

Abstract
Endothelial dysfunction occurs in conduit and cerebral resistance arteries with advancing age. Lifelong caloric restriction (CR) can prevent the onset of age-related dysfunction in many tissues, but its effects on cerebral resistance artery function, as compared with conduit artery function, have not been determined. We measured endothelium-dependent dilation (EDD) in the carotid artery and middle cerebral artery (MCA) from young (5–7 months), old ad libitum fed (AL, 29–32 months), and old lifelong CR (CR, 40 % CR, 29–32 months) B6D2F1 mice. Compared with young, EDD for old AL was 24 % lower in the carotid and 47 % lower in the MCA (p < 0.05). For old CR, EDD was not different from young in the carotid artery (p > 0.05), but was 25 % lower than young in the MCA (p < 0.05). EDD was not different between groups after NO synthase inhibition with Nω-nitro-l-arginine methyl ester in the carotid artery or MCA. Superoxide production by the carotid artery and MCA was greater in old AL compared with young and old CR (p < 0.05). In the carotid, incubation with the superoxide scavenger TEMPOL improved EDD for old AL (p > 0.05), with no effect in young or old CR (p > 0.05). In the MCA, incubation with TEMPOL or the NADPH oxidase inhibitor apocynin augmented EDD in old AL (p < 0.05), but reduced EDD in young and old CR (p < 0.05). Thus, age-related endothelial dysfunction is prevented by lifelong CR completely in conduit arteries, but only partially in cerebral resistance arteries. These benefits of lifelong CR on EDD result from lower oxidative stress and greater NO bioavailability.
Keywords: Endothelium-dependent dilation, Aging, Oxidative stress, Nitric oxide, Middle cerebral artery, Carotid
Introduction
Advancing age is a major risk factor for cardiovascular and cerebrovascular diseases (Wolf et al. 1991; Lakatta 2003), and impairments in arterial function with age contribute to these increases in risk (Choi et al. 1998; Lakatta 2003). Furthermore, Alzheimer’s is a disease of aging and impaired cerebral artery function is thought to be an important contributor to Alzheimer’s disease progression (Humpel 2011). As such, it is important to identify interventions and therapeutic targets that could prevent the arterial impairments that occur with advancing age. Lifelong caloric restriction (CR) increases lifespan and reduces the onset of age-related diseases including prevention of cardiovascular disease risk factors and cognitive decline (Halagappa et al. 2007; Carter et al. 2009; Weindruch and Sohal 1997; Masoro 2005). It is likely that these beneficial effects of lifelong CR result from maintained function of both conduit and cerebral resistance arteries with age. However, the effect of lifelong CR on cerebral arterial function has not been fully elucidated.
A key feature of age-related arterial dysfunction is reduced endothelium-dependent dilation (EDD). Older humans and rodents exhibit impaired EDD in conduit arteries as well as cerebral arteries (Lesniewski et al. 2009; Csiszar et al. 2007a; Blackwell et al. 2004; Modrick et al. 2009; Hatake et al. 1990; Eskurza et al. 2004; Celermajer et al. 1994). It has been shown previously that lifelong CR can prevent the age-related decline in EDD in the aorta (Csiszar et al. 2009), but the effect of CR on resistance artery EDD has not been studied. With advancing age, endothelial dysfunction primarily results from reduced nitric oxide (NO) bioavailability (Taddei et al. 2001). Not only is NO the key dilator produced by the endothelium (Vanhoutte 2003), but endothelial-derived NO can also decrease the expression of amyloid precursor proteins (Austin et al. 2010), the accumulation of which is a characteristic feature of Alzheimer’s disease (O’Brien and Wong 2011). Reactive oxygen species, particularly superoxide, reduce NO bioavailability and oxidative stress has been implicated as a major factor leading to impaired EDD with aging (Taddei et al. 2001). Expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a key producer of superoxide in vascular tissue, is increased in conduit and cerebral arteries with advancing age (Miller et al. 2005; Durrant et al. 2009), and inhibiting NADPH oxidase in conduit arteries from old animals improves EDD (Durrant et al. 2009; Rippe et al. 2010). Lifelong CR prevents the increase in oxidative stress with aging in a number of tissues including large arteries (Csiszar et al. 2009; Sohal and Weindruch 1996). However, the effect of lifelong CR on oxidative stress in resistance arteries has not been explored.
We hypothesize that old ad libitum (AL) fed mice would have impaired EDD in conduit (carotid) and cerebral resistance (middle cerebral artery, MCA) arteries compared with young AL fed controls, and the magnitude of this dysfunction would be similar between arteries. We further hypothesized that lifelong CR would prevent the decline in EDD with age, such that carotid and MCA EDD would not be different between old CR and young control mice. We hypothesized that CR would maintain EDD with age by preventing the decline in NO bioavailability and the onset of NADPH oxidase-mediated oxidative stress.
Methods
Animals
Young (5–7 months) and old AL (29–32 months) and old lifelong CR (CR, 29–32 months) male B6D2F1 mice were obtained from the National Institute on Aging rodent colony. In NIH lifelong CR mice, calorie restriction is initiated at 14 weeks of age at ~10 % below AL, increased to ~25 % restriction at 15 weeks and to ~40 % restriction at 16 weeks, and maintained throughout the life of the animal. All mice were housed for at least 2–3 months in an animal care facility at the University of Utah on a 12:12 light:dark cycle at 24 °C and fed the appropriate NIH 31(young or old AL) or NIH 31 fortified diet (old CR). All animal procedures conformed to the Guide to the Care and Use of Laboratory Animals (version 8, revised 2011) and were approved by the Utah Animal Care and Use Committee.
Endothelium-dependent dilation: effect of NO and oxidative stress
Measurements of EDD and endothelial independent dilation (EID) in isolated carotid arteries and MCAs studied ex vivo were performed using a method previously described in detail (Lesniewski et al. 2009; Donato et al. 2009, 2011). Briefly, mice were euthanized by exsanguination via cardiac puncture while under isoflurane anesthesia. Carotid arteries and MCAs were excised and placed in myograph chambers (DMT) with physiological salt solution (PSS) that contained 145.0 mM NaCl, 4.7 mM KCl, 2.0 mM CaCl2, 1.17 mM MgSO4, 1.2 mM NaH2PO4, 5.0 mM glucose, 2.0 mM pyruvate, 0.02 mM EDTA, 3.0 mM MOPS buffer, and 1 g/100 ml BSA, pH 7.4 at 37 °C, cannulated onto glass micropipettes and secured with nylon (11-0) suture. Once cannulated, arteries were warmed to 37 °C, pressurized and allowed to equilibrate for ~1 h. All arteries were submaximally preconstricted with phenylephrine (2 μM) and increases in luminal diameter in response to increasing concentrations of the endothelium-dependent dilator, acetylcholine (ACh: 1 × 10-9 to 1 × 10-4 M) and endothelium-independent dilator, sodium nitroprusside (SNP: 1 × 10-10 to 1 × 10-4 M) were determined. Responses to ACh were repeated in the presence of the NO synthase (NOS) inhibitor, Nω-nitro-l-arginine methyl ester (l-NAME, 0.1 mM, 30-min incubation) to determine the approximate contribution of NO. To determine the effect of superoxide (oxidative stress) on EDD, responses to ACh were measured following a 60-min incubation in the presence of the superoxide scavenger, TEMPOL (1 mM; Didion et al. 2006; Zhang et al. 2003; Qamirani et al. 2005), in different carotid or MCA segments than those initially incubated with l-NAME. Measurement of ACh responses were repeated in TEMPOL-treated arteries after l-NAME addition. The contribution of superoxide produced by NADPH oxidase to EDD was assessed by preincubation with apocynin (1 mM, 60 min), a NADPH oxidase inhibitor (Durrant et al. 2009; Rippe et al. 2010). EDD and EID results are expressed as the percent of possible dilation (Lesniewski et al. 2009; Durrant et al. 2009).
Arterial superoxide production
Production of superoxide in the carotid artery and MCA was measured by electron paramagnetic resonance (EPR) spectrometry using the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH, Alexis Biochemicals). Stock solutions of CMH were prepared in ice-cold deoxygenated Krebs–HEPES buffer (mmol L-1: NaCl 99.01, KCl 4.69, CaCl2 2.50, MgSO4 1.20, K2HPO4 1.03, NaHCO3 25.0, glucose 11.10, Na–HEPES 20.00; pH 7.4) containing 0.1 mmol L-1 diethylenetriamine-penta-acetic acid, 5 μmol L-1 sodium diethyldithiocarbamate and pretreated with Chelex (Sigma) to minimize auto-oxidation of the spin probe. A 3-mm section of the carotid artery and a 5-mm section of the MCA were excised, separately incubated for 60 min at 37 °C in 200 μL Krebs–HEPES buffer containing 0.5 mmol L-1 CMH, and analyzed immediately on an EMX Plus EPR spectrometer (Bruker, Rheinstetten, Germany). Instrument settings were: microwave frequency 9.83 GHz, centerfield 3480 G, sweep 80 G, modulation amplitude 3.3 G, microwave power 40 mW, microwave attenuation 7, and receiver gain 30. A total of six sweeps were conducted lasting 8.7 s per sweep. The running average of the six sweeps was collected with the double integration (area under and over the baseline) of the triplet used to display the magnitude of the signal. The double integration of each sample was adjusted by subtracting the double integration of a blank control measured the day of analysis. The magnitude of this signal directly relates to the amount of superoxide that has been trapped by the CMH. Data were normalized to the mean of the young samples of the appropriate artery type measured on the day of analysis.
Exogenous NADPH
Carotid arteries and MCAs were prepared as described above for EDD measurements. After preconstriction with phenylephrine (2 μM), the change in lumen diameter was determined in response to increasing concentrations of NADPH (1 × 10-7 to 1 × 10-4 M; Didion and Faraci 2002; Trott et al. 2011). This change in diameter is presented as a percentage of the preconstricted diameter for each artery.
Statistics
For animal and vessel characteristics, group differences were determined by one-way analysis of variance (ANOVA). For all dose responses, group differences were determined by repeated-measures ANOVA. A least significance difference post hoc test for equal variances was used for preplanned comparisons where appropriate. To compare the magnitude of impairment in EDD between arteries (Fig. 2), maximal responses to ACh were normalized to the mean maximal response of the young for that artery. EC50 values were calculated by fitting each dose response to a four-parameter logistic equation. EC50 values for the MCA response to ACh in the presence of l-NAME were not calculated as these responses do not fit a logistic equation. Data are presented as mean ± SEM. Significance was set at p < 0.05.
Fig. 2.

Maximal response to acetylcholine in carotid and middle cerebral arteries (MCA) from young, old ad libitum fed (AL), and old lifelong calorically restricted (CR) mice (n = 9–16 per group). Each maximal response was normalized to the mean response of the young group for that artery. †p < 0.05 vs. old AL carotid, ‡p < 0.05 vs. old CR carotid. Values are mean±SEM
Results
Animal characteristics
Body mass was similar between young and old AL mice (p > 0.05), while old CR mice had a lower body mass than the other groups (p < 0.05, Table 1). Old CR had lower food intake compared with young and old AL (p < 0.05), and old AL had lower food intake compared with young (p < 0.05, Table 1).
Table 1.
Animal characteristics
| Young | Old AL | Old CR | |
|---|---|---|---|
| Age (months) | 5.9 ± 0.1 | 30.0 ± 0.1* | 30.1 ± 0.1* |
| Body mass (g) | 34.0 ± 1.2 | 36.8 ± 1.2 | 28.5 ± 0.6*† |
| Food intake (g day-1) | 4.8 ± 0.1 | 4.1 ± 0.1* | 3.2 ± 0.0*† |
Data are mean±SEM
AL ad libitum, CR caloric restricted
Endothelium-dependent dilation
In the carotid artery, the response to ACh was impaired in old AL compared with young and old CR mice, with the old AL maximal dilation being 24 % lower than young and 19 % lower than old CR (dose response and max: p < 0.05 vs. young and old CR, Fig. 1a). The dilation to ACh was not different between young and old CR in the carotid artery (dose response and max: p > 0.40, Fig. 1a).
Fig. 1.

Endothelium-dependent dilation (EDD) to acetylcholine (ACh) in the absence or presence of nitric oxide synthase inhibitor Nω-nitro-l-arginine methyl ester (l -NAME) measured by pressure myography in carotid (a) and middle cerebral arteries (MCA, b) from young, old ad libitum fed (AL), and old lifelong calorically restricted (CR) mice (n = 5–16 per group). Endothelium-independent dilation to sodium nitroprusside in carotid arteries (c) and MCAs (d) from young, old AL, and old CR mice (n = 5–9 per group). Significance marks denote a time by group interaction or main effect for group from repeated-measures ANOVA analyses. *p < 0.05 vs. young ACh alone, †p < 0.05 vs. old AL ACh alone, ‡p < 0.05 vs. old CR ACh alone. Values are mean±SEM
In the MCA, the maximal dilation to ACh in old AL was 47 % lower than young and 29 % lower than old CR mice (dose response and max: p < 0.05 vs. young and old CR, Fig. 1b). In contrast to the carotid, the MCA response to ACh was impaired for old CR, with a 25 % lower maximal dilation in old CR vs. young (dose response and max: p < 0.05 vs. young, Fig. 1b).
The response to endothelium-independent dilator SNP did not differ between groups for either artery (Fig. 1c, d; p > 0.05). In addition, there were no group differences for either artery in the sensitivity to ACh (EC50, p > 0.05, Table 2), maximal artery diameter (p > 0.05, Table 2), or the amount of preconstriction prior to dose–response assessment (p > 0.05, data not shown). The magnitude of the impairment in ACh-mediated dilation in old AL was greater in the MCA compared with the carotid for arteries (when data were normalized to young, p < 0.05, Fig. 2). The preservation of ACh-mediated dilation in old CR was less in the MCA compared with the carotid (when data were normalized to young, p < 0.05, Fig. 2).
Table 2.
Artery characteristics
| Carotid | MCA | |||||
|---|---|---|---|---|---|---|
| Young | Old AL | Old CR | Young | Old AL | Old CR | |
| Maximal diameter (μm) | 424 ± 5 | 422 ± 7 | 424 ± 5 | 129 ± 4 | 125 ± 5 | 127 ± 4 |
| EC50, log M | ||||||
| ACh | -7.6 ± 0.2 | -7.5 ± 0.1 | -7.3 ± 0.3 | -7.4 ± 0.4 | -7.4 ± 0.3 | -7.5 ± 0.2 |
| ACh+l-NAME | -7.1 ± 0.3 | -7.0 ± 0.2 | -6.4 ± 0.2* | N/A | N/A | N/A |
| ACh+TEMPOL | -7.5 ± 0.2 | -7.5 ± 0.2 | -7.5 ± 0.2 | -7.9 ± 0.4 | -7.5 ± 0.4 | -7.7 ± 0.3 |
| ACh+TEMPOL + l-NAME | -6.6 ± 0.3 | -7.1 ± 0.5 | -7.0 ± 0.6 | N/A | N/A | N/A |
| SNP | -7.6 ± 0.3 | -7.8 ± 0.3 | -8.1 ± 0.2 | -6.4 ± 0.3 | -6.7 ± 0.2 | -6.0 ± 0.4 |
Values are mean±SEM
ACh acetylcholine, l -NAME NG-nitro-l-arginine methyl ester, SNP sodium nitroprusside
*p < 0.05 vs. young
NO contribution to dilation
In the carotid artery, after inhibition of NOS by preincubation with l-NAME, maximal dilation to ACh was 55 % lower in young, 33 % lower in old AL, and 52 % lower in old CR mice compared with ACh alone (dose response and max: all p < 0.05 with vs. without l-NAME within group, Fig. 1a). Compared to the carotid, the reduction in ACh-mediated dilation after l-NAME preincubation was greater in all groups in the MCA, with maximal dilations 84 % lower in young, 66 % lower in old AL, and 76 % lower in old CR (dose response and max: all p < 0.01 with vs. without l-NAME, Fig. 1b). There was no difference in ACh response between groups in the presence of l-NAME in either the carotid artery or MCA (p > 0.05, Fig. 1a, b).
Oxidative stress
Superoxide production by old AL arteries was about twofold greater in the carotid and about threefold greater in the MCA compared with both young and old CR mice (carotid: p < 0.01 vs. young and old CR, MCA: p < 0.05 vs. young and old CR, Fig. 3a, b). Superoxide production was similar between young and old CR for both arteries (carotid: p = 0.90, MCA: p = 0.86, Fig. 3a, b).
Fig 3.

Superoxide production measured by electron paramagnetic resonance using a CMH spin probe in carotid arteries (a) and middle cerebral arteries (MCA, b) from young, old ad libitum fed (AL), and old lifelong calorically restricted (CR) mice (n = 4–8 group-1). Representative spectra shown below each bar (scale for all carotid spectra is intensity:-1.0 to 1.0; scale for all MCA spectra is intensity: -0.4 to 0.4). *p < 0.05 vs. young, †p < 0.05 vs. old AL. Values are mean±SEM
In old AL mice, TEMPOL resulted in increased ACh-mediated maximal dilation by 26 % in the carotid and 46 % in the MCA (carotid and MCA, max and dose response: p < 0.05; with vs. without TEMPOL, Figs. 4b and 5b). In old AL, the ACh-mediated dilation in the presence of l-NAME and TEMPOL together was not different from the ACh-mediated dilation in the presence of l-NAME alone (p > 0.05).
Fig. 4.
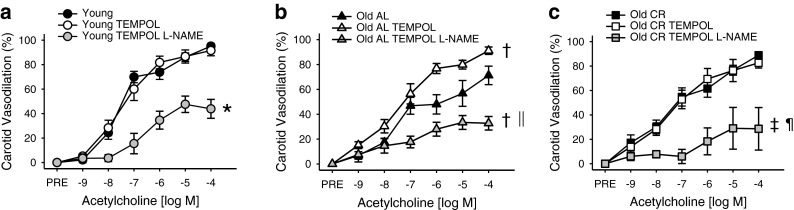
Carotid artery EDD to acetylcholine (ACh) in the absence or presence of superoxide dismutase mimetic TEMPOL, alone or in combination with NOS inhibitor Nω-nitro-l-arginine methyl ester (l -NAME), measured by pressure myography in arteries from young (a), old ad libitum fed (AL, b), and old lifelong calorically restricted (CR, c) mice (n = 4-12 per group). Significance marks denote a time by condition interaction or main effect for condition from repeated-measures ANOVA analyses. *p < 0.05 vs. young ACh alone, †p < 0.05 vs. old AL ACh alone, ‡p < 0.05 vs. old CR ACh alone, §p < 0.05 vs. young ACh with TEMPOL, ‖ p < 0.05 vs. old AL ACh with TEMPOL, ¶p < 0.05 vs. old CR ACh with TEMPOL. Values are mean±SEM
Fig. 5.

Middle cerebral artery (MCA) EDD to acetylcholine (ACh) in the absence or presence of superoxide dismutase mimetic TEMPOL, alone or in combination with NOS inhibitor Nω-nitro-l-arginine methyl ester (l -NAME), measured by pressure myography in arteries from young (a), old ad libitum fed (AL, b), and old lifelong calorically restricted (CR, c) mice (n = 3–16 per group). Significance marks denote a time by condition interaction or main effect for condition from repeated-measures ANOVA analyses. *p < 0.05 vs. young ACh alone, †p < 0.05 vs. old AL ACh alone, ‡p < 0.05 vs. old CR ACh alone, ‖p < 0.05 vs. old AL ACh with TEMPOL. Values are mean±SEM
For young and old CR, scavenging superoxide with TEMPOL did not affect the response to ACh in the carotid (p > 0.05, Fig. 4a, c). In contrast, TEMPOL reduced maximal dilation by 55 % for young and 52 % in old CR in the MCA (max: all p < 0.01, dose response: all p < 0.05, with vs. without TEMPOL within group, Fig. 5a, c).
NADPH oxidase
In the MCAs from old AL mice, apocynin incubation led to a 48 % increase in maximal dilation (p = 0.03, Fig. 6) to ACh. In the MCAs of young mice and old CR mice, apocynin incubation led to a 44 % and 36 % decrease in maximal ACh-mediated dilation, respectively (p < 0.05, Fig. 6). We have previously established with certainty that apocynin improves ACh-mediated dilation in carotid arteries from old AL mice, but has no effect on ACh-mediated dilation in carotid arteries from young mice (Durrant et al. 2009; Rippe et al. 2010). In old CR mice, apocynin incubation had no effect on the ACh response in carotid arteries (max response: 88.8 ± 2.2 % vs. 83.9 ± 8.1 %; ACh alone vs. ACh with apocynin, respectively; p = 0.42). The response to exogenous NADPH was not different between groups (p > 0.05, Fig. 7a, b) and on average resulted in ~10 % constriction in the carotid artery and ~15 % constriction in the MCA.
Fig. 6.

Maximal middle cerebral artery (MCA) EDD to acetylcholine (ACh) in the absence or presence of NADPH oxidase inhibitor apocynin (APO), alone or in combination with NOS inhibitor Nω-nitro-l-arginine methyl ester (l -NAME), measured by pressure myography in arteries from young, old ad libitum fed (AL), and old lifelong calorically restricted (CR) mice (n = 3–16 per group). *p < 0.05 vs. young ACh alone, †p < 0.05 vs. old AL ACh alone, ‡p < 0.05 vs. old CR ACh alone, ‖p < 0.05 vs. old AL ACh with APO, ¶p < 0.05 vs. old CR ACh with APO. Values are mean±SEM
Fig. 7.

Change in diameter in response to exogenous nicotinamide adenine dinucleotide phosphate (NADPH) in the carotid (a) and middle cerebral arteries (MCA, b) from young, old ad libitum fed (AL), and old lifelong calorically restricted (CR) mice (n = 5–11 per group). Values are mean±SEM
Discussion
This study is the first to determine the effect of lifelong CR on cerebral artery function. We demonstrate that lifelong CR completely prevents the age-related decline in EDD in conduit arteries, but only partially prevents the age-related decline in cerebral arteries. The effects of CR are mediated by greater NO bioavailability resulting from lower oxidative stress.
Endothelium-dependent dilation
In the present study, we find that aging leads to impaired conduit and cerebral artery EDD, as indicated by a lower response to ACh in arteries from old mice with no differences between young and old arteries in SNP response. This is in accordance with previous results reported by us and others (Lesniewski et al. 2009; Csiszar et al. 2007a; Blackwell et al. 2004; Modrick et al. 2009; Hatake et al. 1990). Lifelong CR completely prevented this age-related decline in EDD in conduit arteries (carotid), but only partially prevents the decline in cerebral resistance arteries (MCA). As previous studies have only focused on the effect of lifelong CR on conduit artery function (Csiszar et al. 2009), this is the first indication that CR has differential effects on arteries of different sizes. The fact that CR does not completely prevent the age-related decline in endothelial function in all arteries suggests that intervention/treatment strategies may not be equally effective at preserving/improving endothelial function with aging in different vascular beds. Future studies are needed to determine if the disparities in efficacy of lifelong CR for preservation of function differs by the artery type (conduit vs. resistance artery) and location, or if partial preservation of function is specific to the cerebral circulation.
NO bioavailability
As with conduit arteries (Lesniewski et al. 2009; Csiszar et al. 2007a), we find that in cerebral arteries, the impairments in EDD with age are a result of reduced NO bioavailability. Furthermore, we demonstrate that greater NO bioavailability is the mechanism for the partial prevention of age-related decline in MCA EDD with life-long CR. Similar to previous studies in cerebral arteries and arterioles (Kitazono et al. 1995; Mayhan 1990; Sobey and Faraci 1997), we find that in the MCA, dilators produced by NOS (NO and perhaps superoxide) account for most of the dilation in response to ACh.
Oxidative stress
We used EPR, the gold standard technique, to measure the superoxide production by arteries. We demonstrate that aging results in increased production of superoxide in both the carotid artery and MCA, but lifelong CR prevented this age-related increase. Although we cannot compare absolute differences in the amount of superoxide production between arteries due to differences in the tissue size collected, previous studies have demonstrated that under basal conditions cerebral arteries produce more superoxide compared to the carotid arteries per mass (Miller et al. 2005, 2009).
Similar to previous studies, we find that scavenging superoxide improves EDD in conduit and cerebral arteries from old mice (Blackwell et al. 2004; Lesniewski et al. 2009; Mayhan et al. 2008; Modrick et al. 2009) and does not affect EDD in conduit arteries from young or old CR mice (Csiszar et al. 2009). However, we demonstrate that scavenging superoxide in the MCA from young and old CR mice results in reduced EDD, which is in accordance with previous studies demonstrating reactive oxygen species contribute to the dilation of resistance arteries in skeletal muscle (Sindler et al. 2013; Trott et al. 2011), cardiac muscle (Miura et al. 2003; Feng et al. 2010; Kang et al. 2011), and cerebral tissue (Drouin et al. 2007). Similarly, brachial artery dilation to handgrip exercise in humans, which is NO-dependent (Wray et al. 2011), is improved in older adults following ingestion of an antioxidant cocktail, but is reduced in young adults after the same antioxidant cocktail (Donato et al. 2010). Thus, in healthy arteries where dilation is largely reduced by NOS inhibition, superoxide appears to contribution to dilation, perhaps as a result of superoxide produced by NOS.
NADPH oxidase
One source of increased reactive oxygen species within arteries with age is NADPH oxidase (Csiszar et al. 2007b; Durrant et al. 2009; Rippe et al. 2010). Cerebral arteries have greater NADPH oxidase activity compared with conduit arteries and this contributes to their greater production of superoxide (Miller et al 2005, 2009). Aging leads to increased NADPH oxidase expression in cerebral arteries (Mayhan et al. 2008) as well as increased NADPH oxidase expression and activity in the aorta (Durrant et al. 2009; Rippe et al. 2010). Inhibiting NADPH oxidase activity with apocynin led to improved EDD in the MCAs of old AL mice, in agreement with our previous findings in carotid arteries (Durrant et al. 2009). Similar to young mice (Durrant et al. 2009; Rippe et al. 2010), inhibiting NADPH oxidase does not affect EDD in the carotid arteries of old CR mice. In contrast, inhibiting NADPH oxidase impairs EDD in the MCAs from young mice and old CR mice. Inhibition of NADPH oxidase also impairs EDD in skeletal muscle arterioles of young mice (Sindler et al. 2013; Trott et al. 2011). These results indicate that reactive oxygen species produced by NADPH oxidase impair EDD with aging, but contribute to functional resistance artery EDD in young and lifelong CR mice.
We demonstrate that increasing NADPH oxidase activity with exogenous NADPH causes dose-dependent constriction of conduit and cerebral arteries, and this response is not affected by age or lifelong CR. Previous ex vivo studies have also found constriction of cerebral arteries from young healthy animals in response to exogenous NADPH (Didion and Faraci 2002). However, in the skeletal muscle circulation, exogenous NADPH causes dilation of feed arteries from young mice and constriction of feed arteries from old mice (Trott et al. 2011). In addition, in vivo cerebral arteriole diameter and cerebral blood flow increase in response to exogenous NADPH in young healthy animals (Paravicini et al. 2004; Park et al. 2004), a response mediated by increased hydrogen peroxide (Park et al. 2004). The contradiction of results between in vivo and ex vivo studies may result from the absence/presence of shear stress in these preparations, which can activate NADPH oxidase (Paravicini et al. 2006).
Our results indicate a dichotomy between the effect of basal and stimulated NADPH oxidase activity. In ex vivo MCAs from young and old CR mice, basal levels of NADPH oxidase activity (i.e., those that are inhibited by apocynin) aid in dilation to ACh, while increased NADPH oxidase activity (i.e., by stimulation with exogenous NADPH, or perhaps as occurs with aging) results in constriction. In the MCA from young mice (and similarly in old CR), the total contribution to dilation by NOS products (84 %) and NADPH oxidase products (44 %) appear to overlap. We are not aware of any effect of apocynin on NOS, any effect of l-NAME on NADPH oxidase, or any interaction of their products that would explain these results. Thus, these findings suggest that a novel interaction between NOS and NADPH oxidase may exist and future studies are needed to elucidate the mechanisms involved.
Limitations
In the present study, we were limited in our ability to explore molecular mechanisms for the effects of aging and CR on endothelial function. The size of the MCA precludes evaluation of enzyme activities, such as the activity of NOS or NADPH oxidase. Future studies will be needed, perhaps using genetic manipulation or in vivo and ex vivo treatments, to further determine the molecular mechanisms that lead to cerebral artery endothelial dysfunction with aging and the improvements with lifelong CR.
Conclusion
We demonstrate for the first time that lifelong CR does not completely prevent the age-related decline in cerebral artery function, while CR does maintain conduit artery function with age. The effects of lifelong CR on cerebral and conduit artery function are mediated by maintaining NO bioavailability and preventing the age-related increase in oxidative stress. The favorable effects we observed of lifelong CR on conduit and cerebral artery function may explain the beneficial effects of CR on cardiovascular and cerebrovascular disease risk as well as cognitive decline.
Acknowledgment
This study was supported by the National Institutes of Health awards AG029337, AG040297, AG033196, and HL007576.
References
- Austin SA, Santhanam AV, Katusic ZS. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res. 2010;107(12):1498–1502. doi: 10.1161/CIRCRESAHA.110.233080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell KA, Sorenson JP, Richardson DM, Smith LA, Suda O, Nath K, Katusic ZS. Mechanisms of aging-induced impairment of endothelium-dependent relaxation: role of tetrahydrobiopterin. Am J Physiol Heart Circ Physiol. 2004;287(6):H2448–H2453. doi: 10.1152/ajpheart.00248.2004. [DOI] [PubMed] [Google Scholar]
- Carter CS, Leeuwenburgh C, Daniels M, Foster TC. Influence of calorie restriction on measures of age-related cognitive decline: role of increased physical activity. J Gerontol A Biol Sci Med Sci. 2009;64(8):850–859. doi: 10.1093/gerona/glp060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celermajer DS, Sorensen KE, Bull C, Robinson J, Deanfield JE. Endothelium-dependent dilation in the systemic arteries of asymptomatic subjects relates to coronary risk factors and their interaction. J Am Coll Cardiol. 1994;24(6):1468–1474. doi: 10.1016/0735-1097(94)90141-4. [DOI] [PubMed] [Google Scholar]
- Choi JY, Morris JC, Hsu CY. Aging and cerebrovascular disease. Neurol Clin. 1998;16(3):687–711. doi: 10.1016/S0733-8619(05)70089-X. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Orosz Z, Xiangmin Z, Buffenstein R, Ungvari Z. Vascular aging in the longest-living rodent, the naked mole rat. Am J Physiol Heart Circ Physiol. 2007;293(2):H919–H927. doi: 10.1152/ajpheart.01287.2006. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol. 2007;170(1):388–398. doi: 10.2353/ajpath.2007.060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Jimenez R, Pinto JT, Ballabh P, Losonczy G, Pearson KJ, de Cabo R, Ungvari Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev. 2009;130(8):518–527. doi: 10.1016/j.mad.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didion SP, Faraci FM. Effects of NADH and NADPH on superoxide levels and cerebral vascular tone. Am J Physiol Heart Circ Physiol. 2002;282(2):H688–H695. doi: 10.1152/ajpheart.00576.2001. [DOI] [PubMed] [Google Scholar]
- Didion SP, Kinzenbaw DA, Schrader LI, Faraci FM. Heterozygous CuZn superoxide dismutase deficiency produces a vascular phenotype with aging. Hypertension. 2006;48(6):1072–1079. doi: 10.1161/01.HYP.0000247302.20559.3a. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Gano LB, Eskurza I, Silver AE, Gates PE, Jablonski K, Seals DR. Vascular endothelial dysfunction with aging: endothelin-1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2009;297(1):H425–H432. doi: 10.1152/ajpheart.00689.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Uberoi A, Bailey DM, Wray DW, Richardson RS. Exercise-induced brachial artery vasodilation: effects of antioxidants and exercise training in elderly men. Am J Physiol Heart Circ Physiol. 2010;298(2):H671–H678. doi: 10.1152/ajpheart.00761.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Magerko KA, Lawson BR, Durrant JR, Lesniewski LA, Seals DR. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J Physiol. 2011;589(Pt 18):4545–4554. doi: 10.1113/jphysiol.2011.211219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin A, Thorin-Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73(1):73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Durrant JR, Seals DR, Connell ML, Russell MJ, Lawson BR, Folian BJ, Donato AJ, Lesniewski LA. Voluntary wheel running restores endothelial function in conduit arteries of old mice: direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J Physiol. 2009;587(Pt 13):3271–3285. doi: 10.1113/jphysiol.2009.169771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskurza I, Monahan K, Robinson J, Seals D. Effect of acute and chronic ascorbic acid augmentation on flow-mediated dilation with physically active and sedentary aging. J Physiol. 2004;556:215–224. doi: 10.1113/jphysiol.2003.057042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Damrauer SM, Lee M, Sellke FW, Ferran C, Abid MR. Endothelium-dependent coronary vasodilatation requires NADPH oxidase-derived reactive oxygen species. Arterioscler Thromb Vasc Biol. 2010;30(9):1703–1710. doi: 10.1161/ATVBAHA.110.209726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2007;26(1):212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Hatake K, Kakishita E, Wakabayashi I, Sakiyama N, Hishida S. Effect of aging on endothelium-dependent vascular relaxation of isolated human basilar artery to thrombin and bradykinin. Stroke. 1990;21(7):1039–1043. doi: 10.1161/01.STR.21.7.1039. [DOI] [PubMed] [Google Scholar]
- Humpel C. Chronic mild cerebrovascular dysfunction as a cause for Alzheimer’s disease? Exp Gerontol. 2011;46(4):225–232. doi: 10.1016/j.exger.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang LS, Chen B, Reyes RA, Leblanc AJ, Teng B, Mustafa SJ, Muller-Delp JM. Aging and estrogen alter endothelial reactivity to reactive oxygen species in coronary arterioles. Am J Physiol Heart Circ Physiol. 2011;300(6):H2105–H2115. doi: 10.1152/ajpheart.00349.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazono T, Heistad DD, Faraci FM. Dilatation of the basilar artery in response to selective activation of endothelin B receptors in vivo. J Pharmacol Exp Ther. 1995;273(1):1–6. [PubMed] [Google Scholar]
- Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: Cellular and molecular clues to heart and arterial aging. Circulation. 2003;107(3):490–497. doi: 10.1161/01.CIR.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- Lesniewski LA, Connell ML, Durrant JR, Folian BJ, Anderson MC, Donato AJ, Seals DR. B6D2F1 Mice are a suitable model of oxidative stress-mediated impaired endothelium-dependent dilation with aging. J Gerontol A Biol Sci Med Sci. 2009;64(1):9–20. doi: 10.1093/gerona/gln049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoro EJ. Overview of caloric restriction and ageing. Mech Ageing Dev. 2005;126(9):913–922. doi: 10.1016/j.mad.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Mayhan WG. Impairment of endothelium-dependent dilatation of basilar artery during chronic hypertension. Am J Physiol. 1990;259(5 Pt 2):H1455–H1462. doi: 10.1152/ajpheart.1990.259.5.H1455. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Arrick DM, Sharpe GM, Sun H. Age-related alterations in reactivity of cerebral arterioles: role of oxidative stress. Microcirculation. 2008;15(3):225–236. doi: 10.1080/10739680701641421. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, Schmidt HH, Sobey CG. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97(10):1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, De Silva TM, Mast AE, Hickey H, Williams JP, Broughton BR, Sobey CG. NADPH oxidase activity is higher in cerebral versus systemic arteries of four animal species: role of Nox2. Am J Physiol Heart Circ Physiol. 2009;296(1):H220–H225. doi: 10.1152/ajpheart.00987.2008. [DOI] [PubMed] [Google Scholar]
- Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92(2):e31–e40. doi: 10.1161/01.RES.0000054200.44505.AB. [DOI] [PubMed] [Google Scholar]
- Modrick ML, Didion SP, Sigmund CD, Faraci FM. Role of oxidative stress and AT1 receptors in cerebral vascular dysfunction with aging. Am J Physiol Heart Circ Physiol. 2009;296(6):H1914–H1919. doi: 10.1152/ajpheart.00300.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravicini TM, Chrissobolis S, Drummond GR, Sobey CG. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke. 2004;35(2):584–589. doi: 10.1161/01.STR.0000112974.37028.58. [DOI] [PubMed] [Google Scholar]
- Paravicini TM, Miller AA, Drummond GR, Sobey CG. Flow-induced cerebral vasodilatation in vivo involves activation of phosphatidylinositol-3 kinase, NADPH-oxidase, and nitric oxide synthase. J Cereb Blood Blow Metab. 2006;26(6):836–845. doi: 10.1038/sj.jcbfm.9600235. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Wang G, Iadecola C. Exogenous NADPH increases cerebral blood flow through NADPH oxidase-dependent and -independent mechanisms. Arterioscler Thromb Vasc Biol. 2004;24(10):1860–1865. doi: 10.1161/01.ATV.0000142446.75898.44. [DOI] [PubMed] [Google Scholar]
- Qamirani E, Ren Y, Kuo L, Hein TW. C-reactive protein inhibits endothelium-dependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2005;25(5):995–1001. doi: 10.1161/01.ATV.0000159890.10526.1e. [DOI] [PubMed] [Google Scholar]
- Rippe C, Lesniewski L, Connell M, LaRocca T, Donato A, Seals D. Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell. 2010;9(3):304–312. doi: 10.1111/j.1474-9726.2010.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindler AL, Reyes R, Chen B, Ghosh P, Gurovich AN, Kang LS, Cardounel AJ, Delp MD, Muller-Delp JM. Age and exercise training alter signaling through reactive oxygen species in the endothelium of skeletal muscle arterioles. J Appl Physiol. 2013;114(5):681–693. doi: 10.1152/japplphysiol.00341.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobey CG, Faraci FM. Effects of a novel inhibitor of guanylyl cyclase on dilator responses of mouse cerebral arterioles. Stroke. 1997;28(4):837–842. doi: 10.1161/01.STR.28.4.837. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, Salvetti A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension. 2001;38(2):274–279. doi: 10.1161/01.HYP.38.2.274. [DOI] [PubMed] [Google Scholar]
- Trott DW, Seawright JW, Luttrell MJ, Woodman CR. NAD(P)H oxidase-derived reactive oxygen species contribute to age-related impairments of endothelium-dependent dilation in rat soleus feed arteries. J Appl Physiol. 2011;110(5):1171–1180. doi: 10.1152/japplphysiol.01037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte PM. Endothelial control of vasomotor function: from health to coronary disease. Circ J. 2003;67(7):572–575. doi: 10.1253/circj.67.572. [DOI] [PubMed] [Google Scholar]
- Weindruch R, Sohal RS. Seminars in medicine of the Beth Israel Deaconess Medical Center. Caloric intake and aging. N Engl J Med. 1997;337(14):986–994. doi: 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf PA, D’Agostino RB, Belanger AJ, Kannel WB. Probability of stroke: a risk profile from the Framingham study. Stroke. 1991;22(3):312–318. doi: 10.1161/01.STR.22.3.312. [DOI] [PubMed] [Google Scholar]
- Wray DW, Witman MA, Ives SJ, McDaniel J, Fjeldstad AS, Trinity JD, Conklin JD, Supiano MA, Richardson RS. Progressive handgrip exercise: evidence of nitric oxide-dependent vasodilation and blood flow regulation in humans. Am J Physiol Heart Circ Physiol. 2011;300(3):H1101–H1107. doi: 10.1152/ajpheart.01115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Hein TW, Wang W, Kuo L. Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ Res. 2003;92(3):322–329. doi: 10.1161/01.RES.0000056759.53828.2C. [DOI] [PubMed] [Google Scholar]