

Abstract
Background
Balanced chromosomal rearrangements occasionally have strong phenotypic effects, which may be useful in understanding pathobiology. However, conventional strategies for characterizing breakpoints are laborious and inaccurate. We present here a proband with a thoracic aortic aneurysm and a balanced translocation t(10;11)(q23.2;q24.2). Our purpose was to sequence the chromosomal breaks in this family to reveal a novel candidate gene for aneurysm.
Methods and results
Intracranial and thoracic aortic aneurysms appear to run in the family in an autosomal dominant manner: After exploring the family history, we observed that the proband’s two siblings both died from cerebral hemorrhage, and the proband’s parent and parent’s sibling died from aortic rupture. After application of a genome-wide paired-end DNA sequencing method for breakpoint mapping, we demonstrate that this translocation breaks intron 1 of a splicing isoform of Neurotrimin (NTM) at 11q25 in a previously implicated candidate region for intracranial (IAs) and aortic aneurysms (AAs) (OMIM 612161).
Conclusions
Our results demonstrate the feasibility of genome-wide paired-end sequencing for the characterization of balanced rearrangements and identification of candidate genes in patients with potentially disease-associated chromosome rearrangements. The family samples were gathered as a part of our recently launched National Registry of Reciprocal Balanced Translocations and Inversions in Finland (n=2575), and we believe that such a registry will be a powerful resource for the localization of chromosomal aberrations, which can bring insight into the etiology of related phenotypes.
Keywords: Molecular genetics, Cardiovascular Medicine, Chromosomal, Clinical Genetics, Genome-wide
INTRODUCTION
Intracranial aneurysms (IA), thoracic aortic aneurysms (TAA), and abdominal aortic aneurysms (AAA) contribute significantly to morbidity, mortality, and medical expenditure despite continued improvement in diagnostic and surgical techniques [1–4]. Aneurysms usually result from accumulated degenerative changes, and over the course of time, medial degeneration in the artery wall can progress to rupture i.e. sudden subarachnoid hemorrhage (SAH), or acute aortic dissection or rupture.
Aneurysms are typically multifactorial, with both genetic and environmental risk factors such as hypertension, atherosclerosis, bicuspid aortic valve, alcohol consumption, smoking, and sex influencing the risk of subsequent aneurysm [5–12]. Pedigree analysis examining the familial forms of TAA suggest that the primary mode of inheritance is autosomal dominant with reduced penetrance and variable expression, whereas in IA and AAA families, autosomal recessive and X-linked modes are also observed [1,13–25]. Clinicians have historically regarded IA, TAA, and AAA as distinct diseases, but recent studies suggest commonalities in some families [26]. Estimation of co-occurrence of IA and aortic aneurysms (AA) within a family is 10.5%, and, in these families, patients often share not only aneurysm morphology but also a susceptibility locus [26–32]. Researchers have applied multiple genetic approaches including candidate gene approaches [33], linkage analysis [27,34–45], and GWAS [11,46–50], and revealed multiple loci predisposing to aneurysm [27]. Identification of the causative genes has important clinical implications - not only allowing identification of individuals at risk, but also optimizing the timing for surgical repair of such aneurysms.
Interestingly, the chromosome 11q25 region has been implicated in both IA and AA in several independent linkage studies. First, a Japanese study of affected sib pairs with IA revealed significant linkage to 11q25 [36]. Ozturk et al. subsequently reported an analysis of two multigenerational families with IA replicating this susceptibility locus [42]. Mounting evidence came from Worrall et al. who studied families with both IA and AA, and managed to verify the 11q25 region [51].
To date, six genes are known to be associated with familial TAA: TGFBR1 at 9q22 [52], TGFBR2 at 3p24-25 [28], MYH11 at 16p13.13-13.12 [53], ACTA2 at 10q23-24 [54], MYLK at 3q21 [55], and SMAD3 at 15q21-q22 [56]. In addition, the gene implicated in Marfan syndrome, FBN1 at 15q24-26, has variants, which, on rare occasions, can lead to TAA without comorbid Marfan syndrome [48,57,58]. One of these genes, ACTA2 (OMIM 102620) locates near our 10q translocation breakpoint, and is implicated in 14% of TAA cases. Also, two recent studies have identified mutations in TGFB2 at 1q41 associated with TAA and propose haploinsufficiency as the relevant mechanism [59,60]. Family studies have suggested two additional loci: TAAD1 (AAT2) at 5q13-14 [44], and FAA1 (AAT1) at 11q23-24 [27]. FAA1 (OMIM 607086) [27,61] was identified near our 11q translocation breakpoint through genome-wide linkage analysis (Zmax = 4.4, 1.1 cM telomeric to D11S1356). However, candidate gene analysis has thus-far failed to identify any causative variants on 11q. Together all these known genes and loci explain less than 20% of familial TAA cases [13,14].
In this pilot study, we identified the exact breakpoint on 11q25 on a prior strong positional candidate locus for IA and AA (OMIM 612161) through the application of genome-wide paired-end sequencing. Our study demonstrates the potential power and utility of comprehensive national ascertainment of balanced translocations and inversions for gene identification.
MATERIALS AND METHODS
National Registry of Reciprocal Balanced Translocations and Inversions in Finland
We have created a comprehensive National Registry of Reciprocal Balanced Translocations and Inversions in Finland by ascertaining all known carriers (n=2575) from every Medical Genetics Department and Clinical Genetics Laboratory in the country. This is feasible because the Finnish health care system retains comprehensive lifelong medical records of every individual. One physician (TV) has examined all the rearrangement carriers’ medical records, summarized their medical histories, and photocopied the specific medical records containing clinically relevant phenotypic information. Where multiple family members were known to carry the same rearrangement, we recorded the pedigree structure (n=584) together with the aforementioned clinical information. Our database allows for examination of all clinical aspects of all cases, such that we can use the information for diagnostic purposes, for genetic counseling, and also for subsequent follow-up.
Our main scientific objective is to identify the specific chromosomal breakpoints for each translocation, and then to link this information with both data from the medical records as well as clinical data available from Finland’s extensive collection of population health registries. This facilitates the process of determining the specific effects of chromosomal rearrangements on various traits and diseases. Finland’s extensive collection of registries includes the Finnish Cancer Registry, the Social Insurance Institution of Finland registries (listing for all citizens e.g. all compensable prescription medications and all diagnoses which led to >9 sick days away from work), and the National Institute for Health and Welfare registries (nationwide registry of all hospitalized patients and all congenital malformations). All familial relationships back to the 1600s are likewise known and searchable at the Finnish population registries [62].
Permission for sampling and analyzing the genetic material was granted by the Ethical Committees of the Joint Authority for the Hospital District of Helsinki and Uusimaa, and the Office of the Data Protection Ombudsman in connection of the Ministry of Justice is appropriately notified. The Ministry of Social Affairs and Health has approved our search for additional clinical information from the national health registries described above.
Sample collection
In August 2010 we launched nationwide sampling from the Department of Medical Genetics, Väestöliitto, The Family Federation of Finland. To date, we have collected blood samples from 91 carriers of balanced translocations or inversions. Extracted DNA, RNA and established lymphoblastoid cell lines are stored at −20°C, −80°C, and −150°C respectively.
Case report
Cytogenetic analysis
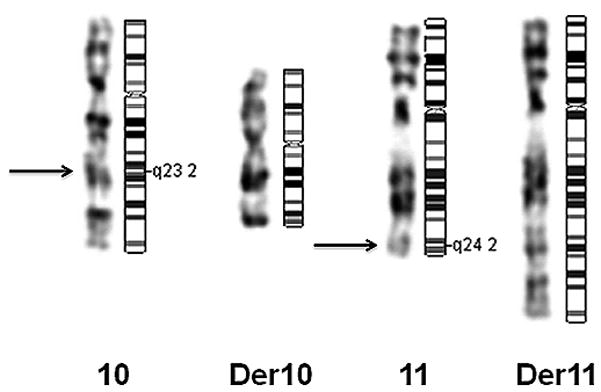
We performed standard cytogenetic analyses to verify earlier karyotyping (figure 1). Blood samples and informed consent were obtained from the proband and proband’s child. Peripheral-blood lymphocytes were isolated from whole blood and primed in vitro with phytohemagglutinin. Lymphocytes were harvested and chromosomes were prepared and stained using standard techniques. G-banding karyotype analysis was performed at an approximately 650 band resolution according to established protocols. Chromosomal aberrations were described according to the ISCN 2009 guidelines [63].
Figure 1.
Copy number analysis
To study submicroscopic deletions or duplications associated with the translocation breakpoints, genotyping was performed with the HumanHap300 BeadChip platform developed by Illumina Inc. (San Diego, CA, USA) according to the manufacturer’s protocol. The data were normalized using a commercially available proprietary algorithm implemented in the BeadStudio 3.1.0 software provided by Illumina Inc. [64]. Genotype clusters were created with BeadStudio 3.1.0. from a large panel of normal population-based samples. A genotype success rate of >95% was required for SNPs and for samples in each study set. The B allele frequency and log R ratio were visualized with Illumina BeadStudio 3.1.0. Illumina Genome Viewer (IGV) was utilized to inspect each breakpoint region for CNVs.
ACTA2 sequencing
We sequenced ACTA2 using the conventional Sanger sequencing method. Primer sequences were designed using UCSC Genome Browser (http://www.genome.ucsc.edu) and Primer 3 version 0.4.0 (http://frodo.wi.mit.edu) and their map locations were verified with UCSC In-Silico PCR, Fast PCR software (PrimerDigital Ltd., Helsinki, Finland) and Sequencer 4.8 software (Gene Codes Corporation, Ann Arbor, MI, USA). Primers and PCR conditions are available on request. Coding exons, intron-exon boundaries, introns, and 5′ and 3′ regulatory regions flanking 3 kb up- and downstream from ACTA2 gene were PCR-amplified and cyclic bidirectional sequencing reactions were carried out using BigDye 3.1 terminator technology (Applied Biosystems, CA, USA). Electrophoresis of the sequenced products was conducted with ABI3730 x1 capillary DNA Analyzer (Applied Biosystems), and base calling was performed using Sequencing Analysis 5.2 software (Applied Biosystems). The alignment between sequenced individuals and reference obtained from UCSC Genome Browser database was performed using Sequencher 4.8 software. Sequencing results were checked against dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/).
Genome-wide paired-end sequencing
To characterize the chromosomal breakpoints in the proband and proband’s child, we applied a paired-end sequencing strategy. We randomly sheared 3 μg of genomic DNA using a Covaris-S2 focused sonicator (Covaris, Inc., Woburn, MA, USA) with the following parameters: duty cycle 10%, intensity 5 and 200 cycles per burst. Genomic paired-end libraries were prepared using NEBNext® DNA Sample Prep Master Mix Set 2 (New England BioLabs, Ipswich, MA, USA) according to the manufacturer’s protocol with minor modifications. We generated three libraries with distinct target fragment sizes of ~200 bp, ~500 bp, and ~1 kb with the Caliper LabChip XT –instrument (Caliper Life Sciences, Hopkinton, MA, USA) to increase the genomic coverage and optimize the insert size for translocation detection. We assessed whether the insert size could be increased up to 1 kb without compromising sequence yield, mapping, or evenness of the genomic coverage. The 500 bp library was the most effective of the insert sizes we targeted, yielding the best coverage. Illumina GAII and HiSeq2000 analyzers were used for cluster amplification and to generate three lanes of short sequencing reads from both ends of these DNA fragments using the manufacturer’s protocol. The images were processed using the manufacturer’s software.
Data processing
An in-house developed SAMtools-based variant calling pipeline version 1.0 (VCP 1.0), described in detail by Sulonen et al., was used to align the sequence reads (GRCh37) [65]. We generated a total of ~354 million paired-end reads mapping back to the reference yielding 91% coverage of the reference. The overall mean depth of coverage was 8.7 X, and for NTM gene the average coverage was 6.04 X. We excluded 3.4 million reads as PCR duplicates, 0.8 million read pairs where only one end mapped to the genome, and 6.3 million read pairs because neither end mapped. We used the Integrative Genomics Viewer visualization program (Broad Institute, Cambridge, MA, USA) to identify rearrangements as discordant reads aligning to different chromosomes. Only when ≥2 read pairs spanned the same junction fragment were they used in further analysis. We subsequently targeted our search to paired-end anomalies mapping to chromosome 10 at one end and chromosome 11 at the other.
Confirmatory screening of the junction fragments
We designed primers for capillary sequencing of both derivative chromosomes to encompass the junction fragments. We used different combinations of primer sets located 1 kb outside the paired-end reads, for a maximum product size of 1 kb. Phusion High Fidelity PCR kit (Finnzymes, Vantaa, Finland) and a gradient of 56–65°C for primer annealing were used on patient and normal genomic DNA for each set of primers at least twice. Primers and PCR conditions are available on request. We assessed products from each primer set using gel electrophoresis. Rearrangements were defined as those PCR reactions giving a convincing band in the patient DNA with no matching band in the control, detected in ≥2 separate reactions, together with ambiguously mapping sequence data. PCR products were used as templates for sequencing in both directions using BigDye Terminator 3.1 chemistry (Applied Biosystems) on 3730xl DNA Analyzer (Applied Biosystems). The sequences of the junction fragments were aligned to the human genome reference sequence (GRCh37) using Sequencher 4.8 software (Gene Codes Corporation) to define the breakpoints at base-pair resolution.
RESULTS
Karyotype and exclusion of copy number variation
Conventional G-banding karyotype analysis refined the preexisting translocation breakpoint from t(10;11)(q22;q23) to t(10;11)(q23.2;q24.2) (figure 1). To assess whether the translocation is truly balanced, we performed SNP genotyping of the two translocation carriers to detect CNVs. The raw intensity plots of the breakpoint regions (several megabases up- and downstream) revealed no detectable aberrations, submicroscopic, or large-scale CNVs.
Clinical summary
While building up our comprehensive National Registry, we observed that a proband with translocation t(10;11)(q23.2;q24.2) had suffered from a TAA. This comorbidity was previously not recognized by the clinical geneticists, and only came to light as a byproduct of this project. The pedigree is given in figure 2. The patient first had shortness of breath after exertion after the age of 50. Echocardiography and angiography revealed a TAA: dilatation of the ascending aorta to 68 mm as well as aortic insufficiency grade III – IV. At the age of 55 the proband received a prosthesis of the ascending aorta and valve. Based on death certificates, the proband’s two siblings both died of cerebral hemorrhage at the ages of 69 and 50, respectively. The first sibling’s forensic autopsy confirmed a ruptured aneurysm in the left carotid artery as the cause of death. The second sibling died in hospital care. The cause of death was based on anamnesis, clinical examination and laboratory results. According to the family history, the index case’s parent and parent’s sibling both had the same kind of symptoms and disease as the proband, and very likely had an aortic dilatation: their causes of death were aortic ruptures at the ages of 65 and 69, respectively. Unfortunately, autopsies were not routinely performed in the 1950’s at the time of their death. We excluded Marfan syndrome in the family based on family history and detailed medical records. The index case’s child is now followed regularly every three years with magnetic resonance imaging (MRI) or echocardiography, which have thus-far revealed nothing abnormal by the age of 45.
Figure 2.

The translocation in this family was initially observed a decade ago, when the proband’s child’s family had a routine prenatal screening of their first pregnancy. An otherwise normal ultrasonography repeatedly failed to reveal an air bubble in the ventricle of the fetus. Being suspicious of an esophageal atresia, the gynecologist referred the family to prenatal genetic counseling. After counseling, the parents opted for examination of the fetal chromosomes from a chorionic villi sample, which revealed an unbalanced translocation with a large excess of chromosome 11. The parents chose to terminate the pregnancy. Autopsy of the fetus confirmed the diagnosis of esophageal atresia and esophagotracheal fistula, and revealed dysmorphic features: broad back of the nose, micrognathia, narrow palpebral fissures, and single palmar creases in both hands.
ACTA2 sequencing
We chose ACTA2 located near our 10q breakpoint for systematic bidirectional mutation screening. The coding sequence revealed no previously uncharacterized variants, and no splice mutations. The only detected variants were two single base substitutions verified as common SNPs (rs72809344 and rs72809340) in intron 1. The paired-end sequencing data confirmed these variants as heterozygous SNPs.
Breakpoint mapping
From the ~354 million uniquely mapping paired-end read pairs, we searched for those spanning the breakpoints. We identified five pairs that mapped consistently to chromosomes 10 and 11, suggesting they might cross the translocation breakpoint (figures 3A–B). In addition, we identified one orphan read with only one end mapping to chromosome 10, whereas the other unpaired end was complementary to chromosome 11. The minimal intervals containing the breakpoints defined by these overlapping reads were ~750 bp on chromosome 11 between nucleotides 131778051 and 131778904, and ~40 bp on chromosome 10 between nucleotides 90973021 and 90973118. The junction fragments crossing the translocation were subsequently amplified by PCR and Sanger sequenced. The exact breakpoint positions were finally identified on chr10: 90,973,081-90,973,082 and chr11: 131,778,234-131,778,235 (GRCh37) (figures 4A and 4C). The chromosome 11 breakpoint is in intron 1 of a splicing isoform of the NTM gene and the 5′ regulatory region of another NTM splicing isoform (http://genome.ucsc.edu/, NM_001048209) (figure 4A). In contrast, the breakpoint on chromosome 10q maps to an intergenic region flanked by the 3′ end of LIPA and the 5′ end of CH25H (figure 4C) within a simple tandem repeat consisting of a tract of ‘AAAGA’ (figures 4B and 4D). This repeat structure is not present in the chromosome 11 region, which shares no apparent regional microhomology with the chromosome 10 breakpoint region. We detected no gain or loss of chromosomal material near the translocation breakpoint on the derivative chromosome 11 region. On the derivative chromosome 10 region, we identified three breakpoint flanking microdeletions. Two were 1-bp deletions at 90,973,081 and 90,973,078, and one a 5-bp deletion between nucleotides 90,973,049 and 90,973,055 (GRCh37) (figure 5). To identify interspersed repetitive elements near the breakpoints, we referred to the UCSC Genome Browser (GRCh37), but identified no repeats within 1 kb upstream or downstream. No significantly homologous sequences were observed to flank the two breakpoints.
Figure 3.

Figure 4.


National Registry of Reciprocal Balanced Translocations and Inversions in Finland
We are currently constructing the National Registry of Reciprocal Balanced Translocations and Inversions in Finland. We have ascertained all known Finnish cases (n=2575), of which 2104 are balanced translocations and 471 balanced inversions. To date 584 are known to be familial cases and 102 to carry a de novo mutation. We are currently harmonizing the data and performing the relevant quality control steps and aim to have the Registry accessible to the scientific and clinical community in the near future. An epidemiological study of all known Finnish cases is in progress.
DISCUSSION
Discovery of this translocation described above is a byproduct of a larger project to collect all known balanced translocations and inversions in Finland, giving us momentum towards our goal of characterizing more generally the molecular basis of all translocations in Finland and their associated phenotypic effects. The opportunity in the Nordic countries to link the data to national health registers provides additional openings for future studies. Such a database will benefit not only those of us interested in research, but more importantly will be a useful resource for clinical geneticists and genetic counselors.
We systematically examined and analyzed the medical records from all translocation carriers in our database, and as the first clinical fruit of this endeavor, we were able to identify a translocation carrier with TAA, who had a strong family history of both TAA and IA. Both conditions can lead to sudden death without follow-up examination, therefore we paid immediate clinical attention to this family. For first-degree family members, it will be essential to schedule an angiography to diagnose future IAs and echocardiography or MRI to follow the aortic dilatation expansion rate so that surgical intervention can be correctly timed before a fatal aneurysm occurs. Based on previous reports that the region at chromosome 11q25 was involved in both IA and AA [51], we focused on the translocation breakpoint to search for a novel genetic disruption potentially underlying the risk of these aneurysms. As shown here, sequencing the breakpoint regions in a patient with an aneurysm and reciprocal translocation has facilitated the identification of one potential positional candidate gene, which had not been previously associated with TAA or IA. However, our preliminary finding requires extensive validation, hence follow-up studies will be essential to establish conclusively whether this gene-disruption is itself causative of aneurysm, and to exclude other potential genetic factors nearby.
Balanced chromosomal rearrangements occasionally have strong phenotypic effects, which may be useful in understanding pathobiology. To delineate translocation breakpoints in a timely and cost-effective fashion, we employed next-generation sequencing. Genome-wide paired-end sequencing enabled us to map the chromosomal breakpoints to a <1000 bp region, pointing to a single PCR primer pair: The identified translocation disrupts intron 1 of a splicing isoform of Neurotrimin (NTM) at 11q25 in a strong candidate region for IA and AA (OMIM 612161). This susceptibility locus has previously emerged in three independent linkage studies suggesting a susceptibility gene for IA somewhere between 125.8 and 133.7 Mb on 11q [36,42,51].
The NTM gene encodes a member of the IgLON cell adhesion molecule family. The encoded protein has been previously implicated in the promotion of neurite outgrowth and adhesion. NTM is mainly expressed in the central nervous system, but also in lung and heart. Studies have confirmed that it can mediate cell-cell interactions, indicating that it may have additional functions in the human cardiovascular system [66]. Our study suggests that the disrupted NTM gene is one potential positional candidate gene for the IA/TAA phenotype in this family. Unfortunately, we lacked biological samples from the deceased relatives, which would be needed to confirm that they carry the translocation.
The reciprocal breakpoint on chromosome 10q is located in an intergenic region flanked by the 5′ end of CH25H and the 3′ end of LIPA. The LIPA gene encodes lipase A, the lysosomal acid lipase, which catalyzes the hydrolysis of cholesteryl esters and triglycerides in the lysosomes. Mutations in LIPA cause Wolman disease and cholesteryl ester storage disease (OMIM 278000). CH25H is also involved in cholesterol and lipid metabolism. Without further insight we are reluctant to speculate that either of these genes on 10q are plausible functional candidates for IA or TAA. A novel gene for familial TAA, ACTA2 also lies at 10q23-24 (49). However, early on in our study, we sequenced this candidate gene, and excluded its causative mutations.
Repetitive elements have been implicated as one cause of chromosomal instability. However, in this case, we detected no interspersed repeats or common overlapping microhomology flanking our breakpoints, other than a simple tandem repeat of ‘AAAGA’ within the der(10) breakpoint. Small cryptic submicroscopic changes (microdeletions and duplications) have also been associated with balanced translocations (56). While the SNP array revealed no suspicious CNVs in our patients, the subsequent Sanger sequencing identified three microdeletions on der(10) regions: a 1-bp deletion located precisely at the genomic breakpoint at position 90,973,082, a 1-bp deletion at 90,973,079, and a 5-bp deletion (‘AAAGA’) between 90,973,049 and 90,973,055 (GRCh37). The microdeletions identified are consistent with previous studies indicating that small cryptic submicroscopic changes, which may also lead to phenotypic abnormalities in dosage-sensitive genomic regions, can be associated with balanced translocations [67,68]
Previous studies suggest that balanced rearrangements might be risk factors for many complex and late-onset diseases like diabetes, schizophrenia, and dyslexia [69–72]. Our comprehensive National Registry of Reciprocal Balanced Translocations and Inversions in Finland will, in the future, link translocations and inversions with all lifelong registry diagnoses from all Finnish national health registries, and it will therefore become a powerful resource for complex disease research as well as clinical practice.
Acknowledgments
We are grateful to the Finnish families for providing the data for this study. We thank genetic nurses Marjatta Sipponen and Ulla Parisaari for excellent clinical assistance. We are grateful to Anne Nyberg, Elli Kempas, and Seija Puomilahti for their assistance and expertise during the sample collection. We thank Maija Lepistö, Sari Hannula and Tiina Hannunen at the FIMM Technology Centre for performing sequencing experiments. We would like to acknowledge Ilse Klein at Caliper Life Sciences for paired-end library preparation. We thank Anna-Maija Sulonen, Henrikki Almusa and Timo Miettinen for their technical support. We wish to thank Olli Pietiläinen for his contribution to the copy number analysis. Jukka Illikainen, Leyla Avsar and Teemu Masalin are recognized for their valuable contribution to the figure editing. Finally, we would like to cordially acknowledge the overwhelming enthusiasm and support of the late Academician Leena Peltonen-Palotie.
FUNDING
This work was supported by the State Appropriations of Helsinki University Central Hospital grant number TLE82G0016 (to MP and TV), FiDiPro grant from the Academy of Finland grant number 118351 (to JT), the National Institutes of Health grant number MH084995 (to JT), the Wellcome Trust grant number 098051 (to AP), the Academy of Finland grant number 251704 (to AP), the Academy of Finland, Center of Excellence in Complex Disease Genetics grants number 213506 and 129680 (to AP), the European Community’s Seventh Framework Programme SYNSYS grant number 242167 (to AP) and the Sigrid Juselius Foundation, Finland (to AP).
Footnotes
COMPETING INTERESTS
None.
CONTRIBUTORS
The nine authors are justifiably credited with authorship, according to the authorship criteria. In detail: TL – design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, final approval given; MP – design, critical revision of manuscript, final approval given; AP – design, critical revision of the manuscript, final approval given; PE – design, analysis and interpretation of data, critical revision of the manuscript, final approval given; SL - design, acquisition of data, analysis and interpretation of data, critical revision of the manuscript, final approval given; JL – design, critical revision of the manuscript, final approval given; JT – design, critical revision of the manuscript, final approval given; RS – design, acquisition of data, critical revision of the manuscript, final approval given; TV - conception, design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, final approval given.
References
- 1.Lilienfeld DE, Gunderson PD, Sprafka JM, Vargas C. Epidemiology of aortic aneurysms: I. Mortality trends in the United States, 1951 to 1981. Arteriosclerosis. 1987;7:637–643. doi: 10.1161/01.atv.7.6.637. [DOI] [PubMed] [Google Scholar]
- 2.Sarti C, Tuomilehto J, Salomaa V, Sivenius J, Kaarsalo E, Narva EV, Salmi K, Torppa J. Epidemiology of subarachnoid hemorrhage in Finland from 1983 to 1985. Stroke. 1991;22:848–853. doi: 10.1161/01.str.22.7.848. [DOI] [PubMed] [Google Scholar]
- 3.Pande RL, Beckman JA, Creager MA, Stone DH, Cronenwett JL, Eagleton MJ, Upchurch GR., Jr . Aortic Aneurysm. In: Creager MA, Dzau VJ, Loscalzo J, editors. Vascular Medicine. Philadelphia, Pa: Elsevier Inc; 2006. [Google Scholar]
- 4.Kuivaniemi H, Platsoucas CD, Tilson MD., 3rd Aortic aneurysms: an immune disease with a strong genetic component. Circulation. 2008;117:242–252. doi: 10.1161/CIRCULATIONAHA.107.690982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connolly ES, Jr, Choudhri TF, Mack WJ, Mocco J, Spinks TJ, Slosberg J, Lin T, Huang J, Solomon RA. Influence of smoking, hypertension, and sex on the phenotypic expression of familial intracranial aneurysms in siblings. Neurosurgery. 2001;48:64–69. doi: 10.1097/00006123-200101000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Nahed BV, DiLuna ML, Morgan T, Ocal E, Hawkins AA, Ozduman K, Kahle KT, Chamberlain A, Amar AP, Gunel M. Hypertension, age, and location predict rupture of small intracranial aneurysms. Neurosurgery. 2005;57:676–683. [PubMed] [Google Scholar]
- 7.Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Pannu H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu Rev Genomics Hum Genet. 2008;9:282–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 8.Tromp G, Kuivaniemi H, Hinterseher I, Carey DJ. Novel genetic mechanisms for aortic aneurysms. Curr Atheroscler Rep. 2010;12:259–266. doi: 10.1007/s11883-010-0111-x. [DOI] [PubMed] [Google Scholar]
- 9.Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, Jones GT, Rinkel GJ, Blankensteijn JD, Ronkainen A, Jaaskelainen JE, Kyo Y, Lenk GM, Sakalihasan N, Kostulas K, Gottsater A, Flex A, Stefansson H, Hansen T, Andersen G, Weinsheimer S, Borch-Johnsen K, Jorgensen T, Shah SH, Quyyumi AA, Granger CB, Reilly MP, Austin H, Levey AI, Vaccarino V, Palsdottir E, Walters GB, Jonsdottir T, Snorradottir S, Magnusdottir D, Gudmundsson G, Ferrell RE, Sveinbjornsdottir S, Hernesniemi J, Niemela M, Limet R, Andersen K, Sigurdsson G, Benediktsson R, Verhoeven EL, Teijink JA, Grobbee DE, Rader DJ, Collier DA, Pedersen O, Pola R, Hillert J, Lindblad B, Valdimarsson EM, Magnadottir HB, Wijmenga C, Tromp G, Baas AF, Ruigrok YM, van Rij AM, Kuivaniemi H, Powell JT, Matthiasson SE, Gulcher JR, Thorgeirsson G, Kong A, Thorsteinsdottir U, Stefansson K. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–224. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 10.Bown MJ, Jones GT, Harrison SC, Wright BJ, Bumpstead S, Baas AF, Gretarsdottir S, Badger SA, Bradley DT, Burnand K, Child AH, Clough RE, Cockerill G, Hafez H, Scott DJ, Futers S, Johnson A, Sohrabi S, Smith A, Thompson MM, van Bockxmeer FM, Waltham M, Matthiasson SE, Thorleifsson G, Thorsteinsdottir U, Blankensteijn JD, Teijink JA, Wijmenga C, de Graaf J, Kiemeney LA, Assimes TL, McPherson R, Folkersen L, Franco-Cereceda A, Palmen J, Smith AJ, Sylvius N, Wild JB, Refstrup M, Edkins S, Gwilliam R, Hunt SE, Potter S, Lindholt JS, Frikke-Schmidt R, Tybjaerg-Hansen A, Hughes AE, Golledge J, Norman PE, van Rij A, Powell JT, Eriksson P, Stefansson K, Thompson JR, Humphries SE, Sayers RD, Deloukas P, Samani NJ CARDIoGRAM Consortium, Global BPgen Consortium, DIAGRAM Consortium, VRCNZ Consortium. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am J Hum Genet. 2011;89:619–627. doi: 10.1016/j.ajhg.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gretarsdottir S, Baas AF, Thorleifsson G, Holm H, den Heijer M, de Vries JP, Kranendonk SE, Zeebregts CJ, van Sterkenburg SM, Geelkerken RH, van Rij AM, Williams MJ, Boll AP, Kostic JP, Jonasdottir A, Jonasdottir A, Walters GB, Masson G, Sulem P, Saemundsdottir J, Mouy M, Magnusson KP, Tromp G, Elmore JR, Sakalihasan N, Limet R, Defraigne JO, Ferrell RE, Ronkainen A, Ruigrok YM, Wijmenga C, Grobbee DE, Shah SH, Granger CB, Quyyumi AA, Vaccarino V, Patel RS, Zafari AM, Levey AI, Austin H, Girelli D, Pignatti PF, Olivieri O, Martinelli N, Malerba G, Trabetti E, Becker LC, Becker DM, Reilly MP, Rader DJ, Mueller T, Dieplinger B, Haltmayer M, Urbonavicius S, Lindblad B, Gottsater A, Gaetani E, Pola R, Wells P, Rodger M, Forgie M, Langlois N, Corral J, Vicente V, Fontcuberta J, Espana F, Grarup N, Jorgensen T, Witte DR, Hansen T, Pedersen O, Aben KK, de Graaf J, Holewijn S, Folkersen L, Franco-Cereceda A, Eriksson P, Collier DA, Stefansson H, Steinthorsdottir V, Rafnar T, Valdimarsson EM, Magnadottir HB, Sveinbjornsdottir S, Olafsson I, Magnusson MK, Palmason R, Haraldsdottir V, Andersen K, Onundarson PT, Thorgeirsson G, Kiemeney LA, Powell JT, Carey DJ, Kuivaniemi H, Lindholt JS, Jones GT, Kong A, Blankensteijn JD, Matthiasson SE, Thorsteinsdottir U, Stefansson K. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42:692–697. doi: 10.1038/ng.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinterseher I, Tromp G, Kuivaniemi H. Genes and abdominal aortic aneurysm. Ann Vasc Surg. 2011;25:388–412. doi: 10.1016/j.avsg.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. 1997;25:506–511. doi: 10.1016/s0741-5214(97)70261-1. [DOI] [PubMed] [Google Scholar]
- 14.Coady MA, Davies RR, Roberts M, Goldstein LJ, Rogalski MJ, Rizzo JA, Hammond GL, Kopf GS, Elefteriades JA. Familial patterns of thoracic aortic aneurysms. Arch Surg. 1999;134:361–367. doi: 10.1001/archsurg.134.4.361. [DOI] [PubMed] [Google Scholar]
- 15.Bromberg JE, Rinkel GJ, Algra A, van Duyn CM, Greebe P, Ramos LM, van Gijn J. Familial subarachnoid hemorrhage: distinctive features and patterns of inheritance. Ann Neurol. 1995;38:929–934. doi: 10.1002/ana.410380614. [DOI] [PubMed] [Google Scholar]
- 16.Chambers WR, Harper BF, Jr, Simpson JR. Familial incidence of congenital aneurysms of cerebral arteries: report of cases of ruptured aneurysms in father and son. J Am Med Assoc. 1954;155:358–359. doi: 10.1001/jama.1954.73690220001007. [DOI] [PubMed] [Google Scholar]
- 17.De Braekeleer M, Perusse L, Cantin L, Bouchard JM, Mathieu J. A study of inbreeding and kinship in intracranial aneurysms in the Saguenay Lac-Saint-Jean region (Quebec, Canada) Ann Hum Genet. 1996;60:99–104. doi: 10.1111/j.1469-1809.1996.tb01181.x. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto I. Familial intracranial aneurysms and cerebral vascular anomalies. J Neurosurg. 1977;46:419–427. doi: 10.3171/jns.1977.46.4.0419. [DOI] [PubMed] [Google Scholar]
- 19.Kim DH, Van Ginhoven G, Milewicz DM. Incidence of familial intracranial aneurysms in 200 patients: comparison among Caucasian, African-American, and Hispanic populations. Neurosurgery. 2003;53:302–308. doi: 10.1227/01.neu.0000073418.34609.35. [DOI] [PubMed] [Google Scholar]
- 20.Leblanc R. Familial cerebral aneurysms. Can J Neurol Sci. 1997;24:191–199. doi: 10.1017/s031716710002179x. [DOI] [PubMed] [Google Scholar]
- 21.Nakagawa T, Hashi K, Kurokawa Y, Yamamura A. Family history of subarachnoid hemorrhage and the incidence of asymptomatic, unruptured cerebral aneurysms. J Neurosurg. 1999;91:391–395. doi: 10.3171/jns.1999.91.3.0391. [DOI] [PubMed] [Google Scholar]
- 22.Raaymakers TW, Rinkel GJ, Ramos LM. Initial and follow-up screening for aneurysms in families with familial subarachnoid hemorrhage. Neurology. 1998;51:1125–1130. doi: 10.1212/wnl.51.4.1125. [DOI] [PubMed] [Google Scholar]
- 23.Ronkainen A, Hernesniemi J, Ryynanen M, Puranen M, Kuivaniemi H. A ten percent prevalence of asymptomatic familial intracranial aneurysms: preliminary report on 110 magnetic resonance angiography studies in members of 21 Finnish familial intracranial aneurysm families. Neurosurgery. 1994;35:208–213. doi: 10.1227/00006123-199408000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Schievink WI, Schaid DJ, Michels VV, Piepgras DG. Familial aneurysmal subarachnoid hemorrhage: a community-based study. J Neurosurg. 1995;83:426–429. doi: 10.3171/jns.1995.83.3.0426. [DOI] [PubMed] [Google Scholar]
- 25.Wang PS, Longstreth WT, Jr, Koepsell TD. Subarachnoid hemorrhage and family history. A population-based case-control study. Arch Neurol. 1995;52:202–204. doi: 10.1001/archneur.1995.00540260108026. [DOI] [PubMed] [Google Scholar]
- 26.Kim DH, Van Ginhoven G, Milewicz DM. Familial aggregation of both aortic and cerebral aneurysms: evidence for a common genetic basis in a subset of families. Neurosurgery. 2005;56:655–661. doi: 10.1227/01.neu.0000156787.55281.53. [DOI] [PubMed] [Google Scholar]
- 27.Vaughan CJ, Casey M, He J, Veugelers M, Henderson K, Guo D, Campagna R, Roman MJ, Milewicz DM, Devereux RB, Basson CT. Identification of a chromosome 11q23. 2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation. 2001;103:2469–2475. doi: 10.1161/01.cir.103.20.2469. [DOI] [PubMed] [Google Scholar]
- 28.Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, Shete S, Willing MC, Raman CS, Milewicz DM. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 29.Milewicz DM, Chen H, Park ES, Petty EM, Zaghi H, Shashidhar G, Willing M, Patel V. Reduced penetrance and variable expressivity of familial thoracic aortic aneurysms/dissections. Am J Cardiol. 1998;82:474–479. doi: 10.1016/s0002-9149(98)00364-6. [DOI] [PubMed] [Google Scholar]
- 30.Norrgard O, Angqvist KA, Fodstad H, Forssell A, Lindberg M. Co-existence of abdominal aortic aneurysms and intracranial aneurysms. Acta Neurochir (Wien) 1987;87:34–39. doi: 10.1007/BF02076012. [DOI] [PubMed] [Google Scholar]
- 31.Hasham SN, Willing MC, Guo DC, Muilenburg A, He R, Tran VT, Scherer SE, Shete SS, Milewicz DM. Mapping a locus for familial thoracic aortic aneurysms and dissections (TAAD2) to 3p24–25. Circulation. 2003;107:3184–3190. doi: 10.1161/01.CIR.0000078634.33124.95. [DOI] [PubMed] [Google Scholar]
- 32.Cannon Albright LA, Camp NJ, Farnham JM, MacDonald J, Abtin K, Rowe KG. A genealogical assessment of heritable predisposition to aneurysms. J Neurosurg. 2003;99:637–643. doi: 10.3171/jns.2003.99.4.0637. [DOI] [PubMed] [Google Scholar]
- 33.Ruigrok YM, Rinkel GJ, Wijmenga C. Genetics of intracranial aneurysms. Lancet Neurol. 2005;4:179–189. doi: 10.1016/S1474-4422(05)01015-X. [DOI] [PubMed] [Google Scholar]
- 34.Shibamura H, Olson JM, van Vlijmen-Van Keulen C, Buxbaum SG, Dudek DM, Tromp G, Ogata T, Skunca M, Sakalihasan N, Pals G, Limet R, MacKean GL, Defawe O, Verloes A, Arthur C, Lossing AG, Burnett M, Sueda T, Kuivaniemi H. Genome scan for familial abdominal aortic aneurysm using sex and family history as covariates suggests genetic heterogeneity and identifies linkage to chromosome 19q13. Circulation. 2004;109:2103–2108. doi: 10.1161/01.CIR.0000127857.77161.A1. [DOI] [PubMed] [Google Scholar]
- 35.Van Vlijmen-Van Keulen CJ, Rauwerda JA, Pals G. Genome-wide linkage in three Dutch families maps a locus for abdominal aortic aneurysms to chromosome 19q13. 3. Eur J Vasc Endovasc Surg. 2005;30:29–35. doi: 10.1016/j.ejvs.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 36.Onda H, Kasuya H, Yoneyama T, Takakura K, Hori T, Takeda J, Nakajima T, Inoue I. Genomewide-linkage and haplotype-association studies map intracranial aneurysm to chromosome 7q11. Am J Hum Genet. 2001;69:804–819. doi: 10.1086/323614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olson JM, Vongpunsawad S, Kuivaniemi H, Ronkainen A, Hernesniemi J, Ryynanen M, Kim LL, Tromp G. Search for intracranial aneurysm susceptibility gene(s) using Finnish families. BMC Med Genet. 2002;3:7. doi: 10.1186/1471-2350-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Voet M, Olson JM, Kuivaniemi H, Dudek DM, Skunca M, Ronkainen A, Niemela M, Jaaskelainen J, Hernesniemi J, Helin K, Leinonen E, Biswas M, Tromp G. Intracranial aneurysms in Finnish families: confirmation of linkage and refinement of the interval to chromosome 19q13. 3. Am J Hum Genet. 2004;74:564–571. doi: 10.1086/382285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamada S, Utsunomiya M, Inoue K, Nozaki K, Inoue S, Takenaka K, Hashimoto N, Koizumi A. Genome-wide scan for Japanese familial intracranial aneurysms: linkage to several chromosomal regions. Circulation. 2004;110:3727–3733. doi: 10.1161/01.CIR.0000143077.23367.18. [DOI] [PubMed] [Google Scholar]
- 40.Mineharu Y, Inoue K, Inoue S, Yamada S, Nozaki K, Hashimoto N, Koizumi A. Model-based linkage analyses confirm chromosome 19q13. 3 as a susceptibility locus for intracranial aneurysm. Stroke. 2007;38:1174–1178. doi: 10.1161/01.STR.0000259657.73682.03. [DOI] [PubMed] [Google Scholar]
- 41.Nahed BV, Seker A, Guclu B, Ozturk AK, Finberg K, Hawkins AA, DiLuna ML, State M, Lifton RP, Gunel M. Mapping a Mendelian form of intracranial aneurysm to 1p34.3-p36. 13. Am J Hum Genet. 2005;76:172–179. doi: 10.1086/426953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ozturk AK, Nahed BV, Bydon M, Bilguvar K, Goksu E, Bademci G, Guclu B, Johnson MH, Amar A, Lifton RP, Gunel M. Molecular genetic analysis of two large kindreds with intracranial aneurysms demonstrates linkage to 11q24-25 and 14q23-31. Stroke. 2006;37:1021–1027. doi: 10.1161/01.STR.0000206153.92675.b9. [DOI] [PubMed] [Google Scholar]
- 43.Verlaan DJ, Dube MP, St-Onge J, Noreau A, Roussel J, Satge N, Wallace MC, Rouleau GA. A new locus for autosomal dominant intracranial aneurysm, ANIB4, maps to chromosome 5p15.2-14. 3. J Med Genet. 2006;43:e31. doi: 10.1136/jmg.2005.033209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo D, Hasham S, Kuang SQ, Vaughan CJ, Boerwinkle E, Chen H, Abuelo D, Dietz HC, Basson CT, Shete SS, Milewicz DM. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13-14. Circulation. 2001;103:2461–2468. doi: 10.1161/01.cir.103.20.2461. [DOI] [PubMed] [Google Scholar]
- 45.Ruigrok YM, Rinkel GJ. Genetics of intracranial aneurysms. Stroke. 2008;39:1049–1055. doi: 10.1161/STROKEAHA.107.497305. [DOI] [PubMed] [Google Scholar]
- 46.Bilguvar K, Yasuno K, Niemela M, Ruigrok YM, von Und Zu Fraunberg M, van Duijn CM, van den Berg LH, Mane S, Mason CE, Choi M, Gaal E, Bayri Y, Kolb L, Arlier Z, Ravuri S, Ronkainen A, Tajima A, Laakso A, Hata A, Kasuya H, Koivisto T, Rinne J, Ohman J, Breteler MM, Wijmenga C, State MW, Rinkel GJ, Hernesniemi J, Jaaskelainen JE, Palotie A, Inoue I, Lifton RP, Gunel M. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet. 2008;40:1472–1477. doi: 10.1038/ng.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yasuno K, Bilguvar K, Bijlenga P, Low SK, Krischek B, Auburger G, Simon M, Krex D, Arlier Z, Nayak N, Ruigrok YM, Niemela M, Tajima A, von und zu Fraunberg M, Doczi T, Wirjatijasa F, Hata A, Blasco J, Oszvald A, Kasuya H, Zilani G, Schoch B, Singh P, Stuer C, Risselada R, Beck J, Sola T, Ricciardi F, Aromaa A, Illig T, Schreiber S, van Duijn CM, van den Berg LH, Perret C, Proust C, Roder C, Ozturk AK, Gaal E, Berg D, Geisen C, Friedrich CM, Summers P, Frangi AF, State MW, Wichmann HE, Breteler MM, Wijmenga C, Mane S, Peltonen L, Elio V, Sturkenboom MC, Lawford P, Byrne J, Macho J, Sandalcioglu EI, Meyer B, Raabe A, Steinmetz H, Rufenacht D, Jaaskelainen JE, Hernesniemi J, Rinkel GJ, Zembutsu H, Inoue I, Palotie A, Cambien F, Nakamura Y, Lifton RP, Gunel M. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet. 2010;42:420–425. doi: 10.1038/ng.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lemaire SA, McDonald ML, Guo DC, Russell L, Miller CC, 3rd, Johnson RJ, Bekheirnia MR, Franco LM, Nguyen M, Pyeritz RE, Bavaria JE, Devereux R, Maslen C, Holmes KW, Eagle K, Body SC, Seidman C, Seidman JG, Isselbacher EM, Bray M, Coselli JS, Estrera AL, Safi HJ, Belmont JW, Leal SM, Milewicz DM. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21. 1. Nat Genet. 2011;43:996–1000. doi: 10.1038/ng.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elmore JR, Obmann MA, Kuivaniemi H, Tromp G, Gerhard GS, Franklin DP, Boddy AM, Carey DJ. Identification of a genetic variant associated with abdominal aortic aneurysms on chromosome 3p12. 3 by genome wide association. J Vasc Surg. 2009;49:1525–1531. doi: 10.1016/j.jvs.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 50.Jones GT, van Rij AM. Regarding “Identification of a genetic variant associated with abdominal aortic aneurysms on chromosome 3p12. 3 by genome wide association”. J Vasc Surg. 2009;50:1246–1247. doi: 10.1016/j.jvs.2009.07.098. [DOI] [PubMed] [Google Scholar]
- 51.Worrall BB, Foroud T, Brown RD, Jr, Connolly ES, Hornung RW, Huston J, 3rd, Kleindorfer D, Koller DL, Lai D, Moomaw CJ, Sauerbeck L, Woo D, Broderick JP Familial Intracranial Aneurysm Study Investigators. Genome screen to detect linkage to common susceptibility genes for intracranial and aortic aneurysms. Stroke. 2009;40:71–76. doi: 10.1161/STROKEAHA.108.522631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tran-Fadulu V, Pannu H, Kim DH, Vick GW, 3rd, Lonsford CM, Lafont AL, Boccalandro C, Smart S, Peterson KL, Hain JZ, Willing MC, Coselli JS, LeMaire SA, Ahn C, Byers PH, Milewicz DM. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46:607–613. doi: 10.1136/jmg.2008.062844. [DOI] [PubMed] [Google Scholar]
- 53.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 54.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 55.Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87:701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, Vriend G, Pattynama PM, Collee M, Majoor-Krakauer D, Poldermans D, Frohn-Mulder IM, Micha D, Timmermans J, Hilhorst-Hofstee Y, Bierma-Zeinstra SM, Willems PJ, Kros JM, Oei EH, Oostra BA, Wessels MW, Bertoli-Avella AM. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 57.Milewicz DM, Regalado E. Thoracic Aortic Aneurysms and Aortic Dissections. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993. [Google Scholar]
- 58.Milewicz DM, Michael K, Fisher N, Coselli JS, Markello T, Biddinger A. Fibrillin-1 (FBN1) mutations in patients with thoracic aortic aneurysms. Circulation. 1996;94:2708–2711. doi: 10.1161/01.cir.94.11.2708. [DOI] [PubMed] [Google Scholar]
- 59.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, Bjeda D, Oswald G, Elias AF, Levy HP, Anderlid BM, Yang MH, Bongers EM, Timmermans J, Braverman AC, Canham N, Mortier GR, Brunner HG, Byers PH, Van Eyk J, Van Laer L, Dietz HC, Loeys BL. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, Varret M, Prakash SK, Li AH, d’Indy H, Braverman AC, Grandchamp B, Kwartler CS, Gouya L, Santos-Cortez RL, Abifadel M, Leal SM, Muti C, Shendure J, Gross MS, Rieder MJ, Vahanian A, Nickerson DA, Michel JB, Jondeau G, Milewicz DM National Heart, Lung, and Blood Institute (NHLBI) Go Exome Sequencing Project. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pannu H, Tran-Fadulu V, Milewicz DM. Genetic basis of thoracic aortic aneurysms and aortic dissections. Am J Med Genet C Semin Med Genet. 2005;139C:10–16. doi: 10.1002/ajmg.c.30069. [DOI] [PubMed] [Google Scholar]
- 62.Varilo T. The age of the mutations in the Finnish disease heritage; a genealogical and linkage disequilibrium study. Helsinki: National Public Health Institute and University of Helsinki; 1999. [Google Scholar]
- 63.Brothman AR, Persons DL, Shaffer LG. Nomenclature evolution: Changes in the ISCN from the 2005 to the 2009 edition. Cytogenet Genome Res. 2009;127:1–4. doi: 10.1159/000279442. [DOI] [PubMed] [Google Scholar]
- 64.Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, Cheung SW, Shen RM, Barker DL, Gunderson KL. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sulonen AM, Ellonen P, Almusa H, Lepisto M, Eldfors S, Hannula S, Miettinen T, Tyynismaa H, Salo P, Heckman C, Joensuu H, Raivio T, Suomalainen A, Saarela J. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011;12:R94. doi: 10.1186/gb-2011-12-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu J, Li G, Peng X, Liu B, Yin B, Tan X, Fan M, Fan W, Qiang B, Yuan J. The cloning and preliminarily functional analysis of the human neurotrimin gene. Sci China C Life Sci. 2004;47:158–164. doi: 10.1360/03yc0072. [DOI] [PubMed] [Google Scholar]
- 67.Simovich MJ, Yatsenko SA, Kang SL, Cheung SW, Dudek ME, Pursley A, Ward PA, Patel A, Lupski JR. Prenatal diagnosis of a 9q34.3 microdeletion by array-CGH in a fetus with an apparently balanced translocation. Prenat diagn. 2007;27:1112–1117. doi: 10.1002/pd.1841. [DOI] [PubMed] [Google Scholar]
- 68.Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991;49:995–1013. [PMC free article] [PubMed] [Google Scholar]
- 69.Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders--cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gloyn AL, Ellard S, Shepherd M, Howell RT, Parry EM, Jefferson A, Levy ER, Hattersley AT. Maturity-onset diabetes of the young caused by a balanced translocation where the 20q12 break point results in disruption upstream of the coding region of hepatocyte nuclear factor-4alpha (HNF4A) gene. Diabetes. 2002;51:2329–2333. doi: 10.2337/diabetes.51.7.2329. [DOI] [PubMed] [Google Scholar]
- 71.Taipale M, Kaminen N, Nopola-Hemmi J, Haltia T, Myllyluoma B, Lyytinen H, Muller K, Kaaranen M, Lindsberg PJ, Hannula-Jouppi K, Kere J. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc Natl Acad Sci U S A. 2003;100:11553–11558. doi: 10.1073/pnas.1833911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bache I, Hjorth M, Bugge M, Holstebroe S, Hilden J, Schmidt L, Brondum-Nielsen K, Bruun-Petersen G, Jensen PK, Lundsteen C, Niebuhr E, Rasmussen K, Tommerup N. Systematic re-examination of carriers of balanced reciprocal translocations: a strategy to search for candidate regions for common and complex diseases. Eur J Hum Genet. 2006;14:410–417. doi: 10.1038/sj.ejhg.5201592. [DOI] [PubMed] [Google Scholar]