

Abstract
Farnesoid X receptor (FXR, Nr1h4) and small heterodimer partner (SHP, Nr0b2) are nuclear receptors that are critical to liver homeostasis. Induction of SHP serves as a major mechanism of FXR in suppressing gene expression. Both FXR−/− and SHP−/− mice develop spontaneous hepatocellular carcinoma (HCC). SHP is one of the most strongly induced genes by FXR in the liver and is a tumor suppressor, therefore, we hypothesized that deficiency of SHP contributes to HCC development in the livers of FXR−/− mice and therefore, increased SHP expression in FXR−/− mice reduces liver tumorigenesis. To test this hypothesis, we generated FXR−/− mice with overexpression of SHP in hepatocytes (FXR−/−/SHPTg) and determined the contribution of SHP in HCC development in FXR−/− mice. Hepatocyte-specific SHP overexpression did not affect liver tumor incidence or size in FXR−/− mice. However, SHP overexpression led to a lower grade of dysplasia, reduced indicators cell proliferation and increased apoptosis. All tumor-bearing mice had increased serum bile acid levels and IL-6 levels, which was associated with activation of hepatic STAT3. In conclusion, SHP partially protects FXR−/− mice from HCC formation by reducing tumor malignancy. However, disrupted bile acid homeostasis by FXR deficiency leads to inflammation and injury, which ultimately results in uncontrolled cell proliferation and tumorigenesis in the liver.
Keywords: farnesoid X receptor, small heterodimer partner, tumor malignancy, bile acids, chronic injury, liver tumor
Introduction
Hepatocellular carcinoma (HCC) accounts for 85% of primary liver cancers and represents a major health problem worldwide for its high prevalence and poor prognosis. Chronic viral infections and hepatotoxic agents are the major risk factors contributing to HCC etiology. Although common pathological changes in HCC are well characterized, which consist of intertwined chronic injury and inflammation, the precise molecular mechanisms underlying HCC development are still unclear (Li and Guo, 2011). Emerging evidence strongly indicate that the molecular pathogenesis of HCC is multi-faceted (El-Serag, 2011, and increase in bile acids promotes HCC formation in rodents {Cameron, 1981 #2797) and is associated with HCC development in humans (Chen et al., 2011).
Farnesoid X receptor (FXR, Nr1h4) and small heterodimer partner (SHP, Nr0b2) are ligand-activated nuclear receptors (NRs) and are important in regulating liver functions, including bile acid metabolism, lipid homeostasis, drug disposition, inflammation, cell growth and differentiation, and tumorigenesis (Zhang et al., 2011; Zhu et al., 2011). Bile acids are endogenous ligands of FXR and FXR is essential in regulating bile acid homeostasis (Makishima et al., 1999; Parks et al., 1999; Sinal et al., 2000; Wang et al., 1999). Increase in bile acids causes liver injury, which may serve as one of the mechanisms of HCC formation in FXR−/− mice by activating cell proliferating pathways, such as β-catenin (Wolfe et al., 2011). Furthermore, liver injury leads to increased IL-6 levels, which can activate STAT3, and STAT3 activation is a well-known mechanism responsible for liver carcinogenesis (Yu et al., 2009). Emerging evidence showed FXR deficiency led to STAT3 activation (Meng et al., 2012; Xu et al., 2012), indicating that STAT3 activation may also be involved in liver carcinogenesis with FXR deficiency.
SHP is highly induced by FXR activation in the liver (Li et al., 2010), and both FXR−/− and SHP−/− mice develop spontaneous HCC (Kim et al., 2007; Yang et al., 2007; Zhang et al., 2008). Increasing evidence indicates that SHP possesses potent tumor-suppressive activity by inhibiting cell proliferation and promoting apoptosis (He et al., 2008; Zhang et al., 2010; Zhang et al., 2008). However, the mechanisms whereby FXR suppresses liver carcinogenesis are not completely clear. Understanding the mechanisms will provide critical scientific basis to help understand HCC carcinogenesis, which may be used to improve the HCC therapeutic strategy. Therefore, we hypothesize that deficiency of SHP contributes to HCC development in FXR−/− mice and increase in SHP expression in FXR−/− mice reduces liver tumorigenesis. In the current study, we have generated FXR−/−/SHPTg mice in which SHP was overexpressed in hepatocytes of FXR−/− mice. With this unique mouse model, we have determined the contribution of SHP to liver tumor development in FXR−/− mice.
Materials and Methods
Animals and treatments
All mice were housed and bred in pathogen-free animal facilities in the Laboratory of Animal Research under a standard 12-hr light/dark cycle with free access to food and autoclaved tap water. All protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kansas Medical Center. Four mouse strains in C57BL/6J genetic background were bred in house and used in this study: wild-type (WT), FXR−/−, SHP-hepatocyte transgenic (SHPTg) and FXR−/−/SHPTg mice. The FXR−/−/SHPTg mice were created by cross-breeding FXR−/− and SHPTg mice. For genotyping, tail DNA extraction and standard PCR amplification were performed using a Sigma RED Extract-N-Amp Tissue PCR Kit obtained from Sigma-Aldrich (St. Louis, MO). The sequence of the primers used for genotyping the FXR allele and SHP transgenic allele were presented in supplemental table 1. Both male and female mice were sacrificed at 3, 6, 12 and 18 months of age with n=14–20/gender/genotype/time point.
Mouse liver tissue processing
Liver tissues were either snap-frozen in liquid nitrogen followed by RNA and protein preparations or fixed in 4% formaldehyde-PBS solution, embedded in paraffin, and cut into 5-μm sections for hematoxylin and eosin (H&E) staining or immunohistochemistry. The pathology analysis was performed by Dr. Ossama Tawfik who is a board-certified pathologist with liver experience at the Department of Pathology and laboratory of Medicine at the University of Kansas Medical Center.
5-bromo-2-deoxyuridine (BrdU) staining
To assess liver cell proliferation, 3- and 12-month-old WT, FXR−/−, SHPTg, and FXR−/−/SHPTg mice were injected intraperitoneally with 50 mg/kg of BrdU (BD Biosciences, San Jose, CA) in PBS daily for 5 days. The livers were harvested 2 hrs after the last injection. BrdU staining results were quantitated by assessing at least 1000 cells per tissue section to calculate the percentage of positive nuclei (brown nuclei) at 400×magnification.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay
To determine cell apoptosis, the TUNEL assay was performed on liver sections using a TUNEL kit (In Situ Cell Death Detection Kit AP, Roche Diagnostics, Indianapolis, USA) according to the manufacturer’s recommendations. The numbers of apoptotic hepatocytes were counted in 10 randomly chosen 400X magnification fields using an Olympus BX51 microscope (Hitschfel Instruments, Inc., St. Louis, MO), and the apoptotic TUNEL index was determined as number of apoptotic cells per 1000 cells (‰).
Arginase 1 Immunohistochemistry staining
Immunohistochemistry for arginase 1 was performed using a rabbit polyclonal antibody against arginase-1 (H-52: sc-20150, Santa Cruz, TX) at 1:100 dilution with a labeled streptavidin- biotin-peroxidase complex technique. Briefly, liver tissue sections were deparaffinized and hydrated in xylene and descending grades of alcohol. After rinsing in PBS, antigen retrieval was performed by boring the tissue sections in citrate buffer, pH 6.0 for 10 min. The endogenous peroxidase activity was quenched in 3% hydrogen peroxide for 10 min and endogenous biotin was blocked with avidin/biotin blocking kit (VECTASTAIN, SP-2001) for 30 min before blocking by 5% goat serum for 30min, and subsequently incubated with primary antibody for 1.5 hrs at room temperature. The antibody reaction was detected with the avidin-biotin detection kit using diaminobenzidine (DAB) as chromogen. Sections were counterstained with hematoxylin for 3 min before checked under microscope.
Liver IL-6 levels and serum ALT activity, total bile acids, and IL-6 level
Serum ALT activity and bile acid levels were determined using commercially available kits (Pointe Scientific, Canton, MI and Bio-Quant, San Diego, CA, respectively) according to the manufacturers’ instructions. Serum and liver IL-6 levels were measured with a custom Milliplex MAP kit for mouse cytokines (Millipore Corporation, Billerica, MA) using the Bio-Plex 200 System (Bio-Rad Laboratories).
RNA isolation and quantitative real-time PCR (Q-PCR)
Total RNA was isolated from frozen livers by TRIzol reagent. The mRNA expression levels of genes were quantified by Q-PCR using SYBR green chemistry (Fermentas, Glen Burnie, MD) and normalized to Gapdh mRNA levels. The names of the genes and the primers are listed in Supplemental Table 1.
Western blot analysis
Proteins were isolated and quantitated by standard method and the Western blot analysis was performed as previously described on total, nuclear and cytoplasmic proteins (Maran et al., 2009). Antibodies are described in Supplemental Table 2.
Statistical analysis
All data were presented as mean ± SD, and were analyzed by one-way analysis of variance followed by the Student-Newman-Keuls test. P < 0.05 was considered statistically significant.
Results
Generating FXR−/−/SHPTg mice
We have generated the FXR−/−/SHPTg mice in which SHP was overexpressed in hepatocytes of FXR−/− mice. The successful generation of the FXR−/−/SHPTg mice was confirmed by PCR-based genotyping (Figure 1A) and by measuring mRNA levels of FXR and SHP (Figure 1B). The liver/body weight ratio remained similar among all four strains at 3 and 6 months of age. Starting from 12 months, the liver/body weight ratio was much higher in the FXR−/− and FXR−/−/SHPTg mice (Figure 1C).
Figure 1. Generation of the FXR−/−/SHPTg mice.

FXR−/−/SHPTg mice were generated by cross-breeding FXR−/− and SHPTg mice. (A) The correct genotypes of the mice were confirmed by PCR-based genotyping method. (B) The mRNA expression levels of Fxr and Shp in the livers of these mice. (C) The liver/body weight rate of these mice. An asterisk means P < 0.05, and a double asterisk means P < 0.01.
Overexpression of SHP did not reduce tumor incidence or size, but decreased tumor malignancy
Liver tumor development was examined in WT, FXR−/−, SHPTg, and FXR−/−/SHPTg mice at 3, 6, 12 and 18 months of age. Grossly identifiable liver tumors were not found in WT or SHPTg mice at all ages examined, but tumors were found in FXR−/− and FXR−/−/SHPTg mice at 12 and 18 months of age with no gender difference (Figure 2A). Interestingly, the tumor incidence in FXR −/− mice [9 of 16 (56.3%)] is slightly lower than that in FXR −/−/SHPTg mice [9 of 14 (64.3%)] at 12-month-old age. However, both strains develop 100% tumors at 18-month-old age (Table 1). The overexpression of SHP cannot prevent the tumor development in FXR−/− mice. Liver pathology in 18-month old, tumor-bearing mice included portal and parenchymal inflammation, hepatocellular dysplasias and HCC (Figure 2B), as well as steatosis, bile ductular proliferation, patchy parenchymal necrosis, bile duct adenoma, and bile duct carcinoma (data not shown). As mentioned earlier, the liver/body weight ratio increased dramatically in FXR−/− and FXR−/−/SHPTg, but not in WT and SHPTg mice at 12 and 18 months of age. However, hepatocellular dysplasias, portal and parenchymal inflammatory cell infiltration were found ~20–40% less in FXR−/−/SHPTg mice than in FXR−/− mice, indicating that SHP overexpression likely reduces tumor malignancy (Table 2). Arginase immunohistochemistry staining showed that arginase-1 immunostaining was stronger in FXR−/− mouse livers than in FXR−/−/SHPTg mouse livers at 18-month old (Supplemental Figure 1).
Figure 2. Liver tumor development in WT, FXR−/−, SHPTg and FXR−/−/SHPTg mice.

(A) Representative liver morphology of WT, FXR−/−, SHPTg and FXR−/−/SHPTg mice at 18-month-old shows grossly identified liver tumor nodules (arrows). The scale bar indicates 1cm. (B) Liver H&E staining indicates portal inflammation (yellow arrows), hepatocyte dysplasia (green arrows), and HCC (black arrows). The representative pictures were from FXR−/−/SHPTg mice at 18-month old. Magnification, 200X and 400X. (C) Quantification of the hepatocyte BrdU incorporation index from livers of 3- and 12-month-old mice of each genotype. The representative pictures are from livers of 3-month-old FXR−/− and FXR−/−/SHPTg mice. An asterisk or pound indicates P < 0.05 between WT and FXR−/− or FXR−/−/SHPTg groups at the same age. A dollar sign means P < 0.05 between 3- and 12-month-old mice of the same genotype. (D) Quantification of TUNEL assay results from livers of 3- and 12-month-old mice from each genotype. The representative pictures are for livers of 3-month-old FXR−/− and FXR−/−/SHPTg mice. An asterisk indicates P < 0.05 between WT and other groups of the same age. A pound sign indicates P < 0.05 between 3-month and 12-month-old mice of the same genotype. A dollar sign indicates P < 0.05 between FXR−/− and FXR−/−/SHPTg mice of the same age.
Table 1.
Tumor incidence in 12- and 18-month old mice
| Group | 12-month old | 18-month old | ||
|---|---|---|---|---|
| Mouse number | Tumor incidence (%) | Mouse number | Tumor incidence (%) | |
| WT | 16 | 0 | 18 | 0 |
| FXR−/− | 16 | 56.3 | 20 | 100 |
| SHPTg | 16 | 0 | 18 | 0 |
| FXR−/−/SHPTg | 14 | 64.3 | 18 | 100 |
Note: Mice in each group were sacrificed at 12 and 18 months of age with n=14–20 per group.
Table 2.
Summarized results of pathology for 18-month old male mice
| Groups | Rate of HCC or dysplasia (tumor early stage) | Rate of inflammation (portal) | Rate of inflammation (parenchymal) | Rate of bile ductular proliferation | Rate of necrosis |
|---|---|---|---|---|---|
| Wild Type (n=6) | N/A | N/A | N/A | N/A | N/A |
| FXR Knockout (n=10) | HCC (5/10), high grade dysplasia (5/10) | portal inflammation (6/10): mild (2/10), moderate (1/10), or marked (3/10) | parenchymal inflammaiton (3/10): mild (1/10) or marked (2/10) | 2/10 | 3/10 |
| SHP Transgenic (n=8) | N/A | N/A | N/A | N/A | N/A |
| FXR Knockout/SHP Transgenic (n=9) | HCC (5/9), high grade dysplasia (1/9), low grade dysplasia (3/9) | portal inflammation (4/9): mild (2/9), moderate (1/9), or marked (1/9) | N/A | 1/9 | 1/9 |
Cell proliferation and apoptosis
Hepatocyte proliferation, assessed by BrdU incorporation, was examined in 3- and 12-month old mice. At 3-month old, BrdU staining markedly increased in FXR−/− and FXR−/−/SHPTg mice, but not in WT and SHPTg mice (Figure 2C). At 12 months of age, hepatocyte proliferation was slightly increased only in FXR−/− mice, but not in other strains. We then determined the degree of apoptosis in livers of these mice at 3 and 12 months of age by TUNEL assay (Figure 2D). In agreement with the previous report (Zhang et al., 2010), SHP overexpression increased the degree of apoptosis in both 3- and 12-month old mice. Interestingly, both FXR−/− and FXR−/−/SHPTg mice markedly increased apoptosis at 3 months of age, but this trend did not continue with aging, as the 12-month old FXR−/− and FXR−/−/SHPTg mice had a lower degree of apoptosis than the 3-month old mice had, albeit still higher than WT mice at this age.
Serum ALT and BA levels
Both serum ALT activity and bile acid levels were normal in WT and SHPTg mice in all ages examined, but were markedly increased in FXR−/− and FXR−/−/SHPTg mice in an age-dependent manner (Figure 3).
Figure 3. Serum ALT activity and serum bile acid (BA) levels.
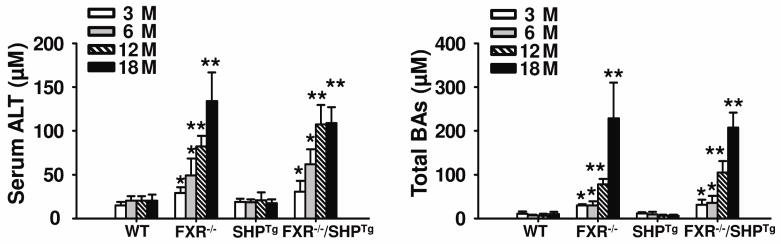
ALT activity (A) and BA levels (B) were determined by commercially available kits in WT, FXR−/−, SHPTg and FXR−/−/SHPTg mice at 3-, 6-, 12- and 18-month old. An asterisk indicates P < 0.05 and double asterisks indicate P < 0.01 between WT and FXR−/− or FXR−/−/SHPTg mice.
Cyclin D1 levels
Cyclin D1 is critical for cell proliferation and has been shown to be a negative target gene of SHP (Zhang et al., 2008). Levels of CyclinD1 mRNA were increased in livers of 12- and 18-month old FXR−/− and FXR−/−/SHPTg mice, but not in younger mice (Figure 4A). Levels of CyclinD1 protein were determined in livers of 3- and 18-month old mice, and the results showed that no difference was found at 3-month old, but at 18-month old, FXR−/− and FXR−/−/SHPTg mice had higher CyclinD1 protein levels (Figure 4B).
Figure 4. The mRNA and protein expressions of Cyclin D1.

(A) The Cyclin D1 mRNA levels were determined by the Q-PCR method in WT, FXR−/−, SHPTg and FXR−/−/SHPTg mice at 3-, 6-, 12- and 18-month old. (B) The protein levels of Cyclin D1 were determined in WT, FXR−/−, SHPTg and FXR−/−/SHPTg mice at 3- and 18-month old. An asterisk indicates P < 0.05.
Activation of STAT3 in the liver of FXR−/− and FXR−/−/SHPTg mice
Despite histologic evidence of reduced tumor malignancy in SHP-overexpressing mice, tumor incidence and size was similar in FXR−/− mice regardless of SHP expression levels. This result indicates a cell-proliferative pathway mainly regulated by FXR is responsible for early development of liver tumors. To elucidate the early events during liver carcinogenesis mediated by FXR deficiency, we focused on determining the activation of several signaling pathways that are known to contribute to HCC development in 3-month old mice. STAT3 activation, revealed by Tyr705 and Ser727 phosphorylation, was increased in FXR−/− and FXR−/−/SHPTg mice, whereas total STAT3 protein levels remained unchanged (Figure 5A). Moreover, phosphorylated JAK2, the up-stream activator of STAT3, was increased in FXR−/− and FXR−/−/SHPTg mouse livers and decreased in SHPTg mice (Figure 5A). The protein levels of glycoprotein 130 receptor (GP130), the IL-6 receptor that activates the STAT3 pathway, were slightly decreased in livers of SHPTg mice only (Figure 5A). Protein levels of total and phosphorylated STAT1, total JAK1 (receptor to activate STAT1), total and phosphorylated mTOR and its pathway components (Rictor, Raptor, and GβL), and nuclear and cytoplasm nuclear factor-Kappa B (NF-κB), were not changed in these mice except for reduced phosphorylated JAK1 in FXR deficient mice (Figure 5B, 5C, 5D).
Figure 5. STAT3, but not STAT1, mTOR or NF-κB was activated in livers of FXR−/− and FXR−/−/SHPTg mice.

Western Blot analysis of total hepatic protein levels from 3-month-old WT, FXR−/−, SHPTg and FXR−/−/SHPTg mice. (A) Representative results of phosphorylated STAT3, total STAT3, GP130, phosphorylated JAK2, and total JAK2. (B) Phosphorylated STAT1, total STAT1, phosphorylated JAK1, and total JAK1. (C) Phosphorylated mTORs, total mTOR, Rictor, Raptor, and GβL. (D) Hepatic cytoplasmic and nuclear protein levels of NF-κB in 3-month-old WT, FXR−/−, SHPTg and FXR−/−/SHPTg mice. Each band represents a pooled sample from 3 individual mice with the same genotype, and β-actin levels were used as internal loading control.
It is clear that STAT3 is activated by FXR deficiency. Therefore, we determined the mRNA levels of several STAT3 target genes, c-Myc, p21, Survivin and Socs3 (Yu et al., 2009), in livers of all genotypes at 3, 6, 12 and 18 months. As anticipated, the mRNA levels of c-Myc, p21 and Survivin, but not Socs3, were induced in FXR−/− and FXR−/−/SHPTg mice (Figure 6A). Socs3, a feedback inhibitor of the JAK2/STAT3 pathway, were slightly decreased in livers of FXR−/− and FXR−/−/SHPTg mice. STAT3 is known to be activated by IL-6, and the increased bile acids following FXR deficiency is known to induce inflammation. Thereby, we have measured the IL-6 protein levels in serum in 3-, 6-, 12- and 18-month old WT, SHPTg and FXR−/− mice and the results showed that at all ages, the FXR deficiency increased IL-6 levels in the serum (Figure 6B), and at 3-month old, deficiency of FXR also tended to increase IL-6 levels in the liver without statistical significance (Figure 6B). We then measured the mRNA levels of IL-1β and IL-6, as well as IL-1α. There was no change in the mRNA levels of IL-1α (data not shown), but increased levels of IL-1β and IL-6 were associated with FXR deficiency (Figure 6C).
Figure 6. Activation of JAK2/STAT3 signaling pathway in livers of WT, FXR−/−, SHPTg, and FXR−/−/SHPTg mice at 3-, 6-, 12-, and 18-month old.

(A) Q-PCR analysis of mRNA expression levels of STAT3 downstream target genes, c-Myc, p21, Survivin, and Socs3. (B) Serum IL-6 levels in WT, SHPTg and FXR−/− mice at 3-, 6-, 12- and 18-month old; liver IL-6 protein levels in WT and FXR−/− mice at 3-month old. (C) Q-PCR analysis of mRNA expression levels of IL-1β and IL-6 in livers of each genotype at 3-, 6-, 12- and 18-month old. An asterisk indicates P < 0.05 and double asterisks mean P < 0.01 between WT and FXR−/− or FXR−/−/SHPTg mice.
Discussion
In the present study, we have tested the hypothesis that deficiency of SHP contributes to the HCC development in FXR−/− mice and increase in SHP expression in FXR−/− mouse livers reduces liver tumorigenesis. The results showed that overexpression of SHP did not reduce tumor incidence or tumor size, but reduced tumor malignancy, likely through enhanced apoptosis. FXR deficiency increased bile acid and IL-6 levels, as well as JAK2/STAT3 activation, which is likely a mechanism contributing to HCC development following FXR deficiency. Together, these findings reveal that molecular mechanisms of spontaneous carcinogenesis in livers of FXR−/− mice are not only due to decreased SHP expression, but also due to activation of cell proliferation pathway(s) following cell injury and inflammation.
Both FXR−/− and SHP−/− mice develop spontaneous liver tumors (He et al., 2008; Kim et al., 2007; Yang et al., 2007; Zhang et al., 2008). SHP is a well-known FXR target gene (Guo et al., 2003), and over-expression of SHP inhibits HCC foci formation and arrests HCC tumor growth in xenografted nude mice models (He et al., 2008; Zhang et al., 2008). Therefore, over-expression of SHP in livers of FXR−/− mice was used to determine if SHP deficiency is a major mechanism leading to HCC development in FXR−/− mice. In this study, although the FXR−/−/SHPTg mice had similar tumor incidence and tumor size compared to FXR−/− mice, pathology analysis revealed that FXR−/−/SHPTg mice had fewer dysplasia/HCC cells, less bile ductular proliferation and less portal/parenchymal inflammatory cell infiltration in the liver than FXR−/− mice had. Arginase-1 immunostaining was stronger in FXR−/− mouse livers than in FXR−/−/SHPTg mouse livers at 18-month old. Arginase-1 has proven to be a new immunohistochemical marker in HCC and is highly expressed in HCC progression (Timek et al., 2012; Yan et al., 2010). These results indicate that SHP may partially protect FXR−/− mice from the HCC formation by reducing tumor malignancy. Thus, our current results indicate that overexpression of SHP does not prevent tumor initiation and promotion but likely reduces tumor progression.
The current study showed that CyclinD1 expression was induced only in livers of old FXR−/− mice. Despite that SHP inhibits CyclinD1 expression in vitro (Zhang et al., 2008), our study showed that overexpression of SHP in vivo did not affect CyclinD1 levels. Instead, a previous study reported that Wnt-β-catenin activated CyclinD1 in FXR−/− mice (Wolfe et al., 2011). Therefore, the lack of Cyclin D1 induction may be a reflection of multifaceted regulation of Cyclin D1 following FXR deletion. Consistent with previous observations (Kim et al., 2007; Zhang et al., 2008), increased apoptosis were detected in livers of FXR−/− and SHPTg mice. In addition, despite a similar trend of reduction of apoptosis with aging in livers of FXR−/− and FXR−/−/SHPTg mice, more apoptotic cells were found in aged FXR−/−/SHPTg than in FXR−/− mice. This relatively higher degree of apoptosis likely is a main mechanism contributing to the reduced malignancy of HCC in FXR−/−/SHPTg mice.
However, both FXR−/− and FXR−/−/SHPTg mice showed similarly high tumor incidence (100% at 18 months of age), similar tumor size, marked increase in liver/body weight ratio, and serum ALT activity and bile acid levels. The hepatocyte proliferation index was also similar between FXR−/− and FXR−/−/SHPTg mice. Furthermore, JAK2/STAT3 was specifically activated in livers of FXR−/− and FXR−/−/SHPTg mice as early as 3 months of age, without showing signs of JAK1/STAT1, mTOR, or NF-κB activation. While we were preparing this manuscript, a recent publication showed inhibition of STAT3 activaty following FXR activation in vitro, which confirmed our results (Xu et al., 2012). We further showed that STAT3 target genes, including c-Myc, p21 and Survivin, were induced in livers of FXR−/− mice. These results showed that the JAK2/STAT3 pathway is activated by FXR deficiency, which is not prevented by SHP overexpression. Consistent activation of JAK2/STAT3 signaling pathway may serve as a mechanism of liver carcinogenesis in FXR−/− mice.
Increased inflammatory cytokines, especially IL-6, have been shown to activate STAT3. FXR deficiency is known to increase levels of bile acids, which may lead to cell injury and inflammation. Indeed, mRNA levels of the inflammatory genes, IL-1β and IL-6, were induced in livers of FXR−/− and FXR−/−/SHPTg mice with aging. It is likely that elevated bile acids induce IL-6, which in turn activates STAT3. IL-6 is a potent inflammatory cytokine playing a critical role in carcinogenesis (Naugler and Karin, 2008), and its levels were dramatically increased in HCC patients with higher IL-6 levels associated with primary liver cancer progression (Porta et al., 2008). Our results showed no activation of JAK1/STAT1, but the JAK2/STAT3 signaling pathway was consistently activated in livers of FXR−/− mice. STAT3, which is most commonly activated by IL-6, serves as a crucial regulator of inflammation and carcinogenesis (Wang et al., 2011). Moreover, STAT3 works as a point of convergence of numerous oncogenic signaling pathways during HCC development (Wang et al., 2011; Yu et al., 2009). Upon activation, STAT3 induces the transcription of target genes crucial for cell proliferation. We showed that c-Myc, P21 and survivin, target genes of STAT3, were highly induced, especially in livers of aging FXR−/− mice. These results demonstrate that increased bile acids and IL-6 may contribute to activating the JAK2/STAT3 pathway in FXR−/− mice. Survivin belongs to the inhibitor of apoptosis protein (IAP) family, which inhibits caspases and blocks cell death. Studies have found that survivin is highly expressed in cancer and is associated with a poorer clinical outcome. In FXR−/− mice, survivin expression has been increased with aging, and overexpression of SHP could reduce the expression levels of survivin. These results indicate that SHP may induce apoptosis through inhibition the activation of survivin.
Collectively, our results demonstrate that SHP does not prevent FXR deficiency-induced HCC incidence and size, but significantly reduces tumor malignancy, indicating that overexpression of SHP may reduce tumor progression. Furthermore, the mechanism by which SHP protects from liver tumor progression may likely due to enhanced apoptosis. FXR deficiency increases bile acid levels, which may be a fundamental factor to induce inflammation, cell proliferation, and liver tumorigenesis.
Supplementary Material
The representative staining for arginase-1 on the liver of WT, FXR−/− and FXR−/−/SHPTg mice at 18-month-old (original magnification ×400).
Highlight.
Overexpression of SHP does not prevent HCC incidence and size in FXR KO mice but reduces malignancy
Increased SHP promotes apoptosis
Increased bile acid and inflammation maybe fundamental factors leading to HCC formation with FXR deficiency
Acknowledgments
This study was supported by the NIH fund [DK081343 & DK090036 (GLG)].
List of Abbreviations
- ALT
alanine aminotransferase
- GP130
glycoprotein 130
- JAK
Janus kinases
- FXR
farnesoid X receptor
- FXR−/−
FXR knockout
- HCC
hepatocellular carcinoma
- IL-1α
interleukin-1α
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- Q-PCR
quantitative real-time PCR
- SOCS3
suppressor of cytokine signaling 3
- SHP
small heterodimer partner
- SHP−/−
SHP knockout
- SHPTg
SHP transgenic
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cameron R, Imaida K, Ito N. Promotive effects of deoxycholic acid on hepatocarcinogenesis initiated by diethylnitrosamine in male rats. Gann. 1981;72:635–636. [PubMed] [Google Scholar]
- Chen T, Xie G, Wang X, Fan J, Qiu Y, Zheng X, Qi X, Cao Y, Su M, Wang X, Xu LX, Yen Y, Liu P, Jia W. Serum and Urine Metabolite Profiling Reveals Potential Biomarkers of Human Hepatocellular Carcinoma. Molecular & Cellular Proteomics. 2011:10. doi: 10.1074/mcp.M110.004945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, Kliewer SA, Gonzalez FJ, Sinal CJ. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem. 2003;278:45062–45071. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- He N, Park K, Zhang Y, Huang J, Lu S, Wang L. Epigenetic inhibition of nuclear receptor small heterodimer partner is associated with and regulates hepatocellular carcinoma growth. Gastroenterology. 2008;134:793–802. doi: 10.1053/j.gastro.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007;28:940–946. doi: 10.1093/carcin/bgl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Guo GL. Role of Class II nuclear receptors in liver carcinogenesis. Anti-cancer agents in medicinal chemistry. 2011;11:529–542. doi: 10.2174/187152011796011064. [DOI] [PubMed] [Google Scholar]
- Li G, Thomas AM, Hart SN, Zhong X, Wu D, Guo GL. Farnesoid X receptor activation mediates head-to-tail chromatin looping in the Nr0b2 gene encoding small heterodimer partner. Mol Endocrinol. 2010;24:1404–1412. doi: 10.1210/me.2010-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Maran RRM, Thomas A, Roth M, Sheng Z, Esterly N, Pinson D, Gao X, Zhang Y, Ganapathy V, Gonzalez FJ, Guo GL. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther. 2009;328:469–477. doi: 10.1124/jpet.108.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Wang X, Gan Y, Zhang Y, Zhou H, Ness CV, Wu J, Lou G, Yu H, He C, Xu R, Huang W. Deletion of IFNgamma enhances hepatocarcinogenesis in FXR knockout mice. J Hepatol. 2012;57:1004–1012. doi: 10.1016/j.jhep.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109–119. doi: 10.1016/j.molmed.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- Porta C, De Amici M, Quaglini S, Paglino C, Tagliani F, Boncimino A, Moratti R, Corazza GR. Circulating interleukin-6 as a tumor marker for hepatocellular carcinoma. Ann Oncol. 2008;19:353–358. doi: 10.1093/annonc/mdm448. [DOI] [PubMed] [Google Scholar]
- Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- Timek DT, Shi J, Liu H, Lin F. Arginase-1, HepPar-1, and Glypican-3 are the most effective panel of markers in distinguishing hepatocellular carcinoma from metastatic tumor on fine-needle aspiration specimens. American journal of clinical pathology. 2012;138:203–210. doi: 10.1309/AJCPK1ZC9WNHCCMU. [DOI] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- Wang H, Lafdil F, Kong X, Gao B. Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target. International journal of biological sciences. 2011;7:536–550. doi: 10.7150/ijbs.7.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther. 2011;338:12–21. doi: 10.1124/jpet.111.179390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Huang G, Gong W, Zhou P, Zhao Y, Zhang Y, Zeng Y, Gao M, Pan Z, He F. FXR ligands protect against hepatocellular inflammation via SOCS3 induction. Cellular Signalling. 2012;24:1658–1664. doi: 10.1016/j.cellsig.2012.04.015. [DOI] [PubMed] [Google Scholar]
- Yan BC, Gong C, Song J, Krausz T, Tretiakova M, Hyjek E, Al-Ahmadie H, Alves V, Xiao SY, Anders RA, Hart JA. Arginase-1: a new immunohistochemical marker of hepatocytes and hepatocellular neoplasms. Am J Surg Pathol. 2010;34:1147–1154. doi: 10.1097/PAS.0b013e3181e5dffa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67:863–867. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2011;1812:893–908. doi: 10.1016/j.bbadis.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Soto J, Park K, Viswanath G, Kuwada S, Abel ED, Wang L. Nuclear receptor SHP, a death receptor that targets mitochondria, induces apoptosis and inhibits tumor growth. Mol Cell Biol. 2010;30:1341–1356. doi: 10.1128/MCB.01076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu P, Park K, Choi Y, Moore DD, Wang L. Orphan receptor small heterodimer partner suppresses tumorigenesis by modulating cyclin D1 expression and cellular proliferation. Hepatology. 2008;48:289–298. doi: 10.1002/hep.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Li F, Guo GL. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacological Research. 2011;63:259–265. doi: 10.1016/j.phrs.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The representative staining for arginase-1 on the liver of WT, FXR−/− and FXR−/−/SHPTg mice at 18-month-old (original magnification ×400).