method for isolation of tubulins may exclude some forms of tubulin that would poorly assemble in the presence of Taxol. This is unlikely because the Taxol concentration used is about 10-times higher than the lowest Ka measured for Taxol–tubulin binding, and we did not observe a specific loss of a particular tubulin isotype during the procedure. Obviously, if some tubulin does not assemble in the presence of superstoichiometric concentrations of Taxol, it is likely that it represents a pool of tubulin that is not competent to assemble. Such a pool may co-purify with assembly competent tubulin from extracts using ion-exchange chromatography (Sackett, 1995; Sloboda and Belfi, 1998). Note that purification of tubulin by cycles of assembly–disassembly excludes tubulin species incorporated in cold-stable microtubules (Detrich and Overton, 1986; Pirollet et al., 1983; Williams et al., 1985). Because of the high stability of Taxol–microtubules, our protocols cannot be used to isolate tubulin for subsequent in vitro assays, but is suitable for the analysis of the tubulin content in a cell line or tissue that will be used as a source of soluble and functional tubulin, or in diluted tubulin-containing fractions. The glutamate that is added in protocol 2 provides favorable conditions for tubulin assembly in high salt that removes MAPs and contaminants from the microtubule pellets (Hamel and Lin, 1981; Sackett, 1995). The MES/glutamate buffer was not used to prepare cell line extracts but could be an alternative to the MME buffer used in protocol 1.
B. Electrophoretic Separation of Tubulin Isotypes and Posttranslational Modifications
Electrophoresis is an effective method to separate tubulin isotypes for further analysis. For some experiments, it is necessary only to separate α- and β-tubulin from one another. However, since α- and β-tubulins are of similar molecular weight (∼50 kDa), some modifications to the Lamelli method must be made in order to separate the subunits by SDS-PAGE. It has been demonstrated that a mixture containing SDS as well as longer chain alkyl sulfates is best for separation of α- from β-tubulin (Best et al., 1981; Stephens, 1998). In the case of mammalian tubulin, the α-tubulin subunit will run at an apparent higher molecular weight than the β-subunit. Isoelectric focusing (IEF) can be employed for high-resolution separation of individual tubulin isotypes. The tubulin isotypes have distinct isoelectric points (pIs) (Table I) that fall within a narrow pI range which makes them amenable to separation by high-resolution IEF. The β-tubulin isotypes have predicted pIs from 4.77 to 5.05, with the majority of β-tubulin isotypes pIs falling between 4.77 and 4.79. The α-tubulins have more basic pIs in the range of 4.94–4.98, except for α-like 3, which has a predicted pI of 5.68. PTMs, such as glutamylation and phosphorylation, which add a negative charge to the tubulin isotype, shift the modified isotype to a slightly more acidic pI. Conversely, removal of the C-terminal tyrosine and also the penultimate glutamic acid from α-tubulin shifts the isotype to a slightly higher pI. As described below, commercially available narrow-range immobilized pH gradient (IPG) gels can be employed for high-resolution separation of tubulin isotypes and their PTMs. An example of the separation of tubulin isotypes by high-resolution IEF is shown in Fig. 1A.
Table I. Characteristics of Human Tubulin Isotypes.
| Tubulin Isotype a | Gene | Accession number | pl | Protein mass (Da)b | Mass (Da) of CNBr C-terminal peptidec | CNBr C-terminal peptide |
|---|---|---|---|---|---|---|
| α1A | TUBA1A | NP_006000 | 4.94 | 50135.6 | 2860.19 | AALEKDYEEVGVDSVEGEGEEEGEEY |
| α1B | TUBA1B | NP_006073 | 4.94 | 50151.6 | 2860.19` | AALEKDYEEVGVDSVEGEGEEEGEEY |
| α1C | TUBA1C | NP_116093 | 4.96 | 49895.3 | 2590.04 | AALEKDYEEVGADSADGEDEGEEY |
| α4A | TUBA4A | NP_005991 | 4.95 | 49924.4 | 2633.07 | AALEKDYEEVGIDSYEDEDEGEE |
| α3C | TUBA3C | NP_005992 | 4.98 | 49959.6 | 4150.77 | EEGEFSEAREDLAALEKDYEEVGVDSVEAEAEEGEEY |
| α3D | TUBA3D | NP_525125 | 4.98 | 49959.6 | 4150.77 | EEGEFSEAREDLAALEKDYEEVGVDSVEAEAEEGEEY |
| α3E | TUBA3E | NP_997195 | 4.97 | 49916.6 | 4090.71 | EEGEFSEAREDLAALEKDYEEVGVDSVEAEAEEGEEY |
| α8 | TUBA8 | NP_061816 | 4.94 | 50093.6 | 4156.72 | EEGEFSEAREDLAALEKDYEEVGTDSFEEENEGEEF |
| α-like 3 | TUBAL3 | NP_079079 | 5.68 | 49908.7 | 3058.40 | EEAEFLEAREDLAALERDYEEVAQSF |
| βI | TUBB | NP_821133 | 4.78 | 49670.8 | 3366.33 | NDLVSEYQQYQDATAEEEEDFGEEAEEEA |
| βII | TUBB2B | NP_821080 | 4.78 | 49953.1 | 3466.36 | NDLVSEYQQYQDATADEQGEFEEEEGEDEA |
| βIII | TUBB3 | NP_006077 | 4.83 | 50432.7 | 1624.64 | YEDDEEESEAQGPK |
| βIVa | TUBB4 | NP_006078 | 4.78 | 49585.8 | 3350.38 | NDLVSEYQQYQDATAEEGEFEEEAEEEVA |
| βIVb | TUBB2C | NP_006079 | 4.79 | 49831.0 | 3479.42 | NDLVSEYQQYQDATAEEEGEFEEEAEEEVA |
| βV | TUBB6 | NP_115914 | 4.77 | 49857.1 | 3551.41 | NDLVSEYQQYQDATANDGEEAFEDEEEEIDG |
| βVI | TUBB1 | NP_110400 | 5.05 | 50326.9 | 810.35 | EPEDKGH |
α-Tubulin nomenclature reflects the revised nomenclature for the α-tubulin gene family (Khodiyar, V. K., et al. Genomics, 2007, 90, 285–289).
Protein mass is the average mass.
CNBr peptide mass is reported as the monoisotopic neutral mass.
Fig. 1.
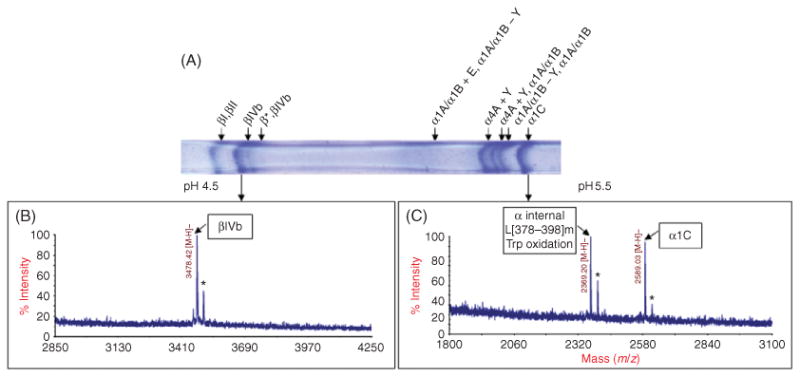
Combination of isoelectric focusing and MALDI-TOF MS analysis of CNBr C-terminal tubulin peptides. (A) Region of a 24-cm pH 4.5–5.5 IPG strip containing all tubulin isotypes from a tubulin pellet of a normal rat liver. Eight bands were excised from the gel as indicated by the arrows and cleaved with CNBr. The isotype(s) observed in each band is listed above the gel. “*” represents a novel IVb posttranslational modification identified in the rat (Miller et al., 2008). The CNBr C-terminal peptides for α1A and α1B isotypes have the same mass and cannot be distinguished from one another. A “+E” indicates glutamylation and “±Y” indicates tyrosination/detyrosination. (B) Mass spectrum obtained after CNBr cleavage of band indicated by the arrow. IVb was observed in this band. The * indicates a +28 Da peak which is due to formylation. (C) Mass spectrum obtained after CNBr cleavage of the band indicated by the arrow. The α1C isotype was observed in this band along with an internal α-tubulin peptide. The “*” indicates a +28 Da peak which is due to formylation.
1. Materials
Narrow-range IPG gels and ampholyte-containing buffer can be obtained from GE Healthcare Life Sciences (Piscataway, NJ) or Bio-Rad (Hercules, CA). We obtained the maximal resolution using 24-cm gels from GE Healthcare Life Sciences (Piscataway, NJ) with a linear gradient from pH 4.5 to 5.5 (Verdier-Pinard et al., 2005; Verdier-Pinard et al., 2003b). Unfortunately, this pH range is no longer available from this company, and alternatively, we are now using 24-cm gel strips with a linear pH gradient from 4.7 to 5.9 or 3.9 to 5.1 from Bio-Rad (Hercules, CA) or a 24-cm gel strip with a nonlinear pH gradient of 3.0–5.6 from GE Healthcare Life Sciences. An Ettan IPGphor II IEF system, also from GE Healthcare Life Sciences, was used for IEF. The IEF gels are stained with Pierce GelCode Blue Stain (Rockford, IL). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO). Images of the gels were obtained on a Microtek ScanMaker 9800 XL (Carson, CA).
2. Protocol
Taxol-stabilized microtubule pellets (previous protocols) should be thoroughly resuspended in 500 µl of solubilization buffer containing 7 M urea, 2 M thiourea, 4% 3-[3-(cholamidopropyl)dimethylammonio]-1-propanesulfonate, 0.5% Triton X-100, 0.5% ampholyte-containing buffer, 20 mM DTT, and a small amount of bromophenol blue. A solution containing everything except the ampholytes, DTT, and bromophenol blue can be prepared ahead of time and stored at −20°C for 6 months. The ampholyte-containing buffer should be in the same pI range as the IPG gel.
The sample is spread out evenly at the bottom of the IPG strip holder. The volume of sample must be adapted to the length of the gel strip (e.g., 450 µl for a 24-cm GE Healthcare gel strip; note that for a long IEF running time, the volume is increased by 10 µl). The IPG strip is then placed in the holder with the gel-side down in contact with the sample and electrodes and the “+”-pointed end toward the pointed end of the strip holder. The entire IPG gel is covered with mineral oil.
For a narrow-range 24-cm gel the typical running parameters include a 12-h rehydration step at 50 V and 20°C. After this active rehydration of the gel, the protein is separated with a program including 1 h at 500 V, 1 h at 1000 V, and the final step of 8 h and 15 min at 8000 V. The 24-cm IPG gels typically run for a total of 65,000 Vh. These parameters should be adjusted following the manufacturer's instructions for the specific length of the gel.
Following electrophoresis, the excess oil is carefully removed from the gel by blotting onto a KimWipe and the gel is fixed for 30 min in a 20% (w/v) trichloroacetic acid. Following the fixation step, the gel is washed in ddH2O for 30 min and then stained with Coomassie blue for 1.5–2h. The gel is then destained overnight.
After sufficient destaining a high-resolution image of the gel is obtained for documentation prior to further processing using a flatbed scanner.
Note: It is important to ensure that all gels are kept free of contaminants, such as keratins, which can interfere with downstream MS analysis. Therefore, one should always wear gloves when handling the gel and thoroughly clean sample tubes before use.
3. Discussion
This method can be used to detect as little as 0.5 µg protein and can resolve isotypes differing in pI by 0.01 units. Typically >15 µg total tubulin pellet is loaded onto the gels. Gels have been successfully stained with other stains, including GE Healthcare Deep Purple fluorescent stain following manufacturer's protocols and acid violet 17 (Williams et al., 1999). Fluorescent staining can be used to visualize small amounts of tubulin protein, which may not be detected by MS.
C. Identification of Tubulin Isotypes and Posttranslational Modifications
MS is an ideal tool for the characterization of tubulin isotypes and PTMs since each isotype has a unique amino-acid sequence and PTMs result in mass differences to the tubulin isotypes (Table II) (Verdier-Pinard et al., 2009). Most tubulin isotype analysis involves cleavage of the protein into smaller peptides which are more amenable to mass spectrometric characterization. However, some MS work has been done on intact tubulin. In our laboratory, we have developed MS-based methods for the characterization of tubulin isotypes at the peptide and the protein levels and both are discussed below.
Table II. Tubulin posttranslational modifications, mass changes, and location.
| Modification | Δm | Location |
|---|---|---|
| Tyrosination/detyrosination | ±163.06 Da | C-terminal tail, α-tubulin |
| Δ2-Tubulin | −292.10 Da | C-terminal tail, α-tubulin |
| Glutamylation | +129.04 Da | C-terminal tail, α- and β-tubulin |
| Glycylation | +57.02 Da | C-terminal tail, α- and β-tubulin |
| Acetylation | +42.01 Da | α-Tubulin (Lys40) |
| Phosphorylation | +79.97 Da | β-Tubulin (Ser441/444) |
1. Protocols
a. Cyanogen Bromide Cleavage
Cyanogen bromide (CNBr) cleaves C-terminal to methionine—a cleavage which typically results in larger peptides than enzymatic cleavage since methionine is less commonly incorporated into proteins. CNBr cleaves α-tubulin into ∼11 peptides ranging in size from 1,300 to 13,000 Da and β-tubulin into ∼18 peptides in the range of 345–8,000 Da. The C-terminal region of both α- and β-tubulins contain a methionine within the last 40 residues which, when cleaved with CNBr, results in unique isotype identifying peptides from 800 to 4,150 Da, which is an ideal mass range for characterization by MS.
Bands containing tubulin should be cut from the gel. If cutting bands from a plastic-backed IEF gel, use a GeneCatcher (Gel Company, San Francisco, CA) gel excision tip (or similar tip) to cut the band. Place the tip above the band, press down firmly, and then slide the tip along the plastic backing parallel to the other bands. This separates the gel from the backing without disrupting adjacent tubulin bands.
Destain the gel pieces in 150 µl 50% acetonitrile in water at 37°C with shaking for 30 min. Repeat this step until the complete removal of the color from the gel. Evaporate the solvent and dry the bands in a SpeedVac concentrator.
Rehydrate the gel pieces in 100 µl of CNBr cleavage solution (100 mg/ml CNBr in 70% formic acid). Note that this step should be done in a chemical fume hood. Also, it is important to use fresh formic acid of the highest purity to avoid formylation adducts (+28 Da) on the peptides. Alternatively, the cleavage can be performed in 70% trifluoroacetic acid (TFA) to avoid the formylation of peptides.
The cleavage is then allowed to proceed overnight (∼16–20 h) at room temperature in the dark.
Following the overnight cleavage, the CNBr solution is removed from the tube and placed in a clean tube and vacuum dried.
The dried peptides are resuspended in 100 µl of 50% acetonitrile containing 0.3% TFA and again vacuum dried.
The previous wash step (step 6) is repeated, but with 50 µl of 50% acetonitrile containing 0.3% TFA.
The washed peptides are resuspended in 10 µl of H2O containing 0.1% TFA and purified on a C18 ZipTip following the manufacturer's suggested protocol.
b. Enzymatic Digestion
While the majority of diversity of tubulin isotypes is contained within the C-terminal region, there are sequence differences throughout the entire primary sequence. Also, some PTMs, such as acetylation at Lys40 in α-tubulin, and sequence mutations have been detected outside of the C-terminal region. Enzymatic digestion typically results in smaller peptides (less than 5000 Da) that can be readily analyzed by matrix-assisted laser desorption ionization time-of-flight MS (MALDI-TOF MS), thereby providing greater sequence coverage than CNBr cleavage for tubulin isotypes.
The bands containing tubulin as seen by Coomassie blue staining should be cut from the gel.
Destain each band with 300 µl of a solution of 200 mM ammonium bicarbonate/50% acetonitrile at pH 8.9. After vortexing, incubate at 37°C for 30 min with shaking. Centrifuge and remove the solution. Repeat this step until a complete loss of color is achieved.
Add 300µl 100% acetonitrile to the gel band. Vortex the tube and then sonicate for 30 s. Remove the solution and dry the gel band in a SpeedVac concentrator until completely dry.
Rehydrate the gel piece in 300 µl 10 mM DTT in 0.1 M ammonium bicarbonate and incubate at 56°C for 45 min to reduce all disulfide bonds. Cool the tubes to room temperature for 5 min and remove the excess solvent. Note: Whereas the C-terminal peptides do not contain cysteine, the internal tubulin peptides may contain cysteine residues and therefore reduction and alkylation steps are included in this protocol.
Cysteines are alkylated by adding 300µl of freshly made 55 mM iodoacetamide in 0.1 M ammonium bicarbonate to the gel piece. The tube is then incubated for 30 min at room temperature in the dark, after which the excess solvent is removed.
Add 300 µl of a solution containing 1:1 acetonitrile:200 mM ammonium bicarbonate pH 8.9 to the gel band. Vortex and then sonicate the sample for 30 s. Centrifuge and remove the excess solution. Repeat this step.
Dry the gel by adding 300 µl of 100% acetonitrile. Vortex and then sonicate for 30 s. Remove the solution and dry the gel band in a SpeedVac concentrator.
Prepare a solution of trypsin in ammonium bicarbonate (25 ng/µl trypsin in 50 mM ammonium bicarbonate pH 8.9). Add 20 µl trypsin solution to the dried gel piece. Gently mix and centrifuge briefly. Incubate on ice for 45 min.
Remove excess trypsin solution (if any) and add 50µl of digestion buffer (50 mM ammonium bicarbonate, pH8.9). Note:Adigestion enhancer, such as ProteaseMAX surfactant (Promega, Madison, WI), can be added at this point to increase digestion efficiency. This may result in enhanced signal for higher molecular weight peptides.
Digest at 37°C with shaking for 18 h.
Add 1% TFA to a final concentration of 0.1% to stop the digestion.
The peptides are then purified on a C18 ZipTip following manufacturer's instructions.
c. Combined Fragmentation
Combining enzymatic digestion and chemical cleavage of tubulin may result in the highest sequence coverage since the smaller peptides should all be easily released from the gel. This combination of cleavage techniques can also be used to achieve localization of PTMs when tandem MS (MS/MS) data cannot be obtained.
Follow the trypsin digest protocol above through step 10.
Dry the gel pieces in a SpeedVac concentrator.
Wash and dehydrate twice by addition of 100 µl of acetonitrile followed by drying by SpeedVac concentrator.
Add 25µl of CNBr cleavage solution (100 mg/ml CNBr in 70% TFA) to the gel pieces. Incubate in dark for 14 h.
Collect the supernatant.
Extract the peptides twice by sonication for 5 min in 30 µl of 60% acetonitrile, 1% TFA, and 0.1% octyl-β-D-glucopyranoside (OBG). Pool the extraction with supernatant obtained in step 5. The OBG is added to enhance the extraction of larger peptides from the gel.
Dry or concentrate the pooled peptide solution in a SpeedVac concentrator.
d. MALDI-TOF Mass Spectrometry
Matrix-assisted laser desorption ionization (MALDI) coupled to a time-of-flight MS (TOF MS) provides the high sensitivity and mass accuracy required for the analysis of peptides derived from enzymatic or chemical cleavage of tubulin isotypes. Furthermore, peptides harboring PTMs such as tyrosination/detyrosination and glutamylation have a unique mass shift that allows for confident assignment of these tubulin-specific modifications by MS. Sequence information and PTM localization can also be obtained by performing MS/MS on tandem TOF instruments (MALDI-TOF/TOF). Examples of MALDI mass spectra of tubulin C-terminal peptides generated by CNBr cleavage are shown in Fig. 1 B and C.
The ZipTip purified peptides are mixed 1:1 with a matrix solution containing 5 mg/ml α-cyano-4-hydroxycinnamic acid in 50% acetonitrile with 0.1% TFA. This peptide/matrix mix is then spotted onto a MALDI target. Traditionally sinapinic acid is recommended for negative ion mode MS, but we have found that α-cyano-4-hydroxycinnamic acid works equally well in negative and positive ion mode and therefore use it for all MALDI experiments.
The C-terminal peptides generated by CNBr cleavage are highly acidic and are best analyzed in negative ion mode (Jai-nhuknan and Cassady 1998). However, in order to obtain the highest sequence coverage, both negative and positive ion mode spectra should be acquired. Since some of the C-terminal peptides are >4000 Da, it is important to ensure that the mass range covered in each spectrum is well above this to account for potential PTMs.
For enzymatic digestion, positive ion mode typically provides the best signal for all peptides. Be certain that the m/z range is sufficient to account for any PTMs that may occur.
In order to assign the peptides obtained via cleavage, a list of theoretical tubulin peptides specific for the enzyme or CNBr cleavage needs to be generated. Since most programs do not include the tubulin PTMs (detyrosination, glutamylation, glycylation), it is necessary to add these masses manually to each of the potential modification sites.
D. Analysis of Intact Tubulins by Electrospray Ionization Mass Spectrometry
A complementary approach to tubulin peptide analysis, as discussed above, is the study of intact tubulin proteins. Analysis of the intact proteins provides a direct method to determine the tubulin isotype composition and PTMs of a sample. Also, some sequence mutations that result in a mass shift may be detected by this method. While the cleavage of the protein into peptides is more amenable to routine mass spectrometric analysis, measuring the intact protein mass allows for identification of mass changes that lie outside of the C-terminus which may not be detected at the peptide level.
1. Protocol
Dissolve purified microtubule pellets (∼10 µg) in 20 µl of 70% formic acid and load onto a 1.0×150 mm C3 column with a flow rate of 50 µl/min. The mobile phases employed are 5% acetonitrile containing 0.1% FA (solvent A) and 95% (v/v) acetonitrile containing 0.1% FA (solvent B).
Wash the sample for 45 min. in 5% solvent B and then use the following gradient to separate the tubulin isotypes: 5–30% B in 3 min., 30–45% B in 15 min., 45–55% B in 120 min., 55–75% B in 15 min., 75–95% B in 5 min., 95–5% B in 5 min.
The eluent from the column is directly coupled to an electrospray ionization (ESI) MS. Operate the MS in normal MS scan mode and detect ions in the m/z range of 600–1800.
The resulting mass spectra are deconvoluted to obtain the molecular weight for each tubulin species. When assigning tubulin isotypes based on intact mass measurements, it is important to bear in mind any potential PTMs and how they can shift the mass of each isotype.
In order to obtain more detailed information about the tubulin isotypes, the column effluent flow can be split and a portion of the high-performance liquid chromatography (HPLC) eluent collected for trypsin or CNBr cleavage. Reduce the volume of the fractions to ∼10 µl by vacuum drying and then follow the procedures above to cleave the protein into peptides.
2. Discussion
We have employed LCQ quadrupole ion trap and LTQ linear ion trap MSss (ThermoFinnigan, Riviera Beach, FL) for the intact analysis of tubulin, but any ESI-MS can be used. With the LCQ, we were able to determine the masses of the tubulin isotypes within 10 Da, and with the LTQ, we measure masses within 5 Da of the predicted values. We have used C4 columns to separate tubulin isotypes, but find that C3 columns typically provide better performance.
E. Relative Quantitation of Tubulin Isotypes
The quantitation of specific tubulin isotypes is commonly achieved by Western blot analysis with isotype-specific antibodies. However, validated antibodies are not available for all tubulin isotypes. MS-based methods can be employed for the relative quantitation of known tubulin isotypes as well as any novel tubulin forms or PTMs. While an extensive discussion of these methods is out of the scope of this chapter, we provide a brief overview of a couple of methods below and list a few references to which one can refer for detailed protocols.
One method that has been successfully employed for tubulin quantitation in cell lines is stable isotope labeling by amino acids in cell culture (SILAC) (Ong et al., 2002; Ong and Mann, 2007). With SILAC, one cell line is grown in media lacking an essential amino acid and is supplemented with the same “heavy” amino acid that is labeled with 2H, 15N, or 14C. This cell line is then mixed in an equal amount with a control cell line grown in normal, nonlabeled media. Incorporation of the heavy amino acid into proteins generates a mass shift from the unlabeled version. The ratio of the heavy to the light version of the peptides can then be determined by comparing peak intensities in the mass spectrum. Using this method we were able to measure the relative levels of β-tubulin isotypes between an ovarian (Hey) and a lung (A549) cancer cell line (Verdier-Pinard et al., 2005).
Since not all samples can be directly labeled with heavy isotopes as described above, an alternative strategy must be used for quantitation. In our laboratory, we developed a method that employed two 15N-labeled standards for relative quantitation of tubulin isotypes in tissue samples (Miller et al., 2008). The first 15N-labeled peptide was common to all β-tubulin isotypes observed in the experiment, and the second 15N-labeled peptide was specific for the C-terminal CNBr peptide for the β-tubulin isotype that was quantitated. The tubulin sample was run on SDS-PAGE and the band containing all tubulin isotypes was cut from the gel. The same amount of a stock solution containing both of the 15N-labeled peptides was added to each sample before CNBr cleavage, which was performed as described in protocol 3.1. Following cleavage with CNBr, the samples were analyzed in negative and positive ion mode MALDI-TOF/TOF. The ratio of the sample to the 15N-labeled standard was determined for both the C-terminal peptide and the internal peptide by comparing the intensity of the isotopes in the mass spectrum. The internal peptide was used to standardize the amount of β-tubulin in each sample. After normalizing all the samples, the relative amount of the isotype-specific C-terminal peptide could be determined across the different samples. With this methodology, it is important to synthesize peptides that will be equivalent to those generated by CNBr cleavage. Therefore, for internal peptides it may be necessary to add a couple of extra residues past the methionine so that the homoserine lactone is generated upon CNBr cleavage. Also, before synthesizing the internal standards, it is important to determine which internal peptides are detected in each cleavage reaction and if they are common to all observed isotypes. This method can be used to quantitate the relative amount of any α- or β-tubulin isotype. Alternatively, but following these principles, a label-free approach for tubulin isotype quantitation has been recently described (Winefield et al., 2009).
F. Microtubule Interactions with Drugs and MAPs
1. Hydrogen–Deuterium Exchange
HDX-MS has emerged as a rapid and powerful experimental tool to investigate many aspects of protein architecture/dynamics including (1) determining both the sites of ligand binding and associated conformational changes, (2) mapping the contact sites between proteins, and (3) investigating the effects of single-point mutations or of amino-acid substitutions in isotypes on the regional and global dynamics of a protein (Chik and Schriemer, 2003; Stokasimov and Rubenstein, 2009; Wang et al., 1998a,b, 2000; Wang et al., 2001). This technology provides us with an avenue to obtain crucial knowledge of tubulin structure/dynamics that is not available from other biophysical methods (Bennett et al., 2009; Huzil et al., 2008; Xiao et al., 2006).
a. Materials
Tubulin isolated from the marginal bands of chicken erythrocytes by the method of Murphy (Murphy, 1991) was selected for the HDX method to study interactions between tubulin and its stabilizing drugs. Chicken erythrocyte tubulin contains a single α- and β-isotype, α1 and βVI, whose amino-acid sequences are 95% and 84% identical to their human orthologs, respectively. Purity was 99% as evaluated by SDS/PAGE and Coomassie staining, and isotype content was checked by high-resolution IEF (Verdier-Pinard et al., 2005; Verdier-Pinard et al., 2003b). The tubulin stock solution at 15 mg/ml was stored at −80°C. Pepsin was purchased from Sigma-Aldrich, D2O (99.9%) from Cambridge Isotope Laboratories (Andover, MA), TFA from Applied Biosystems (Foster City, CA), and acetonitrile from Fisher Scientific (Pittsburgh, PA). Guanosine diphosphate (GDP) and GTP were purchased from Roche Applied Science (Indianapolis, IN). All other chemicals were of highest grade commercially available from Sigma-Aldrich. Guanosine-5′-[(αβ)-methylene] triphosphate (GMPCPP) was purchased from Jena Bioscience (Jena, Germany).
b. Protocols
a.1. Hydrogen—Deuterium Exchange (HDX) in Tubulin
All tubulin samples are clarified by centrifugation at 100,000×g at 4°C for 10 min before assembly.
For GTP- or GMPCPP-induced assembly, tubulin is incubated at 6.0 mg/ml (10 times the critical concentration for assembly) in MEM buffer (0.1 M 2-morpholinoethane sulfonic acid/1 mMEGTA/0.5 mMMgCl2, pH 6.9) at 37°C in the presence of 1 mMGTP or GMPCPP for 30 min.
For drug-binding experiments, Taxol and discodermolide (or other drugs) are added in three increments of increasing concentrations (10 µM, 100 µM, and 1 mM), and allowed to incubate for 10 min after the first two additions and 15 min after the last one.
GDP–tubulin dimers are prepared in the same MEM buffer, by inducing polymerization into microtubules (hydrolysis of GTP to GDP in most heterodimers) at 37°C and resuspension of microtubules in cold buffer containing 1 mMGDP (depolymerization of microtubules). The solution containing tubulin–GDP dimers is incubated at room temperature for 30 min.
HDX on tubulin was initiated by diluting each sample 20-fold in 0.1 M deuterated MEM buffer, pH 6.9 at 37°C. Exchange was allowed to proceed for certain time, e.g., 5, 10, 20, 30, and 60 min, after which point the aliquot exchange solutions were quenched with equal volumes of prechilled 0.5 M ammonium phosphate buffer (pH 2.5, 0°C). To minimize the back exchange during HPLC, the solvent precooling coil, static mixing tee, Rheodyne injector, and column were immersed in an ice bath.
For global HDX, 7 µl of quenched reaction mixture is injected onto a 1.0 mm ID × 50 mm C4 column (Waters Inc., Milford, MA). After desalting with 5% of solvent B [95% (v/v) acetonitrile containing 0.2% FA and 0.01% TFA] for 5 min, intact tubulin is eluted with a 2-min gradient composed of 5–95% solvent B. The effluent is directly delivered to the LTQ MS (Thermo Scientific, Waltham, MA) for mass analysis.
For local HDX, 5 µl of pepsin solution is added into the quenched exchange aliquot (pepsin:protein is 1:1 molar ratio). After 5 min digestion, 20 µl of chilled digest is injected onto a 1.0 mm ID × 50 mm C-8 column (Waters Inc.). It should be noted that denaturant guanidinium hydrochloride (1.5 M–6 M) and reducing agent tris (2-carboxyethyl)phosphine (2.5 mM) can be added to optimize pepsin digestion.
Allowing a 5-min desalting with 5% solvent B, the peptic peptides are eluted with a 0.5-min gradient from 5 to 10% solvent B, followed by an 8-min gradient from 10 to 50% solvent B. The 50 µl/min nonsplit effluent is delivered into a LTQ MS (Thermo Electron Corporation) or Fourier transform ion cyclotron resonance (FTICR) MS.
a.2. Data Analysis
The peptides are identified by a combination of accurate masses and MS/MS, first with nondeuterated buffer. The extent of the deuterium incorporation of each peptic peptide is determined by MS from the centroid mass difference between deuterated and nondeuterated samples. The effect of deuterium gain or loss does not need to be taken into account when comparing the difference between distinct forms of tubulins under identical conditions.
Typically, triplicates are performed in HDX experiments. Average changes in deuterium incorporation (DHDX) ± standard deviations are determined from these three separate experiments.
Peptides that exhibit significant changes in deuterium incorporation are mapped onto the tubulin dimer structure and onto a structure of a chicken erythrocyte microtubule protofilament pair previously constructed in our laboratory.
c. Discussion
Tubulin isolated from chicken erythrocytes is composed of only one α- and one β-tubulin isotype, i.e., α1 and βVI. PTMs to erythrocytes are minimal; the α-isotype is almost completely detyrosinated, while ∼10% of the β-isotype is phosphorylated on Ser441 in the C-terminal domain (Rudiger and Weber, 1993). These characteristics of chicken erythrocyte tubulin make it ideal to study using MS as it eliminates any ambiguity in the assignment of measured masses and potential conformational differences between different tubulin isotypes. Nevertheless, it could be argued that visualizing the average conformation changes rather than those occurring specifically for a given tubulin heterodimer is also important. Such analysis of a more complex mixture of tubulin can actually be facilitated by primary results on a unique tubulin heterodimer. GMPCPP, the nonhydrolysable analog of GTP, produces much more stable microtubules than GTP does. This GMPCPP mode of stabilization still needs to be elucidated using a proper control, but Taxol or other drug-specific effects can be accurately assessed against GMPCPP-stabilized microtubules.
2. Analysis of Photoaffinity-Labeled Microtubules
Photoaffinity labeling is a powerful method to determine the binding site of drugs such as Taxol on microtubules. Direct binding of the natural drug Taxol was not successful due to the low extent of photoincorporation of the drug. To improve the photoincorporation, Taxol analogs, [3H]3′-(p-azidobenzamido)Taxol, [3H]2-(m-azidobenzoyl)Taxol, and [3H]7-BzDC-Taxol, were used to photolabel microtubules. These three analogs were found to label the microtubules on β-tubulin, each at a distinct site on the microtubule. [3H]3′-(p-azidobenzamido)-Taxol photolabels the N-terminal 31 amino acids of β-tubulin, [3H]2-(m-azidobenzoyl)Taxol photolabels the peptide β217–233, and [3H]7-BzDC-Taxol cross-links to Arg282 of β-tubulin (Rao et al., 1999; Rao et al., 1994; Rao et al., 1995). Similarly, an analog of discodermolide, C19-[3H]BPC-discodermolide, was located at a binding site on β-tubulin at peptide 355–359 (Xia et al., 2006).
a. Materials
Microtubule protein (MTP) is purified from calf brain by two cycles of temperature-dependent assembly–disassembly. The concentration of tubulin in MTP is based on a tubulin content of 85%. MTP (final concentration 1.0–1.5 mg/ml) is assembled by suspending in assembly buffer consisting of 0.1 M 2(N-morpholino)-ethanesulfonic acid (MES), 1 mM ethylene glycol bis (aminoethyl)-N,N′-tetraacetic acid (EGTA), 0.5 mM MgCI2, and 3 M glycerol, pH 6.6. Assembly at 35°C is monitored spectrophotometrically at 350 nm by following changes in the turbidity signal that are representative of polymer mass.
[3H]3′-(p-azidobenzamido)Taxol, [3H]2-(m-azidobenzoyl)Taxol, and [3H]7-BzDC-Taxol were used at a specific radioactivity of 1.7–2.8 Ci/mmol, 0.12 Ci/mmol, and 50 Ci/mmol, respectively. C19-[3H]BPC-discodermolide was used with radioactivity of 4.1 Ci/mmol.
b. Protocols
1. Photoaffinity Labeling of Tubulins
The drug analog, for example, 3H-3′-(p-azidobenzamido)Taxol (10 µM, 1.7–2.8 Ci/mmol) is added to MTP (10 µM tubulin) in assembly buffer and incubated at 37°C for 30 min. Aliquots (250 µl) are placed in a multiwell plate (1.7 cm in diameter), which is kept at 4°C and irradiated for 30 min at 254 nm with a Mineralight lamp (model R52G, UVP Inc., San Gabriel, CA) at a distance of 7 cm. The extent of photoincorporation is calculated, based on one drug-binding site per dimer, by a filter-binding assay after precipitation of photolabeled tubulin with cold acetone. The photolabeled samples are analyzed on 9% SDS-PAGE gels. For fluorography the analytical gels were stained with Coomassie R-250, destained, treated with EN3HANCE (PerkinElmer, Waltham, MA), and exposed to PerkinElmer X-Omat AR film at −70°C. Typically, it takes 1–8 days to obtain a clear image by fluorography In the case of tubulin photolabeling, it takes as long as 30 days to obtain an optimal image of photolabeled tubulin due to the low extent of photoincorporation.
2. Purification of Radiolabeled β-Tubulin
Gel-Based Separation
For the purification of β-tubulin, preparative SDS-PAGE gels (9%, 3 mm) are immersed in ice-cold 20 mM KCl for 5 min to visualize the α- and β-tubulin subunits. The β-tubulin band is excised, washed six to eight times with H2O, and electroeluted for 16 h with an electroeluter (model 422, Bio-Rad, Richmond, CA) at 10 mA per electroelution tube. The recovery of β-tubulin is between 60 and 80%. SDS is removed from electroeluted β-tubulin using Extracti-gel D (Pierce, Rockford, IL), and the eluate is treated with trichloroacetic acid (final concentration 12% w/v) to precipitate β-tubulin and remove SDS. The precipitate is washed twice with ice-cold acetone to remove residual trichloroacetic acid. The radiolabeled β-tubulin is reduced and carboxymethylated prior to enzymatic cleavage.
HPLC-Based Separation
Acetone precipitation is performed on photolabeled tubulin to remove free unbound drug (1 vol of protein solution plus 4 vol of cold acetone, −20°C, overnight). Samples are then denatured with guanidine hydrochloride, and the cysteine residues are first reduced with 5 mM DTT and then carboxymethylated with 55 mM iodoacetamide. α- and β-tubulin subunits are separated by reverse-phase HPLC on an Aquapore BU-300 (220 × 2.1 mm) C4 column using an HP1090 liquid chromatography station. The protein is eluted with a linear acetonitrile 0.1% TFA gradient (25–55% over 60 min) at a flow rate of 200 µl/min. Fractions are collected every minute. The elution of α- and β-tubulin subunits is monitored by Western blot. Fractions containing radiolabeled protein are detected with fluorography Radioactive fractions are collected and dried by SpeedVac concentrator.
Note: An old HPLC system was used here since these experiments are using radiolabeled material. The SpeedVac concentrator, the gel apparatus, and MS equipment should be used cautiously.
3. Different Enzymatic Digestions or Chemical Cleavage of Purified Radiolabeled β-Tubulin
Subtilisin Digestion
Subtilisin is added at a 500:1 (w/w) ratio of protein: enzyme. After digestion for 1 and 12 h, reactions are stopped with 2 mM phenylmethylsulfonyl fluoride. Samples are resolved by SDS-PAGE on a 9% gel followed by fluorography.
Asp-N and Arg-C Enzyme Digestion of β-Tubulin
Samples are digested with either Asp-N or Arg-C (protein:enzyme 100:1, w/w) for 6, 12, or 18 h at 37°C according to the manufacturer's instructions (Roche Applied Bioscience). The digestion buffer for Asp-N was 50 mM sodium phosphate, pH 8.0, containing 1 M urea. Arg-C digestion buffer was 90 mM Tris-HCl, pH 7.5, 8.5 mM CaCl2, 5 mM DTT, 0.5 mM EDTA, and 1 M urea. Digestion products were resolved on a 10–20% Tricine gel and subjected to fluorography.
Trypsin Digestion
The radioactive β-tubulin is dried and dissolved in 50 mM NH4HCO3, 1 mM CaCl2, and 1 M urea, and then treated with trypsin (enzyme: protein w/w 1:40) for 24–48 h at 25°C.
Formic Acid Cleavage
Photolabeled β-tubulins (10–15 nmol) are dissolved in 400 µl of 75% formic acid and incubated at 37°C. After 72-96 h, formic acid is removed by evaporation in a SpeedVac concentrator (Savant, Holbrook, NY). The residues are washed twice with H2O and dried afterwards in SpeedVac concentrator. The formic acid cleavage products are separated on 17.5% SDS-PAGE gels with 0.1 M Tris, 0.1 M Tricine, and 0.1% SDS as the cathode buffer TM and visualized by fluorography.
CNBr Cleavage
β-Tubulin is dissolved in 400 µl of 70% formic acid containing 20 mg CNBr and incubated at 37°C for 48 h. The formic acid is evaporated in a SpeedVac concentrator and the sample is washed twice with water and dried. Samples are resolved by SDS-PAGE on a 15% gel followed by fluorography.
4. Determination of Peptide Mass and Sequence of the Radiolabeled Peptide from Formic-Acid-Cleaved Photolabeled β-Tubulin
Formic acid is known to cleave preferentially Asp-Pro bonds. Because β-tubulin contains two such linkages at positions 31–32 and 304–305, complete formic acid cleavage of β-tubulin will result in three distinct peptide fragments consisting of amino acids 1–31 (A1, Mr = ∼3500), 32–304 (A2, Mr = ∼31,000), and 305–445 (A3, Mr = ∼16,000). Besides SDS-PAGE analysis of these fragments, two alternative methods have also been used for the analysis.
Following formic acid cleavage of photolabeled β-tubulin, the protein is reduced and alkylated. The sample is centrifuged through a Centricon-10 (Amicon, Danvers, MA) microconcentrator and the filtrate is purified by reversed-phase (RP)-HPLC on an Aquapore RP-300 (Applied Biosystems, San Jose, CA) (2.1 × 220 mm) C-8 column using an HP1090 liquid chromatograph. The peptides are eluted with a linear gradient (1%/min) of H2O/0.1% TFA and acetonitrile/0.1% TFA at a flow rate of 200 µl/min. The fraction containing the major UV absorbing (214 nm) material, which is also the major peak of radioactivity, is collected and concentrated in a SpeedVac concentrator. The sample is ionized by electrospray on a PE-Sciex API-III (Ontario, Canada) mass analyzer and the monoisotopic mass of the sample measured. The measured mass of the sample is obtained from the different charge states. Peptide sequence can be obtained by sequencing on an Applied Biosystems 477A sequencer or by MS/MS.
5. Analysis of other Enzymatic Digests
Peptides resulting from the digestions are diluted with an equal volume of 6 M guanidine HCl and chromatographed on an Aquapore RP-300 C-8 column (2.1 × 220 mm) using an HP1090 liquid chromatograph. The peptides are eluted with a linear gradient A from 80% H2O, 0.1% TFA and 20% acetonitrile, 0.1% TFA to 30% H2O, 0.1% TFA and 70% acetonitrile, and 0.1% TFA in 50 min at a flow rate of 0.2 ml/min. Fractions are collected every minute and tested for radioactivity. The radioactive fraction is further purified by C-8 reverse phase HPLC and eluted with a linear gradient B from 70% H2O, 0.1% TFA and 30% acetonitrile, 0.1% TFA to 50% H2O, 0.1% TFA and 50% acetonitrile, and 0.1% TFA in 60 min. Amino-acid sequence of these radioactive fractions can be obtained using Applied Biosystems 477A sequencer. The sequence can also be obtained using MS/MS.
c. Discussion
The most difficult part of this method is to design analogs of microtubule-stabilizing drugs. These analogs must be able to label microtubules with a sufficiently high extent of photoincorporation and maintain biological activity. It is also difficult to detect the photolabeled peptides due to the relatively low degree of photoincorporation. Liquid scintillators such as EN3HANCE (Dupont NEN) is necessary to improve the sensitivity of the fluorography. MS/MS may not provide sufficient sequence information for large peptides such as for peptides 32–304 and 305–445 generated by formic acid. Combination of various enzymatic digestions will give better results for sequence information of the radiolabeled peptides. For instance, CNBr cleavage followed by trypsin digestion yields smaller peptides that are easily sequenced by MS/MS compared to CNBr cleavage alone.
III. Summary
The molecular pharmacology studies describing the localization of drug-binding sites and/or the effect of such binding on tubulin molecular dynamics and those focusing on in vitro function of microtubules are mostly performed with brain tubulin. As we stated in the introduction, this is mainly for practical reasons and therefore tubulin proteomics in terms of the analysis of tubulin isotype composition may not appear that crucial for most investigators, whereas in terms of the identification of which region(s) of tubulin is affected by a drug, i.e., its binding site and allosteric effects, it seems more obvious. We claim that it is actually beneficial to perform experiments with nonneuronal sources of tubulin, because they may be more physiologically and pharmacologically representative of tubulin composition encountered in epithelial cells for instance. In this context, experimental design benefits from the analysis of tubulin content based on MS.
We presented protocols that may be used for analyzing the content of such tubulin preparations, and those relative to drug-binding experiments can be also applied to any tubulin. In nonneuronal tubulins, if the diversity of tubulins constituting dynamic microtubules is likely to be reduced, it is still often a mixture of isotypes. Data from HDX-MS and photolabeling experiments with microtubule interacting agents and a mixture of tubulin sequences may be complex to analyze and interpret. Even if the major species, such as βII-tubulin in brain tubulin and βI-tubulin in most nonneuronal sources, do generate the bulk of statistically significant information, the amount of βIVa- or IVb-tubulin is generally not negligible.
Among the still open questions on tubulin function and pharmacology are the differences in molecular dynamics and drug binding between tubulin isotypes. Besides computer-driven modeling (Keskin et al., 2002; Tuszynski et al., 2006), experimental insights into these isotype-specific functions will be necessary and would require working with homogeneous heterodimer preparations. High-throughput screening of human cell lines, amenable to mid-/large-scale production, using validated antibody arrays would identify appropriate sources for the preparation of monoisotype tubulin heterodimers. Indeed, cell-free systems for the biosynthesis of such tubulin heterodimers have been implemented (Shah et al., 2001). Confirmation for assembly competence using Taxol and for homogeneity by MS can be assessed using our protocols.
Acknowledgments
This work has been supported by NIH grants CA077263 and CA124898, the National Foundation for Cancer Research (S.B.H.), NIH grant R33CA101150 (R.H.A.), and NIH grant CA125923 (L.M.M).
References
- Andreu JM. Large scale purification of brain tubulin with the modified Weisenberg procedure. Methods Mol Med. 2007;137:17–28. doi: 10.1007/978-1-59745-442-1_2. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Roach MC, Trcka P, Luduena RF. Increased microtubule assembly in bovine brain tubulin lacking the type III isotype of beta-tubulin. J Biol Chem. 1990;265(3):1794–1799. [PubMed] [Google Scholar]
- Banerjee A, Roach MC, Trcka P, Luduena RF. Preparation of a monoclonal antibody specific for the class IV isotype of beta-tubulin. Purification and assembly of alpha beta II, alpha beta III, and alpha beta IV tubulin dimers from bovine brain. J Biol Chem. 1992;267(8):5625–5630. [PubMed] [Google Scholar]
- Bellocq C, Andrey-Tornare I, Paunier Doret AM, et al. Purification of assembly-competent tubulin from Saccharomyces cerevisiae. Eur J Biochem. 1992;210(1):343–349. doi: 10.1111/j.1432-1033.1992.tb17427.x. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Chik JK, Slysz GW, et al. Structural mass spectrometry of the alphabetatubulin dimer supports a revised model of microtubule assembly. Biochemistry. 2009;48(22):4858–4870. doi: 10.1021/bi900200q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best D, Warr PJ, Gull K. Influence of the composition of commercial sodium dodecyl sulfate preparations on the separation of alpha- and beta-tubulin during polyacrylamide gel electrophoresis. Anal Biochem. 1981;114(2):281–284. doi: 10.1016/0003-2697(81)90481-4. [DOI] [PubMed] [Google Scholar]
- Castoldi M, Popov AV. Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expr Purif. 2003;32(1):83–88. doi: 10.1016/S1046-5928(03)00218-3. [DOI] [PubMed] [Google Scholar]
- Chik JK, Schriemer DC. Hydrogen/deuterium exchange mass spectrometry of actin in various biochemical contexts. J Mol Biol. 2003;334(3):373–385. doi: 10.1016/j.jmb.2003.09.044. [DOI] [PubMed] [Google Scholar]
- Derry WB, Wilson L, Khan IA, Luduena RF, Jordan MA. Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified beta-tubulin isotypes. Biochemistry. 1997;36(12):3554–3562. doi: 10.1021/bi962724m. [DOI] [PubMed] [Google Scholar]
- Detrich HW, 3rd, Overton SA. Heterogeneity and structure of brain tubulins from coldadapted Antarctic fishes. Comparison to brain tubulins from a temperate fish and a mammal. J Biol Chem. 1986;261(23):10922–10930. [PubMed] [Google Scholar]
- Farrell KW. Isolation of tubulin from nonneural sources. Methods Enzymol. 1982;85(Pt B):385–393. doi: 10.1016/0076-6879(82)85039-8. [DOI] [PubMed] [Google Scholar]
- Fourest-Lieuvin A. Purification of tubulin from limited volumes of cultured cells. Protein Expr Purif. 2006;45(1):183–190. doi: 10.1016/j.pep.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Gaskin F, Cantor CR, Shelanski ML. Biochemical studies on the in vitro assembly and disassembly of microtubules. Ann N Y Acad Sci. 1975;30(253):133–146. doi: 10.1111/j.1749-6632.1975.tb19197.x. [DOI] [PubMed] [Google Scholar]
- Gaskin F, Roychowdhury S. Purification of tubulin by fast-performance liquid chromatography. Ann N Y Acad Sci. 1986;466:622–625. doi: 10.1111/j.1749-6632.1986.tb38437.x. [DOI] [PubMed] [Google Scholar]
- Hamel E, Lin CM. Glutamate-induced polymerization of tubulin: characteristics of the reaction and application to the large-scale purification of tubulin. Arch Biochem Biophys. 1981;209(1):29–40. doi: 10.1016/0003-9861(81)90253-8. [DOI] [PubMed] [Google Scholar]
- Hamel E, Lin CM. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry. 1984;23(18):4173–4184. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- Huzil JT, Chik JK, Slysz GW, et al. A unique mode of microtubule stabilization induced by peloruside A. J Mol Biol. 2008;378(5):1016–1030. doi: 10.1016/j.jmb.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jai-nhuknan J, Cassady CJ. Negative ion postsource decay time-of-flight mass spectrometry of peptides containing acidic amino acid residues. Anal Chem. 1998;70(24):5122–5128. doi: 10.1021/ac980577n. [DOI] [PubMed] [Google Scholar]
- Keskin O, Durell SR, Bahar I, Jernigan RL, Covell DG. Relating molecular flexibility to function: a case study of tubulin. Biophys J. 2002;83(2):663–680. doi: 10.1016/S0006-3495(02)75199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV. Purification of yeast tubulin by self-assembly in vitro. Biochemistry. 1981;20(12):3629–3633. doi: 10.1021/bi00515a050. [DOI] [PubMed] [Google Scholar]
- Lee JC. Purification and chemical properties of brain tubulin. Methods Cell Biol. 1982;24:9–30. doi: 10.1016/s0091-679x(08)60645-7. [DOI] [PubMed] [Google Scholar]
- Lu Q, Luduena RF. In vitro analysis of microtubule assembly of isotypically pure tubulin dimers. Intrinsic differences in the assembly properties of alpha beta II, alpha beta III, and alpha beta IV tubulin dimers in the absence of microtubule-associated proteins. J Biol Chem. 1994;269(3):2041–2047. [PubMed] [Google Scholar]
- Luduena RF. Multiple forms of tubulin: different gene products and covalent modifications. Int Rev Cytol. 1998;178:207–275. doi: 10.1016/s0074-7696(08)62138-5. [DOI] [PubMed] [Google Scholar]
- MacRae TH, Gull K. Purification and assembly in vitro of tubulin from Trypanosoma brucei brucei. Biochem J. 1990;265(1):87–93. doi: 10.1042/bj2650087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae TH, Luduena RF. Developmental and comparative aspects of brine shrimp tubulin. Biochem J. 1984;219(1):137–148. doi: 10.1042/bj2190137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa S, Sakai H. Characterization and in vitro polymerization of Tetrahymena tubulin. J Biochem. 1978;83(4):1065–1075. doi: 10.1093/oxfordjournals.jbchem.a131995. [DOI] [PubMed] [Google Scholar]
- Miller LM, Menthena A, Chatterjee C, Verdier-Pinard P, Novikoff PM, Horwitz SB, Angeletti RH. Increased levels of a unique post-translationally modified betaIVb-tubulin isotype in liver cancer. Biochemistry. 2008;47(28):7572–7582. doi: 10.1021/bi8005225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morejohn LC, Fosket DE. Higher plant tubulin identified by self-assembly into microtubules in vitro. Nature. 1982;297(5865):426–428. doi: 10.1038/297426a0. [DOI] [PubMed] [Google Scholar]
- Murphy DB. Purification of tubulin and tau from chicken erythrocytes: tubulin isotypes and mechanisms of microtubule assembly. Methods Enzymol. 1991;196:235–246. doi: 10.1016/0076-6879(91)96022-j. [DOI] [PubMed] [Google Scholar]
- Murphy DB, Hiebsch RR. Purification of microtubule protein from beef brain and comparison of the assembly requirements for neuronal microtubules isolated from beef and hog. Anal Biochem. 1979;96(1):225–235. doi: 10.1016/0003-2697(79)90577-3. [DOI] [PubMed] [Google Scholar]
- Newton CN, DeLuca JG, Himes RH, Miller HP, Jordan MA, Wilson L. Intrinsically slow dynamic instability of HeLa cell microtubules in vitro. J Biol Chem. 2002;277(45):42456–42462. doi: 10.1074/jbc.M207134200. [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- Panda D, Miller HP, Banerjee A, Luduena RF, Wilson L. Microtubule dynamics in vitro are regulated by the tubulin isotype composition. Proc Natl Acad Sci U S A. 1994;91(24):11358–11362. doi: 10.1073/pnas.91.24.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paturle L, Wehland J, Margolis RL, Job D. Complete separation of tyrosinated, detyrosinated, and nontyrosinatable brain tubulin subpopulations using affinity chromatography. Biochemistry. 1989;28(6):2698–2704. doi: 10.1021/bi00432a050. [DOI] [PubMed] [Google Scholar]
- Pirollet F, Job D, Fischer EH, Margolis RL. Purification and characterization of sheep brain cold-stable microtubules. Proc Natl Acad Sci U S A. 1983;80(6):1560–1564. doi: 10.1073/pnas.80.6.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, He L, Chakravarty S, Ojima I, Orr GA, Horwitz SB. Characterization of the Taxol binding site on the microtubule. Identification of Arg(282) in beta-tubulin as the site of photoincorporation of a 7-benzophenone analogue of Taxol. J Biol Chem. 1999;274(53):37990–37994. doi: 10.1074/jbc.274.53.37990. [DOI] [PubMed] [Google Scholar]
- Rao S, Krauss NE, Heerding JM, et al. 3′-(p-azidobenzamido)taxol photolabels the N-terminal 31 amino acids of beta-tubulin. J Biol Chem. 1994;269(5):3132–3134. [PubMed] [Google Scholar]
- Rao S, Orr GA, Chaudhary AG, Kingston DG, Horwitz SB. Characterization of the taxol binding site on the microtubule. 2-(m-Azidobenzoyl)taxol photolabels a peptide (amino acids 217–231) of beta-tubulin. J Biol Chem. 1995;270(35):20235–20238. doi: 10.1074/jbc.270.35.20235. [DOI] [PubMed] [Google Scholar]
- Rudiger M, Weber K. Characterization of the post-translational modifications in tubulin from the marginal band of avian erythrocytes. Eur J Biochem. 1993;218(1):107–116. doi: 10.1111/j.1432-1033.1993.tb18357.x. [DOI] [PubMed] [Google Scholar]
- Sackett DL. Rapid purification of tubulin from tissue and tissue culture cells using solid-phase ion exchange. Anal Biochem. 1995;228(2):343–348. doi: 10.1006/abio.1995.1361. [DOI] [PubMed] [Google Scholar]
- Shah C, Xu CZ, Vickers J, Williams R. Properties of microtubules assembled from mammalian tubulin synthesized in Escherichia coli. Biochemistry. 2001;40(15):4844–4852. doi: 10.1021/bi002446y. [DOI] [PubMed] [Google Scholar]
- Shelanski ML, Gaskin F, Cantor CR. Microtubule assembly in the absence of added nucleotides. Proc Natl Acad Sci U SA. 1973;70(3):765–768. doi: 10.1073/pnas.70.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloboda RD, Belfi LM. Purification of tubulin and microtubule-associated proteins by membrane ion-exchange chromatography. Protein Expr Purif. 1998;13(2):205–209. doi: 10.1006/prep.1998.0902. [DOI] [PubMed] [Google Scholar]
- Stephens RE. Electrophoretic resolution of tubulin and tektin subunits by differential interaction with long-chain alkyl sulfates. Anal Biochem. 1998;265(2):356–360. doi: 10.1006/abio.1998.2909. [DOI] [PubMed] [Google Scholar]
- Stokasimov E, Rubenstein PA. Actin isoform specific conformational differences observed with HD exchange and mass spectrometry. J Biol Chem. 2009;284(37):25421–25430. doi: 10.1074/jbc.M109.013078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski JA, Carpenter EJ, Huzil JT, Malinski W, Luchko T, Luduena RF. The evolution of the structure of tubulin and its potential consequences for the role and function of microtubules in cells and embryos. Int J Dev Biol. 2006;50(2–3):341–358. doi: 10.1387/ijdb.052063jt. [DOI] [PubMed] [Google Scholar]
- Vallee RB. A taxol-dependent procedure for the isolation of microtubules and microtubule-associated proteins (MAPs) J Cell Biol. 1982;92(2):435–442. doi: 10.1083/jcb.92.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier-Pinard P, Pasquier E, Xiao H, et al. Tubulin proteomics: towards breaking the code. Anal Biochem. 2009;384(2):197–206. doi: 10.1016/j.ab.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier-Pinard P, Shahabi S, Wang F, et al. Detection of human betaV-tubulin expression in epithelial cancer cell lines by tubulin proteomics. Biochemistry. 2005;44(48):15858–15870. doi: 10.1021/bi051004p. [DOI] [PubMed] [Google Scholar]
- Verdier-Pinard P, Wang F, Burd B, Angeletti RH, Horwitz SB, Orr GA. Direct analysis of tubulin expression in cancer cell lines by electrospray ionization mass spectrometry. Biochemistry. 2003a;42(41):12019–12027. doi: 10.1021/bi0350147. [DOI] [PubMed] [Google Scholar]
- Verdier-Pinard P, Wang F, Martello L, Burd B, Orr GA, Horwitz SB. Analysis of tubulin isotypes and mutations from taxol-resistant cells by combined isoelectrofocusing and mass spectrometry. Biochemistry. 2003b;42(18):5349–5357. doi: 10.1021/bi027293o. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Gaertig J. The tubulin code. Cell Cycle. 2007;6(17):2152–2160. doi: 10.4161/cc.6.17.4633. [DOI] [PubMed] [Google Scholar]
- Wang L, Li Y, Abildgaard F, Markley JL, Yan H. NMR solution structure of type II human cellular retinoic acid binding protein: implications for ligand binding. Biochemistry. 1998a;37(37):12727–12736. doi: 10.1021/bi9808924. [DOI] [PubMed] [Google Scholar]
- Wang F, Li W, Emmett MR, et al. Conformational and dynamic changes of Yersinia protein tyrosine phosphatase induced by ligand binding and active site mutation and revealed by H/D exchange and electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Biochemistry. 1998b;37(44):15289–15299. doi: 10.1021/bi981481q. [DOI] [PubMed] [Google Scholar]
- Wang F, Miles RW, Kicska G, Nieves E, Schramm VL, Angeletti RH. Immucillin-H binding to purine nucleoside phosphorylase reduces dynamic solvent exchange. Protein Sci. 2000;9(9):1660–1668. doi: 10.1110/ps.9.9.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Shi W, Nieves E, Angeletti RH, Schramm VL, Grubmeyer C. A transition-state analogue reduces protein dynamics in hypoxanthine-guanine phosphoribosyltransferase. Biochemistry. 2001;40(27):8043–8054. doi: 10.1021/bi010203f. [DOI] [PubMed] [Google Scholar]
- Weatherbee JA, Luftig RB, Weihing RR. Purification and reconstitution of HeLa cell microtubules. Biochemistry. 1980;19(17):4116–4123. doi: 10.1021/bi00558a033. [DOI] [PubMed] [Google Scholar]
- Williams RC, Jr, Correia JJ, DeVries AL. Formation of microtubules at low temperature by tubulin from Antarctic fish. Biochemistry. 1985;24(11):2790–2798. doi: 10.1021/bi00332a029. [DOI] [PubMed] [Google Scholar]
- Williams RC, Jr, Lee JC. Preparation of tubulin from brain. Methods Enzymol. 1982;85(Pt B):376–385. doi: 10.1016/0076-6879(82)85038-6. [DOI] [PubMed] [Google Scholar]
- Williams RC, Jr, Shah C, Sackett D. Separation of tubulin isoforms by isoelectric focusing in immobilized pH gradient gels. Anal Biochem. 1999;275(2):265–267. doi: 10.1006/abio.1999.4326. [DOI] [PubMed] [Google Scholar]
- Winefield RD, Williams TD, Himes RH. A label-free mass spectrometry method for the quantification of protein isotypes. Anal Biochem. 2009;395(2):217–223. doi: 10.1016/j.ab.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S, Kenesky CS, Rucker PV, Smith AB, 3rd, Orr GA, Horwitz SB. A photoaffinity analogue of discodermolide specifically labels a peptide in beta-tubulin. Biochemistry. 2006;45(39):11762–11775. doi: 10.1021/bi060497a. [DOI] [PubMed] [Google Scholar]
- Xiao H, Verdier-Pinard P, Fernandez-Fuentes N, et al. Insights into the mechanism of microtubule stabilization by Taxol. Proc Natl Acad Sci U S A. 2006;103(27):10166–10173. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]