

Introduction
Endometrial cancer remains the most common gynecologic malignancy in the United States and the incidence of new cases has increased over the last three decades. In 2013, the American Cancer Society estimates there will be 49,560 new cases and 8,190 deaths from endometrial cancer1. Although many are cured, the subset of patients with advanced stage endometrial cancer at diagnosis or recurrent disease presents a significant therapeutic challenge. The use of chemotherapy has been extensively evaluated but has shown only modest benefit. The optimal treatment approaches yield response rates from 40–78% in the primary advanced setting and 15–30% in the recurrent setting. Furthermore, among the optimal chemotherapy regimens for this group, median progression-free survival is only 6 months and median overall survival reaches 12 months2,3. This underscores the great need for novel treatment strategies to improve outcomes for this population of women with endometrial cancer.
Targeted therapy is a promising strategy as molecular alterations of endometrial cancer are becoming better described and multiple potential targets amenable to biologic therapies are already in development. While single-agent biologic agents may have only a modest clinical impact, augmented results may be anticipated in combination with traditional cytotoxic agents, as well as, other novel biologic agents targeting complementary activated pathways. Targeted therapy carries new and different side effect profiles and toxicities, and patient selection remains one of the largest challenges in effectively incorporating new biologic agents. The use of biologic agents in an unselected patient population has the potential to contribute to morbidity without benefit. Further, this can potentially lead to incorrect classification of a drug as inactive for reasons such as lack of expression of the relevant target or presence of a mutation which confers resistance to the agent 4. At this time, no accurate predictive biomarkers exist for most newly developed targeted agents in endometrial cancer.
Endometrial carcinomas exhibit distinct molecular alterations which hold potential druggable targets. The PI3K/AKT/mTOR pathway is the most frequently altered signaling pathway in endometrial carcinoma, including loss of function of the tumor suppressor PTEN, which is seen in up to 83% of endometrioid carcinomas and 55% of precancerous lesions. Loss of function of the tumor suppressor PTEN has been suggested to be an early event in endometrial tumorigenesis.5 This abberation upregulates signaling through the PI3K/AKT/mTOR pathway, leading to uncontrolled cell proliferation and survival. Activation of the PI3K/AKT/mTOR pathway results in elevated levels of downstream markers such as phosphorylated-S6 ribosomal protein (pS6rp) 6,7. In addition, KRAS mutations are found in up to 30% of endometrial cancers 8,9. K-Ras mutations are also seen in 6–16% of endometrial atypical hyperplasia and thus, are considered one of the earliest molecular events in endometrial cancer.10–12 Preliminary analysis in advanced solid tumors indicates that mutations in KRAS may convey resistance to PI3K-directed therapy, especially among endometrial cancer patients. 13. Due to its prominent role in endometrial carcinogenesis, the PI3K/AKT/mTOR pathway has received significant attention for agent development. Several compounds have been discovered that selectively target this pathway, including rapamycin analogs, which directly inhibit mTOR. Many of these compounds –everolimus14, temsirolimus15, and ridaforolimus16 - have completed or are currently being evaluated in clinical trials as monotherapy and/or in combination regimens for the treatment of endometrial carcinoma.
The objective of this study was to determine if expression of biomarkers in the mTOR pathway or KRAS mutations would predict response to therapy to everolimus, an oral inhibitor of the mTOR signaling pathway.
Materials and Methods
Patient Samples
Following IRB approval, 35 pretreated patients with recurrent endometrial cancer of endometrioid histology were enrolled in a single institution, open-label, phase II study of everolimus, a selective mTOR inhibitor. Everolimus (10 mg PO daily/28 day cycles) was given until progression or toxicity. In this study, clinical benefit rate (CBR) was defined as objective response plus the proportion of patients with prolonged (≥ 20 weeks) stable disease. There were no confirmed objective responses in this trial so the current analysis focused on those patients with prolonged stable disease14. Primary hysterectomy specimens corresponding to these patients were submitted to the Department of Pathology, M.D. Anderson Cancer Center. The H&E-stained slides were evaluated by a gynecologic pathologist (RRB) to confirm the diagnosis. Immunohistochemical analyses for PTEN and Phospho-(S235/236)S6 ribosomal protein (pS6rp), and KRAS mutational analysis were performed using the primary hysterectomy specimen. Association of each variable with response to therapy was tested with Fisher’s exact test. Positive predictive value (PPV) and negative predictive value (NPV) for each variable was estimated with a 95% confidence interval.
Immunohistochemistry
Formalin-fixed paraffin-embedded (FFPE) sections of endometrium were analyzed by immunohistochemistry for expression of pS6rp and PTEN. Slides were deparaffinized in xylene and then rehydrated in serial graded alcohol. Following antigen retrieval, endogenous peroxidase activity was blocked by submerging slides in 3% H2O2 for 10 minutes. Slides were washed in phosphate-buffered saline (PBS) then blocked with 1% normal goat serum for 15 minutes. Primary antibody was applied and slides were incubated overnight at 4°C in a humidified chamber. The primary antibodies used were anti-phospho-(S235/236)S6 ribosomal protein (S235/236) rabbit monoclonal antibody (Cell Signaling Technology, Beverly, MA) at 1:50 dilution and anti-PTEN mouse monoclonal antibody (Dako North America, Carpinteria, CA) at 1:100 dilution. Following overnight incubation, slides were washed then treated with biotin-labeled affinity isolated goat anti-rabbit and goat anti-mouse immunoglobulins in PBS for 10 minutes followed by a wash in PBS, and then application of streptavidin-horseradish peroxidase solution (Dako North America, Carpinteria, CA) for 10 minutes. Slides were rinsed in PBS and developed with 3,3′-diaminobenzidine, counterstained in 10% hematoxylin, and dehydrated in serial graded alcohol and xylene before mounting coverslips.
Two gynecologic pathologists (B.D. and R.B.) independently reviewed the immunohistochemistry slides and scored the expression of each PTEN and pS6rp as positive or negative. For PTEN staining to be positive, diffuse positive cytoplasmic and nuclear staining needed to be present in >90% of cells. Stromal cells and blood vessels were intensely PTEN positive and served as internal positive controls for all cases. For all negative cases, PTEN positive stromal cells confirmed the success of the immunohistochemical reaction. Phospho-S6rp expression was scored as positive if cytoplasmic staining of 2–3+ intensity in ≥ 10% of cells was present.
KRAS mutational analysis
KRAS mutational testing was carried out using conventional PCR followed by pyrosequencing. An amplicon of 98 bp was amplified using a forward primer, 5′-TATAAACTTGTGGTAGTTGG-3′, and a reverse biotinylated primer, 5′-biotin-ATTGTTGGATCATATTCGT-3′. The PCR master mix contained the forward and reverse primers (each 0.2 μmol/l), 250 μmol/l of dNTP mix, 2.5 mmol/l MgCl2, 1 PCR buffer (Applied Biosystems, Carlsbad, CA, USA), 1 U of AmpliTaq Gold and 200 ng of sample genomic DNA in a total volume of 50 l. PCR-cycling conditions consisted of initial denaturing at 95°C for 2 min; 49 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s; and final extension at 72°C for 10 min. The reactions were carried out on an ABI 2720 Thermocycler (Applied Biosystems). All samples were run in duplicate and patient samples were processed along with a positive control sample (positive for the presence of a GGT-to-GCT mutation at codon 12), a negative control sample and a reagent control. The PCR products were electrophoresed in an agarose gel to confirm successful amplification of the 98-bp PCR product before pyrosequencing. The PCR products (each 15 l) were then sequenced by the Pyrosequencing PSQ96 HS System (Biotage AB, Uppsala, Sweden), following the manufacturer’s instructions using the pyrosequencing primer, 5′-CTTGTGGTAGTTGGAGCT-3′. Each sample was sequenced with two separate programs of nucleotide dispensation orders: 5′-TACGACTGC-3′, designed for detecting mutations at codon 12; and 5′-TGTATCGATCGT-3′, designed for detecting mutations at codon 13. This primer and the dispensation orders enabled us to capture all possible mutations of the wild-type sequence of GGT GGC at codons 12 and 13 of KRAS.
Results
In the phase II study of everolimus, the CBR was 21% (6/28; 95% CI 8.3%–41.0%), all with prolonged stable disease; there were no confirmed objective responses. 14. A total of 24 patients were evaluable for PTEN expression (Table 1). Eighty-three percent (5/6) of the patients with prolonged stable disease on everolimus exhibited intact PTEN expression. The grade distribution for patients with prolonged stable disease included 2 patients with grade 1 disease, 3 patients with grade 2 disease and 1 patient with grade 3 disease. Loss of PTEN expression was associated with a PPV of 13% (95% CI: 0.7%–53.3%) and a NPV of 69% (95%CI: 42%–88%). All 28 patients were evaluable for pS6rp status analysis. Fifty percent of patients exhibited positive pS6rp staining, however, responders were twice as likely to lack pS6rp compared to non-responders (Table 1). Thus, pS6rp positive staining was associated with a PPV 14% (95%CI: 2.5%–43.8%) and a NPV of 71% (95% CI: 42%–90%). There were 7 KRAS mutations found (Table 2). Interestingly, 80% (4/5) of patients with prolonged stable disease were KRAS wildtype (Table 1). KRAS mutations were associated with a PPV of 14% (95% CI: 0.8%–58%) and a NPV of 75% (95% CI: 47%–92%).
Table 1.
Evaluation of biomarker status by response to therapy
| Response (≥ 20 week stable disease) | ||||
|---|---|---|---|---|
| No | Yes | Total | ||
| PTEN expression | Negative | 7 (39%) | 1 (17%) | 8 |
| Positive | 11 (61%) | 5 (83%) | 16 | |
| Total | 18 | 6 | 24 | |
| P=0.62 | ||||
| pS6rp status | Negative | 10 (45%) | 4 (67%) | 14 |
| Positive | 12 (55%) | 2 (33%) | 14 | |
| Total | 22 | 6 | 28 | |
| P=0.65 | ||||
| KRAS mutation | Wildtype | 12 (67%) | 4 (80%) | 16 |
| Positive | 6 (33%) | 1 (20%) | 7 | |
| 18 | 5 | 23 | ||
| P=0.99 | ||||
Table 2.
Distribution of KRAS mutations
| KRAS Mutation | Frequency of Mutation |
|---|---|
| G12A | 2 |
| G12C | 1 |
| G12D | 3 |
| G12V | 1 |
We then evaluated the combination pS6rp positive-staining with KRAS mutational status. Twenty-three patients were available for this analysis. No patients exhibiting pS6rp positive staining and an activating KRAS mutation demonstrated a prolonged response of stable disease while taking everolimus (Table 3). Thus, this combined test had a 100% PPV for no clinical benefit. Conversely, we found 100% specificity for this test, meaning that in patients with prolonged response, the probability of a tumor with positive staining for pS6rp and KRAS mutation was 0%.
Table 3.
Combination test predicting lack of response to therapy
| Combined test: pS6rp+, KRAS+ and Prolonged Response | ||||
|---|---|---|---|---|
| Prolonged Response | ||||
| No | Yes | Total | ||
| pS6rp status and KRAS mutation | No | 14 | 5 | 19 |
| Yes | 4 | 0 | 4 | |
| Total | 18 | 5 | 23 | |
Fisher’s Exact Test p-value = 0.5552
Positive predictive value= 4/4 = 100.0% with 95% CI 39.6% to 100.0%
Negative predictive value= 5/19 = 26.3% with 95% CI 10.1% to 51.4%
Sensitivity = 4/18 = 22.2% with 95% CI 7.4% to 48.1%
Specificity = 5/5 = 100.0% with 95% CI 46.3% to 100.0%
Discussion
The majority of targeted therapies will only work in a subpopulation of patients with a given disease. Thus developing predictive biomarkers to identify patients who will or will not respond is of great importance. To our knowledge, this is one of the first studies to identify predictive biomarkers in women with endometrial cancer treated with an mTOR inhibitor. Although the sample size was small and involved a pretreated population of endometrial cancer patients, the strength of this study includes the robust nature of the data, stemming from independent pathology review and subjected to the rigorous standards of oversight required by a clinical trial. Limiting participants to individuals with endometrioid histology provided a more homogenous treatment group.
The PI3K/AKT/mTOR pathway provides multiple potential biomarkers that may serve to predict response to targeted agents as well as to assist in identifying individuals in biomarker driven clinical trials. Many relevant molecular abnormalities in endometrial cancer are linked, either in a direct or indirect fashion to the PI3K/AKT/mTOR pathway17–19. Potential candidates might include PTEN, phospho-AKT, PIK3CA, phospho-mTOR, pS6rp. Our hypothesis was that pS6rp may be a better biomarker than PTEN because it is a downstream target of mTOR and thus integrates input from not only the PI3K/AKT/mTOR pathway, but other pathways as well. Thus, pS6rp may be a more biologically relevant biomarker for predicting response to rapamycin analogues.
In this study, we describe a novel combination test to help identify who may not benefit from an mTOR inhibitor such as everolimus. This test combined immunohistochemistry testing for pS6rp status as well as KRAS mutation testing. Individuals whose tumors harbored KRAS mutations and had evidence of activation of the mTOR pathway as reflected by positive staining of pS6rp, did not respond to everolimus. The strength of this test is that it is clinically feasible and may be performed using standard formalin fixed and paraffin embedded tumor specimens. Such a test may be helpful to prevent exposure to potentially toxic agents that will likely not provide clinical benefit. Given that 49% of patients in the phase II trial of everolimus required a dose reduction secondary to toxicities, a test that will allow improved drug selection can improve patient outcomes in a clinical context where safety and economy are increasingly important. These findings are hypothesis generating. A prospective trial can further elucidate and validate the performance of this combination test as a reliable predictor of non-response to single agent mTOR inhibitors.
KRAS mutations have been repeatedly found to predict resistance to epidermal growth factor receptor (EGFR) targeted therapy in patients with metastatic colorectal cancer and it is now routinely recommended that KRAS testing be performed and treatment with EGFR inhibitors be limited to those who do not have KRAS mutations 20. To date, no such mandate has been made regarding KRAS mutations and other targeted therapies. However, more recently, KRAS mutations have been associated with resistance to everolimus in colorectal cancer as well as other tumor types 21. Our results corroborate this important finding in patients with metastatic endometrial cancer. In addition, a published series of patients with advanced solid tumor, which included patients with endometrial cancer, a higher than expected response rate (35%) was seen in patient’s whose tumors exhibited PIK3CA mutations when their treatment regimen included inhibitors of the PI3K/AKT/mTOR pathways. This study also suggests that the presence of KRAS mutations confers resistance in certain tumor types in this retrospective study13. This suggested interaction between PIK3CA and KRAS mutations has also been demonstrated with human cancer cells treated with everolimus in vitro. Cells harboring PIK3CA mutations were sensitive to everolimus, while those cells with both PIK3CA and KRAS mutations were resistant21. In a recent study using endometrial cancer cell lines with mutations in the PI3K/PTEN/mTOR and MAPK signaling pathways, metformin was found to displace constitutively active K-Ras from the cell membrane, causing uncoupling of the MAPK signaling pathway. Metformin decreased tumor growth in xenograft models of endometrial cancer, with the greatest response being observed in mutant KRAS cells22. This study provides a rationale for clinical trials using metformin in combination with PI3K-targeted agents for tumors harboring activating K-Ras mutations. A clinical trial at our institution evaluating evirolimus, letrozole and metformin in women with recurrent endometrial cancer is ongoing.
Figure 1.
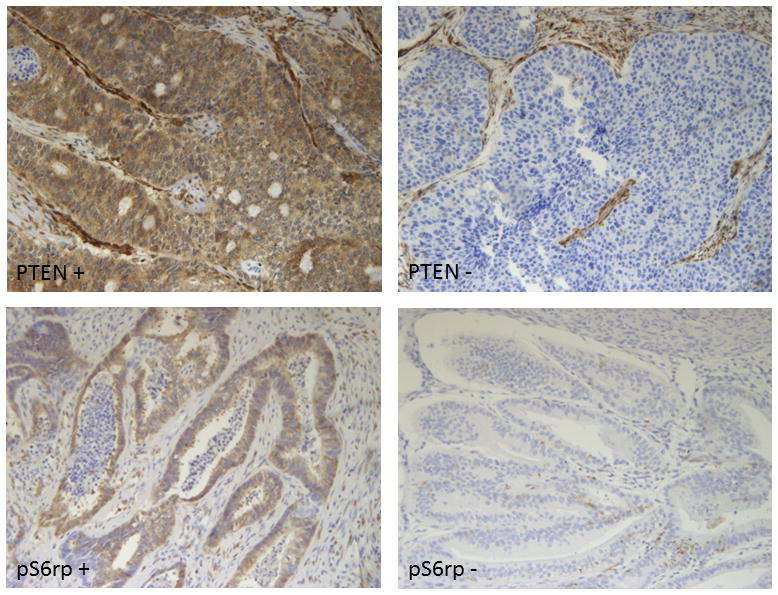
(1a-upper left) Tumors considered positive showed diffuse positive cytoplasmic and nuclear staining in the majority (>90%) of cells. (1b-upper right) Tumors with no or only rare cells staining (<1%) were considered negative for PTEN. (1c-lower left) Tumors considered positive for pS6rp (S235/236) demonstrate cytoplasmic staining. (1d- lower right) lack cytoplasmic staining in the tumor.
Acknowledgments
Funding: NIH 2P50 CA098258-06 SPORE in Uterine Cancer; NIH Training of Academic Gynecologic Oncologists Grant T32-CA101642
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Obel JC, Friberg G, Fleming GF. Chemotherapy in endometrial cancer. Clinical advances in hematology & oncology : H&O. 2006;4:459–68. [PubMed] [Google Scholar]
- 3.Fleming GF, Brunetto VL, Cella D, et al. Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: a Gynecologic Oncology Group Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004;22:2159–66. doi: 10.1200/JCO.2004.07.184. [DOI] [PubMed] [Google Scholar]
- 4.Stewart DJ, Kurzrock R. Cancer: the road to Amiens. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:328–33. doi: 10.1200/JCO.2008.18.9621. [DOI] [PubMed] [Google Scholar]
- 5.Maxwell GL, Risinger JI, Gumbs C, et al. Mutation of the PTEN tumor suppressor gene in endometrial hyperplasias. Cancer research. 1998;58:2500–3. [PubMed] [Google Scholar]
- 6.McCampbell AS, Harris HA, Crabtree JS, Winneker RC, Walker CL, Broaddus RR. Loss of inhibitory insulin receptor substrate-1 phosphorylation is an early event in mammalian target of rapamycin-dependent endometrial hyperplasia and carcinoma. Cancer prevention research. 2010;3:290–300. doi: 10.1158/1940-6207.CAPR-09-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Djordjevic B, Hennessy BT, Li J, et al. Clinical assessment of PTEN loss in endometrial carcinoma: immunohistochemistry outperforms gene sequencing. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2012 doi: 10.1038/modpathol.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mutter GL, Lin MC, Fitzgerald JT, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. Journal of the National Cancer Institute. 2000;92:924–30. doi: 10.1093/jnci/92.11.924. [DOI] [PubMed] [Google Scholar]
- 9.Lax SF, Kendall B, Tashiro H, Slebos RJ, Hedrick L. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer. 2000;88:814–24. [PubMed] [Google Scholar]
- 10.Sasaki H, Nishii H, Takahashi H, et al. Mutation of the Ki-ras protooncogene in human endometrial hyperplasia and carcinoma. Cancer research. 1993;53:1906–10. [PubMed] [Google Scholar]
- 11.Duggan BD, Felix JC, Muderspach LI, Tsao JL, Shibata DK. Early mutational activation of the c-Ki-ras oncogene in endometrial carcinoma. Cancer research. 1994;54:1604–7. [PubMed] [Google Scholar]
- 12.Semczuk A, Berbec H, Kostuch M, Cybulski M, Wojcierowski J, Baranowski W. K-ras gene point mutations in human endometrial carcinomas: correlation with clinicopathological features and patients’ outcome. Journal of cancer research and clinical oncology. 1998;124:695–700. doi: 10.1007/s004320050234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janku F, Tsimberidou AM, Garrido-Laguna I, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Molecular cancer therapeutics. 2011;10:558–65. doi: 10.1158/1535-7163.MCT-10-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slomovitz BM, Lu KH, Johnston T, et al. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer. 2010;116:5415–9. doi: 10.1002/cncr.25515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oza AM, Elit L, Provencher D, Biagi JJ, Panasci L, Sederias J, Dancey JE, Tsao MS, Eisenhauer EA NCIC Clinical Trials Group. A phase II study of temsirolimus (CCI-779) in patients with metastatic and/or locally advanced recurrent endometrial cancer previously treated wtih chemotherapy. Journal of Clinical Oncology. 2008;26:296s. [Google Scholar]
- 16.Colombo N, McMeekin S, Schwartz P, et al. A phase II trial of the mTOR inhibitor AP23573 as a single agent in advanced endometrial cancer. ASCO Meeting Abstracts. 2007;25:5516. [Google Scholar]
- 17.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 18.Bartlett JM. Biomarkers and patient selection for PI3K/Akt/mTOR targeted therapies: current status and future directions. Clin Breast Cancer. 10(Suppl 3):S86–95. doi: 10.3816/CBC.2010.s.017. [DOI] [PubMed] [Google Scholar]
- 19.Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669–73. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- 20.Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:2091–6. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 21.Di Nicolantonio F, Arena S, Tabernero J, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. The Journal of clinical investigation. 2010;120:2858–66. doi: 10.1172/JCI37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iglesias DA, Yates MS, van der Hoeven D, et al. Another Surprise from Metformin: Novel Mechanism of Action via K-Ras Influences Endometrial Cancer Response to Therapy. Molecular cancer therapeutics. 2013;12:2847–56. doi: 10.1158/1535-7163.MCT-13-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]