

Abstract
Various studies have implicated the concave surface of arrestin in the binding of the cytosolic surface of rhodopsin. However, specific sites of contact between the two proteins have not previously been defined in detail. Here, we report that arrestin shares part of the same binding site on rhodopsin as does the transducin Gα subunit C-terminal tail, suggesting binding of both proteins to rhodopsin may share some similar underlying mechanisms. We also identify two areas of contact between the proteins near this region. Both sites lie in the arrestin N-domain, one in the so-called “finger” loop (residues 67–79) and the other in the 160 loop (residues 155–165). We mapped these sites using a novel tryptophan-induced quenching method, in which we introduced Trp residues into arrestin and measured their ability to quench the fluorescence of bimane probes attached to cysteine residues on TM6 of rhodopsin (T242C and T243C). The involvement of finger loop binding to rhodopsin was expected, but the evidence of the arrestin 160 loop contacting rhodopsin was not. Remarkably, our data indicate one site on rhodopsin can interact with multiple structurally separate sites on arrestin that are almost 30 Å apart. Although this observation at first seems paradoxical, in fact, it provides strong support for recent hypotheses that structural plasticity and conformational changes are involved in the arrestin–rhodopsin binding interface and that the two proteins may be able to interact through multiple docking modes, with arrestin binding to both monomeric and dimeric rhodopsin.
The G protein-coupled receptors (GPCRs) are essential mediators for transducing a wide variety of extracellular signals to the inside of the cell.1 Receptor activation leads to G protein-mediated signaling, which is then terminated when the receptor is phosphorylated by a G protein-coupled receptor kinase (GRK) and bound by a protein called arrestin.2 The interaction of an arrestin with a GPCR has been perhaps most extensively explored for the dim light photoreceptor, rhodopsin, with visual arrestin.3 The general surfaces and residues that interact between the two proteins have been identified, and these are briefly reviewed below.4
Mutagenesis, peptide inhibition, electron paramagnetic resonance (EPR), and fluorescence spectroscopic studies implicate residues in the concave sides of both the N- and C-domains of arrestin in receptor binding.5−13 One especially interesting region has been the finger loop of arrestin, a stretch of 13 amino acids in the N-domain (residues 67–79), which sits in the middle of the N- and C-domains on the concave surface of the protein (Figure 1).12,14−16 On rhodopsin, mutagenesis and peptide inhibition studies have shown the receptor C-tail (with the GRK-catalyzed phosphates attached) and intracellular loops 2 and 3 are required for arrestin binding.17−19
Figure 1.
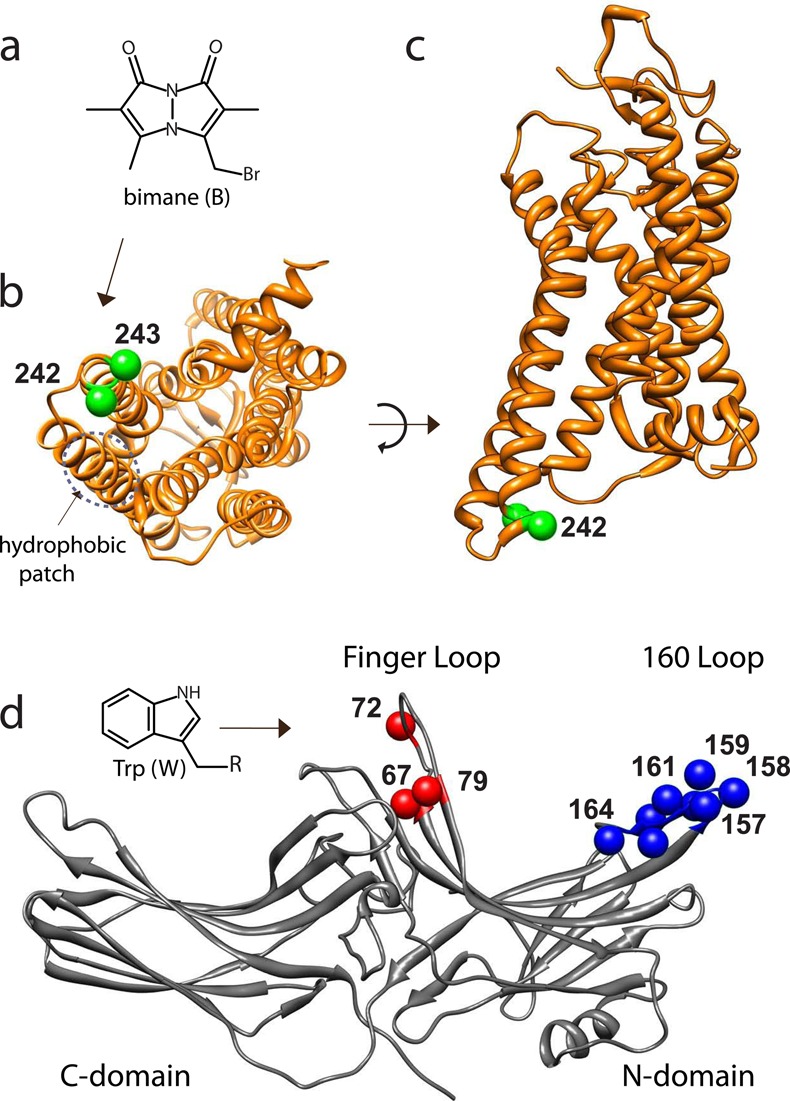
Model showing sites of rhodopsin and arrestin tested for interaction by TrIQ studies. (a) Chemical structure of the fluorophore monobromobimane (mBBr). (b and c) Models showing the cytoplasmic view and transmembrane view, respectively, of opsin M257Y (PDB entry 4A4M). The sites of cysteine residues used to attach the fluorophore bimane (T242 and T243) are indicated (green spheres at the Cα atoms). (d) Model of arrestin (PDB entry 1AYR, chain A). For these studies, a single Trp residue (inset) was introduced at each of the indicated sites, both in the finger loop (residues 67, 72, and 79; red spheres at the Cα) and in the 160 loop (residues 157–164, blue spheres at the Cα), to act as a possible quencher of the bimane fluorescent probes on rhodopsin.
While specific residue–residue interactions between arrestin and a GPCR have not yet been defined, there is mounting evidence that there are likely several types of receptor–arrestin interactions. Specifically, fluorescence studies have implicated residues at the tip of the C-domain implicated in low-affinity interactions with rhodopsin,20 and recent nuclear magnetic resonance (NMR) studies have indicated that arrestin interacts with different states of active rhodopsin differently.21 In this work, we present evidence of a real-time physical interaction between sites on rhodopsin and residues on the N-lobe of arrestin that has not previously been reported to interact with the receptor.
The structure and conformational dynamics of both arrestin and rhodopsin have been extensively studied using site-directed labeling approaches, in which individual cysteine groups are labeled with a spectroscopic reporter group, and these probes then used to glean information about the dynamic changes in the protein at the site of attachment. Spin-labels and EPR spectroscopy [so-called site-directed spin labeling (SDSL)] have been used to map conformational changes in rhodopsin22,23 as well as arrestin.12,24,25 Insights have also been gained from conceptually similar site-directed fluorescence labeling (SDFL) studies, although the latter have not been conducted on both proteins nearly as extensively or systematically.15,16,26−28
The SDSL and SDFL approaches have defined key aspects of GPCR activation and G protein coupling. SDSL studies showed that rhodopsin activation involves an outward movement of TM6,23,29,30 and subsequent SDFL studies showed this movement allowed G protein binding by exposing a “hydrophobic patch” on the receptor that makes critical interactions with the C-terminal tail of the Gα subunit of transducin.31,32 Note that other biochemical studies also suggested TM6 movement during rhodopsin activation,33 as well as the location of the Gtα C-terminal binding site on rhodopsin.34 Ultimately, these interactions were more precisely defined by further SDSL studies35 and crystal structures of active rhodopsin coupled with the Gtα C-tail.36,37
Here, our goal was to assess if exposure of this cleft and “hydrophobic patch” upon TM6 movement during rhodopsin activation might have a similar role in arrestin binding and to define sites of direct contact between the two proteins. Our approach employed a novel SDFL approach that utilizes tryptophan-induced quenching (TrIQ) (discussed below).38 In TrIQ, we assess the ability of Trp residues engineered into arrestin to quench the fluorescence of a bimane fluorophore [monobromobimane (mBBr)] attached to different, engineered cysteine residues on rhodopsin.
To conduct our studies, we introduced the individual cysteine residues into a background construct of rhodopsin that contained no reactive cysteines, called θ,23,26 and also mutations to make it thermostable (N2C/D282C) as well as constitutively active (M257Y).39−41 The use of this rhodopsin mutant provided a number of advantages. The M257Y mutation made it possible to measure stable binding of arrestin to opsin (rhodopsin without retinal), binding that was not lost during MII decay. Also, the absence of retinal prevented any rhodopsin photobleaching by the laser used for fluorescence lifetime measurements. Further, the N2C/D282C mutation stabilized the unliganded opsin in detergent micelles.42 We also used a constitutively active form of arrestin (R175E) to circumvent potential problems of receptor phosphorylation heterogeneity, which could complicate both the arrestin binding studies and receptor purification.40,43−45
Our results show that Trp residues introduced into the arrestin finger loop can quench the fluorescence of a bimane label on TM6 of opsin. Unexpectedly, we find that some Trp residues placed in the arrestin 160 loop (residues 155–165) on the N-lobe can also quench a bimane at site 242 as well as one residue away, at site 243. Analysis of the steady-state and time-resolved fluorescence TrIQ data identifies several instances in which the Trp on an arrestin molecule is statically quenching the bimane probe located on the opsin. These results identify distinct sites of direct physical interaction between the two proteins, further supporting the mounting evidence of structural plasticity in the arrestin–rhodopsin interaction, and hint at how arrestin could bind to an opsin dimer.20,21
Materials and Methods
Materials
All restriction enzymes, ligase, and DNA polymerase were from New England Biolabs. All tissue culture media were purchased from HyClone, except for polyethylenimine (PEI), which was from Polysciences, Inc. n-Dodecyl β-d-maltoside (DM) was purchased from Anatrace. 1,2-Dioleoyl-sn-glycero-3-phospho-l-serine (DOPS) and 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA) were purchased from Avanti Polar Lipids. Monobromobimane was obtained from Invitrogen. The 1D4 antibody was obtained from the Monoclonal Antibody Core at the Vaccine and Gene Therapy Institute of Oregon Health and Science University, and the competing 9-mer peptide (corresponding to the 1D4 epitope) was obtained from the Biotechnology Core Facility Branch of the Centers for Disease Control and Prevention (Atlanta, GA). The BL21-CodonPlus(DE3)-RP strain of Escherichia coli was purchased from Agilent Technologies. Yeast extract and BactoTryptone were from BD Biosciences. Profinity eXact and HiTrap heparin columns were from Bio-Rad and GE Healthcare Life Sciences, respectively. Amicon Ultra protein concentrators (10 kDa cutoff) and nitrocellulose filters (0.45um) were from Millipore. GTP was purchased from Roche, and [35S]GTPγS was from PerkinElmer Life Sciences. Frozen bovine retinas were obtained from Lawson and Lawson, Inc. (Lincoln, NE). GBX red light filters were purchased from Eastman Kodak Co. Band pass filters and long pass filters were purchased from Oriel (Stratford, CT), while cuvettes were purchased from Uvonics (Plainview, NY). All other chemicals and reagents were obtained from Sigma-Aldrich.
The following buffers were used throughout. ROS buffer consisted of 70 mM potassium phosphate and 1 mM magnesium acetate (pH 6.8). PBS consisted of 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 1.46 mM KH2PO4 (pH 7.4). MHE consisted of 5 mM MES, 50 mM HEPES, and 1 mM EDTA (pH 6.8). Wash buffer consisted of 10 mM Tris-HCl (pH 7.5), 0.1 M NaCl, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, and 0.01% DM.
Cloning and Mutagenesis
Opsin Cloning, Expression, Purification, and Fluorescent Labeling
As described previously,47 rhodopsin mutants containing alanine substitutions in the “hydrophobic patch” on TM5 (L226A and V230A) were made in a “Cys-less” background nonreactive construct, called θ, in which the native cysteines C140, C316, C322, and C323 were replaced with serines.48,49 For SDFL studies using opsin, a background construct, termed φ, was created in which the aforementioned M257Y, N2C, and D282C mutations were introduced into θ, using overlap extension polymerase chain reaction (PCR) in the pMT4 vector. The individual cysteine mutations for attaching the bimane fluorophore (T242C and T243C) were subsequently introduced into φ using QuikChange mutagenesis.
Rhodopsin mutants were expressed in COS-1 cells and purified as previously described.26,32 Briefly, COS-1 cells were transfected with 30 μg of plasmid DNA containing mutant rhodopsin per 15 cm plate using 0.1 mg of polyethylenimine/plate. Cells were harvested ∼60 h post-transfection. Cells were solubilized using 0.6 mL of 1% DM in 1× phosphate-buffered saline (1× PBS) per plate of cells. After solubilization for 1 h, the lysates were clarified by centrifugation at 100000g. The clear lysate was applied to 1D4 antibody-coupled Sepharose beads and incubated for 4 h at 4 °C. The beads were then washed sequentially, first with PBS with 1 M NaCl, 3 mM MgCl2, and 0.05% DM, then with 0.05% DM in PBS, and finally with MHE buffer with 0.025% DM. The opsin was then labeled with a 10-fold molar excess of mBBr in the same buffer overnight at 4 °C. The labeling reaction was quenched with 1 mM l-cysteine for 30 min on ice. The beads were washed sequentially with the following buffers: 0.025% DM in MHE buffer, 0.2% DM in MHE buffer, 0.025% DM in MHE buffer, and finally 0.05% DM in 5 mM MES (pH 6). The labeled opsin was eluted with 0.1 mM rhodopsin 9-mer peptide (TETSQVAPA) in 5 mM MES, 140 mM NaCl, and 0.05% DM (pH 6).
Arrestin Cloning, Mutagenesis, Expression, and Purification
The visual arrestin R175E mutant, cloned at the C-terminus of a modified 77-amino acid prodomain region of subtilisin BPN′ (proR8FKAM), in the pG58 vector was a generous gift from K. Ridge.50 All of the arrestin Trp mutants were made by QuikChange mutagenesis. All the mutations were confirmed by DNA sequencing. The mutants were expressed in E. coli BL21(DE3)-RP cells (Stratagene) and purified using a Profinity eXact column (Bio-Rad), followed by ion exchange chromatography using a HiTrap heparin column (GE Healthcare) as described previously.51 Briefly, this involved growing the BL21(DE3)-RP cells transformed with the pG58 expression vector containing the prodomain/arrestin fusion mutants (arrestin R175Q for the pull-down studies and R175E for retinal trapping and SDFL studies) in 1 L of LB medium in the presence of 100 μg/mL ampicillin at room temperature to an A550 of 0.6 and then induced with 30 μM IPTG for 16 h at 16 °C. The cell pellet was resuspended in 50 mM Tris-phosphate (pH 7.2) containing 50 mM NaCl, 5 mM β-mercaptoethanol, 0.1 mM PMSF, and protease inhibitor cocktail (Roche) and then disrupted with a French press. The supernatant obtained after centrifugation of the cell lysate at 100000g for 45 min was loaded onto a 5 mL Profinity eXact column. The column was washed with 20 column volumes of 100 mM sodium phosphate (pH 7.2) and 20 column volumes of 100 mM sodium phosphate and 300 mM sodium acetate (pH 7.2). The cleavage of arrestin from the prosubtilisin tag was initiated by passing 1 column volume of 100 mM sodium phosphate (pH 7.2) containing 100 mM sodium fluoride (elution buffer). The fluoride-mediated cleavage reaction was allowed to occur for 2 h on ice. Tag-free arrestin was eluted off the column by passing 5 column volumes of the elution buffer and further purified by cation exchange chromatography using a 1 mL HiTrap heparin column. The resulting arrestin protein was >95% pure, as assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
Arrestin Functional Pull-Down Assay
Preparation of Rod Outer Segments (ROS) and Rhodopsin Phosphorylation
ROS were isolated from bovine retinas as described previously.16,46 All the steps were conducted at 4 °C under red lights. The rhodopsin concentration was assessed by difference spectra in the presence of hydroxylamine (ε500 = 40800 L cm–1 mol–1). Stocks were snap-frozen and stored at −80 °C. Phosphorylated ROS (ROS-P) was prepared as described previously.16 We consistently obtained between five and six phosphates per rhodopsin using this procedure.
A previously described centrifugal pull-down assay was performed to test the binding of constitutively active arrestin mutant R175Q to wild-type rhodopsin.16,28 In these assays, 12 μM ROS or ROS-P membranes were incubated with 3 μM arrestin in 20 mM HEPES and 140 mM NaCl (pH 7.4) for 15 min at room temperature. The samples were kept in the dark or activated with light using a 150 W fiber-optic light source (>495 nm), and then the reaction was quenched immediately with a 10-fold molar excess of ice-cold buffer. Bound arrestin was separated from free by centrifugation at 100000g for 10 min. The membrane pellets were solubilized in loading dye and subjected to SDS–PAGE, followed by Coomassie staining to visualize the amount of arrestin that bound the receptor-containing membranes. For the pull-down assay in the presence of the Gtα peptide [VLEDLKSVGLF], 100 μM peptide was preincubated with the receptor for 10 min at room temperature before the addition of arrestin.
Retinal Release Assay
The effect of the mutation of rhodopsin “hydrophobic patch” residues on arrestin binding was studied by a retinal release assay, which was based on the observation that arrestin could inhibit the release of retinal from rhodopsin, upon light activation.16,28 All fluorescence measurements were taken using a Photon Technologies QM-1 steady-state fluorescence spectrophotometer with excitation provided by a 295 nm LED (OceanOptics LLS-295). The temperature was held at 20 °C using a water-cooled PTI four-position cuvette turret connected to a circulating water bath (VWR Scientific). Rhodopsin mutants L226A and V230A were expressed in COS-1 cells, regenerated, and purified as described above. Constitutively active arrestin R175E was added at twice the molar concentration of receptor and incubated with 0.25 μM “wild-type” (θ) or the “hydrophobic patch” mutant rhodopsin in a reaction mixture containing 20 mM HEPES, 140 mM NaCl, 0.05% DM, and 0.3 mM DOPA (pH 7.4) in the dark for 30 min on ice. The samples were placed in a 10 mm fluorescence cuvette, and the intrinsic Trp fluorescence was measured using a λex of 295 nm and a λem of 330 nm. A 98% neutral density filter was used to attenuate the excitation light to avoid photobleaching of the samples. After an initial dark-state fluorescence measurement, the samples were irradiated with >500 nm light from a 150 W fiber-optic light source for 20 s, and the subsequent increase in fluorescence was measured over time. After 90 min, 30 mM hydroxylamine (final concentration) was added to cleave the Schiff base and convert all the remaining photoproducts to opsin and free retinaloxime, to yield the maximal Trp fluorescence. From the values of the fluorescence in the dark state (Fo), the fluorescence prior to hydroxylamine addition (F1), and the maximal fluorescence of the sample (F2), the percent retinal trapped was calculated by the expression [(F2 – F1)/(F2 – Fo)] × 100.28
Steady-State Fluorescence Spectroscopy
The fluorescent measurements were taken with the PTI steady-state fluorimeter (described above). Typically, a measurement involved using 0.25 μM bimane-labeled opsin in a 10 mm black-jacketed cuvette, which was excited at 380 nm, and the emitted fluorescence was measured from 400 to 600 nm using 1 nm increments. Each data point was integrated for 0.2 s, and the average of two scans yielded the final spectrum. Arrestin mutants were added at concentrations of both 2 and 5 μM, and the fluorescence was monitored over time. The spectra were obtained every 1 min to monitor the time course of arrestin-induced fluorescence quenching (if any), until there was no further reduction in the bimane fluorescence. The excitation band-pass on the fluorimeter was kept at 1 nm and the emission band-pass at 5 nm. A 20-fold excess of arrestin over opsin was found to be sufficient for complete arrestin binding. For the experiment with the Gtα peptide, 100 μM peptide was added to the opsin mix and incubated with the receptor for 10 min at room temperature before the addition of arrestin. Spectra were acquired and visualized using the PTI software program Felix; the fluorescence spectrum of the buffer was subtracted whenever necessary, and the dilution factor upon addition of arrestin was taken into account during data analysis. The spectral data were plotted and analyzed using SigmaPlot version 11.0.
Statistical analyses of these data were conducted as follows. For T242B quenching, a two-way (dose × mutant) analysis of variance (ANOVA) revealed a significant main effect of the mutant (F11,59 = 86.45; p < 0.001) and dose (F1,59 = 10.63; p = 0.002) but no effect of the dose × mutant interaction (F values of <1). Post hoc analyses identified arrestin Trp mutants 158–163** and 164* as differing significantly from arrestin R175E (*p = 0.002; **p < 0.001), and for T243B quenching (b), a two-way (dose × mutant) ANOVA revealed a significant main effect of the mutant (F11,47 = 18.39; p < 0.001) and no effect of the dose or dose × mutant interaction (F values of <1). Post hoc analyses identified mutants Y67W**, F79W**, V159W*, E160W**, E161W**, and D162W* as differing significantly from R175E (p < 0.05; **p < 0.001).
Time-Resolved Fluorescence Spectroscopy
The same sample in the same 10 mm cuvette that was used for the steady-state fluorescence measurement was used to measure the lifetime of bimane fluorescence using a FluoTime 200 spectrometer (PicoQuant GmbH). The samples were excited using a blue (405 nm) diode laser passed through a neutral density filter to modulate the intensity. The measurements were made at “magic angle” settings (54.7°) to avoid photoselection artifacts. The excitation aperture was set to the minimum, while the emission slits were set to 1 nm. To eliminate scattering, the emission was monitored at 490 nm through two >470 nm long pass filters. The decay spectra were recorded over a range of 0–180 ns using the PicoHarp 300 time-correlated single-photon counting system. The instrument response function was determined from the scatter at 400 nm from a solution of Ludox to be ∼64 ps full width at half-maximum and was used to deconvolute the lifetime decay data with high resolution (<50 ps). The system was controlled using the PicoHarp software, and the obtained spectra were fit using software from the manufacturer (FluoFit). The spectra were fit to a three-exponential decay. The “goodness of fit” was evaluated by plotting the residuals, and a χ2 value of 0.9–1.1 was considered acceptable.52 The amplitude-weighted fluorescence lifetime, ⟨τ⟩, was calculated as ∑αiτi, where αi is the normalized amplitude factor for each lifetime, τi.
Transducin Purification
Transducin was purified from bovine retina as previously described, with slight modifications.53,54 ROS membranes were isolated from bovine retina as mentioned above but were finally suspended in 10 mM Tris-HC1, 0.5 mM MgCl2, 1 mM DTT, 0.1 mM PMSF, and 1× protease inhibitor cocktail (EDTA free) (pH 7.5) supplemented with 0.3 mM EDTA, flash-frozen, and stored at −80 °C. The frozen membranes were thawed, dounced in a tissue homogenizer, and centrifuged at 70000g for 30 min. The pellets were washed with the same buffer twice and then twice with a hypotonic buffer [5 mM HEPES (pH 7.5) with 0.1 mM EDTA and 1 mM DTT]. Gt was then extracted by resuspending the washed pellet with hypotonic buffer containing 200 μM GTP. After three rounds of extraction, the extracts were analyzed by SDS–PAGE. The extracts were pooled, concentrated, and buffer exchanged into 20 mM Tris-HCl, 0.2 M NaCl, and 2 mM MgCl2 (pH 7.5) containing 1 mM DTT and 20 μM GDP, in an Amicon Ultra 15 kDa cutoff centrifugal concentrator. To the concentrated protein was added glycerol to a final concentration of 10%, and the sample was then flash-frozen and stored at −80 °C.
Transducin Inhibition Assay
The binding of arrestin to opsin was assessed by testing its ability to inhibit opsin-induced transducin activation, as measured by a GTPγS incorporation assay.55 Briefly, bimane-labeled opsin mutant was diluted to 0.25 μM in 20 mM HEPES (pH 7.4), 140 mM NaCl, 0.05% DM, 0.3 mM DOPS, 1 mM MgCl2, and 0.1 mM EDTA and incubated with the arrestin mutants (5 μM) for 30 min at room temperature. The reaction was started by adding 0.1 μM Gt and 4.5 μM GTPγS with [35S]GTPγS as a tracer (2000–10000 cpm/pmol). The reaction was allowed to proceed for 10 min at room temperature, following which the free nucleotide was removed from bound by applying the mix to 0.45 μm nitrocellulose filters in duplicate in a Brandel cell harvester attached to a vacuum pump and washing the filter with ice-cold wash buffer.56 The filters were then removed, and the bound radioactivity was measured in a scintillation counter. The data from at least two experiments (each measured in duplicate) were averaged to determine the counts for each sample and then further analyzed in SigmaPlot.
Quantitation of Static and Dynamic Fluorescence Quenching
The fraction of fluorophores undergoing static versus dynamic quenching upon binding of arrestin to bimane-labeled opsin was calculated as described by Mansoor et al.38 Briefly, this approach analyzes the steady-state fluorescence quenching data together with the measured fluorescence lifetimes of the fluorophore (bimane attached to specific cysteines in opsin) in the presence and absence of a quenching tryptophan (introduced at specific sites of arrestin) to determine the fraction of the static quenching component in a fluorophore–quencher pair. For this analysis, we calculated Fw/Fo and τw/τo, where Fw and Fo are the peak fluorescence intensity of the samples with and without the quenching Trp residue, respectively, and τw and τo are the amplitude-weighted fluorescence lifetimes of the fluorophore with and without the quenching Trp residue, respectively.
The relative fraction of Trp–fluorophore pairs not in a static complex was calculated using
| 1 |
The relative fraction of Trp–fluorophore pairs involved in a static complex is given by
| 2 |
while the fraction of those undergoing dynamic quenching is
| 3 |
Model Construction and Docking
In brief, modeling the rhodopsin–arrestin interaction was conducted using DSViewer Pro and Chimera. For modeling the interactions of arrestin with monomeric rhodopsin, coordinates from the M257Y/N2C/D282C rhodopsin mutant were used (PDB entry 4A4M), after the retinal and all other nonprotein components had been deleted. For modeling the interactions of arrestin with dimeric rhodopsin, two models were used. The rhodopsin dimer with opposing TM1/TM4/H8 helices was generated using coordinates from PDB entry 4A4M, which were subjected to the “sym” command in Chimera, followed by slight tilting and movement of one monomer versus the other. The rhodopsin dimer with opposing TM4/TM5 helices was created using the appropriate TM4/TM5 dimer coordinates obtained from PDB entry 1N3M,57 onto which was substituted the structure of PDB entry 4A4M using the “Match Maker” command in Chimera.
Arrestin models used p44 arrestin as a template (PDB entry 4J2Q), onto which the sequence from full length arrestin was modeled (PDB entry 1CF1, chain A) to generate the structure for the missing part of the 160 loop in the p44 structure, and then residues 1–9 and the C-terminus (residues 361 on) were deleted to better match the original p44 structure.
The Modeler function in Chimera was then used to generate other models of the 160 loop for the region of residues 153–167, and the model that most resembled the structure resolved via the EPR DEER studies was then used. Initial guesses for docking of the arrestin to the different rhodopsin models were generated using PatchDock (http://bioinfo3d.cs.tau.ac.il/PatchDock/), followed by manual docking to meet the criteria described in the text, while ensuring little to no steric overlap in the final model.
Results
Our goal was to further define mechanisms for how arrestin interacts with the GPCR rhodopsin and to identify precise sites where these two proteins are in direct physical contact. We focused this investigation on the area around TM6 of rhodopsin, as this region is known to undergo key structural changes required for binding and activation of the G protein, transducin, and we hypothesized a similar mechanism may be involved in arrestin binding. Below, we summarize the results of our studies.
Biochemical Mapping of the Arrestin–Rhodopsin Interaction: Arrestin Utilizes the Same Binding Site on Rhodopsin as the Transducin Gtα C-Terminal Tail
To test the hypothesis that arrestin binding employs the same cavity in rhodopsin exposed by TM6 movement (see above and Figure 2a), we tested if a high-affinity peptide corresponding to the C-terminus of Gtα (Gtα peptide) competed with arrestin in binding to ROS rhodopsin membranes.16,28 The data from these centrifugal pull-down assays show that the Gtα peptide does inhibit the binding of constitutively active arrestin (R175Q) to light-activated rhodopsin (R*), by almost 90% (Figure 2b). The Gtα peptide also reduces the level of binding to the phosphorylated, light-activated receptor (RP*), although to a lesser extent (∼20%), presumably because of the higher affinity for the arrestin imparted by the phosphates in RP*. Together, the data suggest that arrestin, in some way, utilizes the same patch on rhodopsin as the C-terminal tail of transducin.
Figure 2.
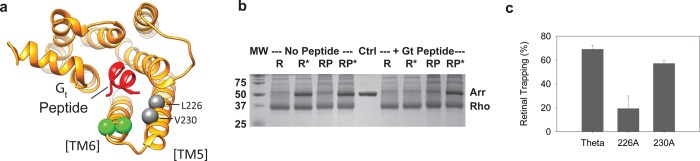
Evidence that arrestin and the Gtα C-terminal tail both bind to the same crevice on rhodopsin. (a) Model showing the location and mechanism of interaction between rhodopsin and the transducin Gtα C-terminus (red). Previous TrIQ studies showed this binding requires a “hydrophobic patch” involving several residues on TM5 of rhodopsin.32 The model was made using coordinates from PDB entry 3DQB. Two of the residues in the “hydrophobic patch” on rhodopsin, L226 and V230, are depicted as gray spheres at the Cα position. The sites of attachment of the bimane fluorophore to rhodopsin (sites 242 and 243) are depicted as green spheres at the Cα position. (b) The ability of a peptide corresponding to the Gtα tail of transducin to compete with the binding of arrestin to rhodopsin was measured. The data show the Gtα tail peptide inhibits the binding of arrestin R175Q to light-activated rhodopsin, R*, measured in a centrifugal pull-down assay. Interestingly, the Gtα peptide does not compete as effectively with RP*. (c) Mutations in the “hydrophobic patch” on TM5 of rhodopsin reduce arrestin-mediated retinal trapping, especially L226A.
We further assessed the role of this region in arrestin binding by altering residues in a “hydrophobic patch” on TM5 of rhodopsin, which becomes exposed by the TM6 movement.32,36,37 Transducin activation and the affinity of the C-terminus of Gtα for rhodopsin are profoundly affected by mutation of residues in this “hydrophobic patch”, L226, T229, and V230,32,58 and the crystal structures of the Gtα peptide bound to active rhodopsin show direct contact of these residues with leucines on the Gtα peptide.31,36,37 We reasoned that if these residues play a similar role in arrestin binding, mutating them should also impair arrestin binding.
Given that the recombinant “hydrophobic patch” mutant receptors are DM-solubilized, and thus less dense than the ROS membrane preparations from bovine retinas, we could not use the traditional centrifugal pull-down assays to measure this interaction. Instead, we monitored the release of retinal from the receptor, which is impeded (∼50% of the retinal is trapped) when arrestin is bound to the receptor.59 We used this approach to measure the binding of arrestin to three different rhodopsin constructs, a minimally reactive cysteine mutant, called theta (θ), and mutants L226A and V230A (each constructed in the θ background). As expected, arrestin R175E caused >60% retinal trapping for the θ “wild-type” rhodopsin (Figure S1 of the Supporting Information and Figure 2c), similar to what is observed for wild-type arrestin–rhodopsin interactions.28 In contrast, both “hydrophobic patch” mutants showed some differences in retinal trapping compared to that of the θ wild type, with the effect of the L226A mutant being the most striking, as indicated by the greater increase in fluorescence during MII decay, and only a slight increase in Trp fluorescence after subsequent hydroxylamine (HA) addition. These results indicate that the ability of arrestin to trap retinal in the rhodopsin “hydrophobic patch” mutant L226A was impaired (presumably because of impaired arrestin binding), as would be expected if arrestin employs part of the same interaction mechanism for binding to rhodopsin as the Gtα C-terminal tail does.
The results described above suggest the cleft exposed in the rhodopsin cytoplasmic face exposed by TM6 movement provides part of the interface for interaction with arrestin. However, these experimental results provide only indirect evidence of this interaction. To more specifically define both where and how arrestin makes contact with this region on rhodopsin, we next turned to studies using an SDFL method, called TrIQ (Trp-induced quenching), as described below.
Identifying Sites of Arrestin–Rhodopsin Interaction Using Tryptophan-Induced Quenching (TrIQ): Two Trp Residues in the Finger Loop of Arrestin Can Quench the Fluorescence of Bimane Fluorophores on TM6 of Opsin
On the basis of the results described above, we decided to introduce the bimane labels onto the receptor at sites close to, but separate from, the “hydrophobic patch” on the base of TM5/TM6 (L226 and V230), so that arrestin binding would not be impaired by either the mutations, the incorporation of the label, or both. The two sites we chose, T242 and T243, are also near the end of TM6 and thus good candidates for efficient labeling because the fluorescent probe used for TrIQ studies, bimane, exhibits significant Förster resonance energy transfer with rhodopsin’s agonist, retinal.26,31,32,60 Thus, to avoid this complication, we used a retinal-free, thermostabilized, constitutively active form of opsin, M257Y, that adopts an active, meta II conformation that can bind and activate transducin.41
We first tested the finger loop on arrestin, which lies in the middle of the two lobes of the protein and marks one extremity on the N-domain (Figure 1d), because we suspected it might bind in the same TM5/TM6 pocket as the Gtα C-terminal tail. The finger loop region has long been thought to be involved in receptor binding,8,12,15 as it undergoes movement upon receptor binding,9,12,16,59 and its flexibility is important for interaction with rhodopsin.15 Interestingly, in agreement with the idea that the arrestin Gtα C-terminal tail and finger loop may bind to the same spot on rhodopsin, we note that the arrestin finger loop (Figure 1d) has a sequence somewhat similar to that of the Gtα C-terminal tail (Figure S7 of the Supporting Information) and, like the Gtα C-terminal tail, has been proposed to adopt a helical structure upon binding rhodopsin.61
These mapping experiments were conceptually straightforward. Arrestin mutants were made with individual Trp residues introduced into the finger loop (Y67W, I72W, and F79W) (Figure 1) and then bound to opsin containing a bimane label on TM6 at site T243C (termed T243B). Evidence of fluorescence quenching (indicated by a decrease in fluorescence upon addition of the arrestin mutant) would indicate proximity between the Trp and fluorophore.
Figure 3 shows representative spectra from these initial studies. Importantly, the control background arrestin mutant, R175E, which contains only one native Trp (W194), caused no quenching of T243B fluorescence (Figure 3a). In contrast, adding arrestin mutant Y67W to opsin T243B caused ∼40% quenching of fluorescence (Figure 3c). This quenching was position-specific, as another finger loop Trp mutant, I72W, showed no quenching of 243B (Figure 3b), and a third Trp mutant in the same loop, F79W, also quenched the bimane fluorescence, by ∼35% (Figure 3d). Importantly, the labeled opsin samples did not contain any free, unbound fluorophore (Figure S2 of the Supporting Information), and each arrestin mutant could bind the receptor, as indicated by the ∼3 nm shift in the observed fluorescence emission maximum (from ∼461 to 458 nm), even when no decrease in fluorescence was observed. These initial studies show clear evidence that the base of the arrestin finger loop can bind near site 243 on opsin.
Figure 3.

Site-specific quenching of opsin T243B fluorescence caused by the arrestin mutant with Trp residues at different positions in the finger loop. Steady-state fluorescence emission spectra of opsin T243B in the absence (black) and presence (red) of the indicated arrestin mutants. (a) The control, arrestin mutant R175E (which has no introduced Trp mutations) causes no quenching of the bimane label on opsin. (b) Arrestin mutant R175E/I72W also causes no quenching. In contrast, the presence of Trp at site 67 (c) or 79 (d) in the arrestin finger loop causes ∼35–40% quenching of fluorescence for the bimane probe at site 243 on opsin. Note that all of the spectra show a slight (∼3 nm) blue shift in the bimane fluorescence emission maxima upon addition of arrestin, indicating that the local environment around the probe has changed, presumably as a result of arrestin binding.
Identifying Sites of Arrestin–Rhodopsin Interaction by TrIQ: Evidence That the Arrestin Finger Loop and the 160 Loop Can Interact with Bimane Fluorophores on TM6 of Opsin
Encouraged by these results, we next tested if the Trp residues on the arrestin finger loop could quench a bimane one residue away on rhodopsin, at position T242 (T242B). Interestingly, no quenching was observed for any of the finger loop Trp mutants for the bimane at site T242B (Figure 4a). There could be two reasons for this: either the bimane at site T242B faces away from the quenching tryptophan, or the arrestin mutants do not bind to the labeled opsin.
Figure 4.

Arrestin finger loop and 160 loop can both interact with probes on TM6 of opsin, as indicated by their ability to quench the steady-state fluorescence and inhibit Gt activation for the bimane-labeled opsin mutants T242B and T243B. Interestingly, both types of arrestin mutants show specific sites of quenching for the bimane labels on the base of TM6 of opsin. (a) Ratio comparing opsin T242B or (b) opsin T243B fluorescence in the presence and absence of finger loop Trp mutants (red bars) and 160 loop Trp mutants (blue bars). Specific sites of quenching are observed at both arrestin concentrations (5 μM, darker bars; 2 μM, lighter bars). (c and d) Ability of arrestin mutants (5 μM) to bind the bimane-labeled opsin mutants, assessed by their inhibition of transducin activation (measured as binding of [35S]GTPγS to transducin). Statistical analyses of these studies are described in Materials and Methods. In brief, a two-way (dose × mutant) ANOVA indicates the results from both arrestin concentrations used above in panels a and b can be compared. Subsequent individual t tests identify the Trp-containing arrestin mutants with significantly greater fluorescence quenching than R175E (*p < 0.05; **p < 0.001) in panels a and b. Individual t tests were also used to identify arrestin mutants that significantly inhibit transducin activation (stimulated [35S]GTPγS binding) in panels c and d (*p ≤ 0.03; **p ≤ 0.007). All experiments shown here used the indicated amounts of arrestin mutants discussed above and 0.25 μM bimane-labeled opsin and in 20 mM HEPES (pH 7.4), 140 mM NaCl, 0.05% DM, 0.3 mM DOPS, 1 mM MgCl2, and 0.1 mM EDTA and were performed after the incubation of opsin and arrestin for 30 min at room temperature.
We also expanded the studies to test if these two sites on opsin interact with the 160 loop on arrestin, as we suspected this region, located at the outer extremity of the concave surface of the N-domain, might also be involved in binding the receptor (see Discussion). The following arrestin mutants with individual Trp residues introduced into the 160 loop (T157W, D158W, V159W, E160W, E161W, D162W, K163W, and I164W) were tested for their ability to quench the bimane probes at positions T242B and T243B on opsin (Table S1 of the Supporting Information).
In contrast to the finger loop mutants, a number of the arrestin 160 loop Trp mutants show significant site-specific quenching of bimane at position 242 on opsin (Figure 4a), with arrestin mutants E160W and E161W showing the most quenching (∼35%). At position 243 on opsin, the arrestin 160 loop Trp mutants exhibit a broader range of ability to quench the bimane fluorescence, with the amount of quenching varying between ∼10 and 25%, and the residues in the middle (E160W and E161W) showing maximal (∼20–25%) quenching (Figure 4b). Formally, it is possible that some small fraction of the quenching could be due to tyrosine (Tyr) residues in arrestin (such as Y67 or Y250), as Tyr can also quench bimane, although it does so much less efficiently than Trp and only at much shorter distances than Trp (A. M. Jones Brunette and D. L. Farrens, manuscript submitted). Moreover, such Tyr quenching would also be systematic and occur in all of the arrestin samples, including the control, R175E. While the bimane label at site 243 does show a small amount of quenching by the R175E mutant in Figure 4b, which might reflect some slight quenching by Y67 on arrestin, the difference is not clearly significant. Thus, our statistical analysis of the TrIQ data was conducted by comparing the Trp arrestin mutants to arrestin 175E, in order to identify which mutants quench significantly more than the control. Interestingly, the diffuse nature of our quenching results for the loop 160 mutants agrees with recent double electron–electron resonance spectroscopy results that show an increased plasticity in the 160 loop region upon binding rhodopsin.62
These data were also subjected to statistical analysis. The results of a two-way (dose × mutant) ANOVA reveals no dose × mutant interaction (F values of <1), thus allowing a comparison of the 2 and 5 μM data. Post hoc analysis of these data was then conducted to compare the effect of the Trp arrestin mutants with the control, arrestin mutant R175E. The significant results identified by this analysis (*p < 0.05; **p < 0.001) are indicated in Figure 4.
Our operational assumption is that the binding of opsin by arrestin in these experiments is at (or near) saturating levels, because experiments using both 2 and 5 μM arrestin show almost identical results (Figure 4a,b). Unfortunately, we were not able to increase the arrestin concentration in the reaction mix above 5 μM, without some precipitation of the protein. Thus, we took care to ensure our analysis does not depend on whether we are at 100% saturation and instead focuses on the instances of substantial quenching.
All of the Arrestin Trp Mutants Can Bind to the Bimane-Labeled Opsin Samples to Some Degree
The positive results (cases that exhibit measurable fluorescence quenching upon arrestin addition) are straightforward to interpret; they indicate that the Trp and bimane are in the proximity of each other. However, a lack of quenching cannot be reliably interpreted as a lack of Trp–bimane proximity, unless it is certain the arrestin mutants have actually bound to the receptor.
Thus, we next tested if the arrestin Trp mutants could bind to the bimane-labeled opsins. Our approach was to measure their ability to block G protein activation.8,63 These experiments used Gt purified from bovine retina (Figure S4 of the Supporting Information) and a well-established G protein activation assay, based on [35S]GTPγS incorporation.55 Importantly, the results from these studies can be directly compared to those of the quenching experiments described above, as they used identical buffers and receptor and arrestin concentrations.
Opsin T242B could bind all the arrestin Trp mutants tested to some degree, as indicated by their inhibition of Gt activation (Figure 4c). The arrestin mutants with Trp residues in the 160 loop clearly inhibited transducin activation to an extent (80–90%) larger than that of the finger loop mutants (∼50%), perhaps because of a lowered affinity for the latter. It is possible that a “weaker” interaction with the finger loop Trp mutants and T242B could contribute to some of the lack of quenching by these mutants. For opsin T243B, essentially all of the arrestin Trp mutants could bind at roughly the same level, as judged by their ability to inhibit the incorporation of [35S]GTPγS into purified transducin [greater than ∼80% (Figure 4d)]. Taken together, these data, although noisy (because of the weakened ability of these opsin mutants to activate transducin58 and the absence of the agonist, all-trans-retinal), suggest that all the arrestin Trp mutants can bind to the receptor to some degree.
The Arrestin 160 Loop Makes Direct Physical Contact with the Base of TM6 in Opsin, As Indicated by the Presence of Static Quenching in the TrIQ Data Analysis
The steady-state TrIQ data (described above) indicate the relative proximity between several sites on arrestin and the bimane-labeled sites on opsin, but they do not explicitly prove the two sites are in direct contact with each other. To better define the proximity of the Trp to the bimane fluorophore, we analyzed the TrIQ data to classify and quantify the types of fluorescence quenching occurring in each case.38 The goal was to identify instances of static quenching, which occurs when a fluorophore and quencher are in physical contact with each other, before (or during) light activation.
Identifying instances of static quenching in a sample requires analysis of both its steady-state fluorescence intensities and its fluorescence decay rates. Thus, we measured the fluorescence lifetimes of the labeled opsin samples in the absence and presence of bound arrestin Trp mutants, using the exact same samples and conditions used for the steady-state fluorescence and G protein activation measurements in Figures 3 and 4a,b. The lifetime data were fit to a three-exponential decay (Figure S5 of the Supporting Information), and these values were then used to calculate the amplitude-weighted lifetime, ⟨τ⟩ (Tables S2 and S3 of the Supporting Information). These ⟨τ⟩ values, in combination with the steady-state fluorescence quenching data, were then used to determine the fraction of dynamic and static quenching for the fluorophore–quencher pairs, as previously described.38
These analyses show that for opsin T242B, the majority of the arrestin 160 loop Trp mutants exhibit some static quenching (Figure 5b). Given the high time resolution (<50 ps) of our lifetime instrument, these data indicate that some fraction (as much as ∼20% in the case of arrestin E161W) of these bimane–Trp pairs is either in contact with each other before (or within 50 ps of) the moment of light excitation. In contrast, for opsin T243B, the majority of quenching by the finger loop Trp residues appears to be dynamic in nature.
Figure 5.
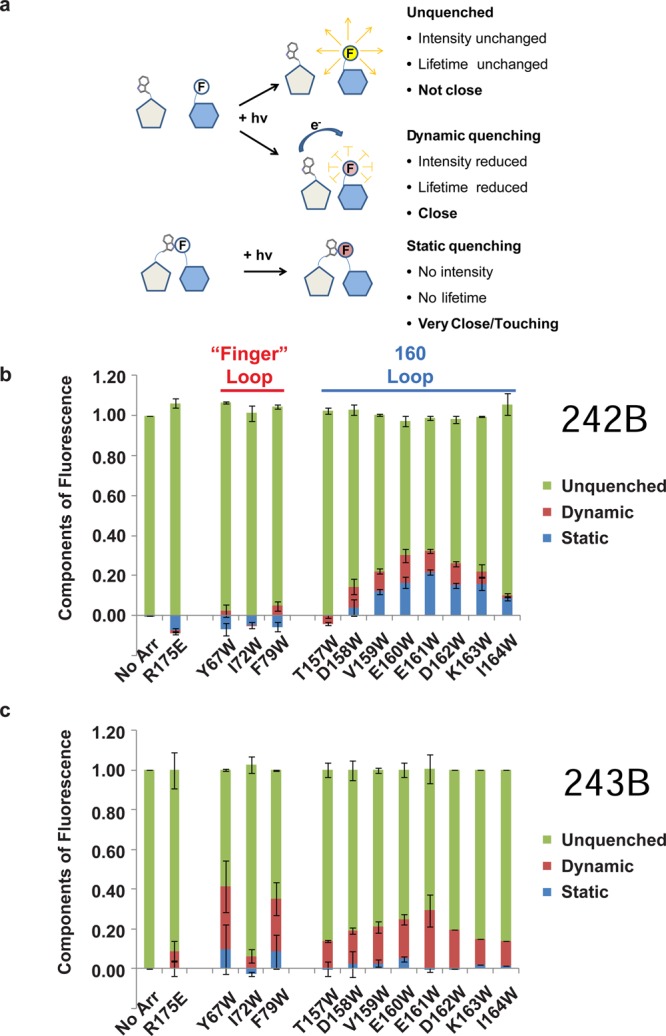
TrIQ analysis indicates the arrestin finger loop and 160 loop can interact with the base of TM6 in opsin. (a) Schematic illustration of the concept of static vs dynamic quenching of fluorescence. As shown, analysis of steady-state quenching and fluorescence lifetime data can be used to identify sites of “static quenching”, i.e., sites of direct contact between the fluorophore and Trp that occur before (or during) light activation. (b) Results from analysis of the steady-state and fluorescence lifetime TrIQ data. The presence of static quenching in the results (blue component in bars) indicates where the Trp residues are making direct contact with the bimane fluorophore on opsin T242B. (c) The same analysis indicates that the arrestin finger loop also interacts with the base of the helix, as seen by the strong quenching of T243B fluorescence, and weaker quenching at other sites.
Discussion
Displacement of TM6 is a key structural change that occurs during rhodopsin activation.29,31 This movement exposes an interhelical cavity, or cleft, that allows the C-terminal tail of the G protein Gα subunit to make critical contacts with a “hydrophobic patch” consisting of residues on the inner face of rhodopsin TM5.32,36,37,64 Here, we present evidence that suggests this same cleft and “hydrophobic patch” play a similar role in permitting arrestin binding. Through use of the TrIQ fluorescence method, we then identified two distinct parts of arrestin that are near or in contact with the base of TM6, the finger loop and the 160 loop. Insights gained from our results are discussed below.
Arrestin Uses Part of the Same Binding Site on Rhodopsin as the Gtα C-Terminal Tail
We first tested if arrestin and transducin share part of the same binding site on TM6 of rhodopsin, by seeing if a peptide corresponding to the C-terminal tail of the transducin Gtα subunit (Gtα peptide) could compete with and block arrestin binding. Indeed, this was observed: the Gtα peptide substantially blocks the binding of arrestin R175Q (a constitutively active arrestin mutant) to light-activated rhodopsin in a pull-down experiment (Figure 2b). This result suggests that arrestin uses at least part of the same binding site on rhodopsin as the Gtα C-terminal tail.
Interestingly, we find the Gtα peptide is less able to block the binding of arrestin to the light-activated, phosphorylated rhodopsin [RP* (Figure 2b)], indicating that only part of the binding affinity is provided by interaction with this region on rhodopsin. Binding of arrestin to RP* has been proposed to involve a multisite interaction between the two proteins and a strong affinity of arrestin for RP*,4 and our data are consistent with this model. Note that the inability of arrestin to bind in the presence of the Gtα peptide is unlikely to be a result of any significant structural change in the receptor caused by the peptide, because structures of metarhodopsin II with and without the peptide are very similar,36 with a root-mean-square deviation (rmsd) of 0.27 Å for all-atom alignment.
Arrestin was also less able to bind and trap retinal in rhodopsin “hydrophobic patch” mutants L226A and V230A (Figure 2c), suggesting that arrestin, like the Gtα tail peptide, requires interactions with this region on TM5 of rhodopsin to bind. A similar impairment of the ability of these rhodopsin mutants to activate transducin was previously observed, because of the resulting ∼3 kcal/mol lower affinity for the Gtα tail peptide caused by the alanine substitutions in the “hydrophobic patch”.32 Together, these two lines of evidence suggest that arrestin and the Gtα C-terminal peptide share a common but not necessarily identical binding site on rhodopsin and that at least some of the arrestin binding affinity may similarly require interaction with the “hydrophobic patch”.
The Arrestin Finger Loop and 160 Loop Bind Close to the “Hydrophobic Patch” on the Base of Opsin TM6
To better localize the interactions mentioned above, we next tried to identify specific sites of interaction between arrestin and opsin, near where the Gtα peptide is known to bind. We used a constitutively active mutant of rhodopsin, M257Y,39−41 and a constitutively active arrestin (R175E) to conduct these studies in the absence of light-sensitive retinal. Although the use of constitutively active mutants for both proteins could conceivably affect some interaction between the two proteins, we propose that the fundamental contacts are still maintained, given the relatively high binding affinity we observe, and the fact that M257Y opsin and active opsin both have been shown to bind a high-affinity peptide corresponding to the C-terminus of Gtα in the same way.41 Our approach used the TrIQ fluorescence method, with which we sought to identify quenching of fluorescence labels on M257Y opsin upon binding by arrestin mutants containing strategically placed Trp residues. We have previously established that TrIQ can detect interactions between a fluorophore and the quenching tryptophan in the 5–15 Å distance range.38,65 Here, our goal was to use distance constraints obtained from our mapping studies to model the physical interaction between arrestin and opsin based on the pattern of site-specific quenching observed.
The two regions on arrestin that we tested, the finger loop and the 160 loop (see Figure 1d), displayed different quenching profiles for the probes on TM6 of opsin. No fluorescence quenching by the arrestin finger loop Trp mutants was seen for opsin T242B, suggesting a lack of proximity between these two sites (Figure 4a). However, one caveat about this specific subset of our data must be noted: the finger loop arrestin Trp mutants also showed less ability to block G protein activation by opsin T242B (Figure 4c), and this might be because they were not bound to the receptor at saturating levels. In fact, this might be expected if there is some steric clash at the interaction interface between the finger loop Trp residues on arrestin and the bimane attached to the receptor at the interaction interface, lowering binding affinity. Nevertheless, these mutants do still show statistically significant (∼50%) impairment of transducin activation by opsin T242B; thus, one would have expected to see some change in fluorescence if they were sufficiently close to cause considerable TrIQ. In contrast, two of the Trp residues in the finger loop of arrestin (Y67W and F79W) appear to be close to position 243, as indicated by the substantial quenching observed, whereas Trp residue I72W shows no quenching (Figures 3 and 4b). This is understandable from a structural standpoint, as both Y67 and F79 are nearby, at the base of the loop, in the arrestin crystal structure, while I72 is farther away (Figure S8 of the Supporting Information). These data provide a clear example of site-specific TrIQ.
Interpreting the data from the arrestin 160 loop Trp mutants is more straightforward. All of these mutants appear to bind both opsin T242B and opsin T243B, as shown by their ability to inhibit opsin-mediated activation of transducin to some degree. Thus, when fluorescence quenching is not observed for one of these mutants, it is reasonable to conclude the result is due to a lack of proximity between the Trp and bimane, and not to a lack of binding. However, we note that because some of the arrestin Trp mutants fail to entirely block G protein activation, we have based our conclusions and modeling of the arrestin–rhodopsin interface only on the results that clearly showed significant fluorescence quenching.
The 160 loop mutants also showed an interesting pattern of quenching, with the maximal TrIQ effect seen for Trp at positions 160 and 161 (Figure 4b). In fact, when we calculate the fraction of quenching due to static interactions, we present clear evidence that several residues in the 160 loop are able to physically contact the fluorescent probe at T242B. Recall that the presence of static quenching indicates that a quencher–probe pair is in a nonfluorescent complex on the time scale of light excitation, in our case ∼50 ps. Overall, the results suggest that arrestin binding places the finger loop and the 160 loop near positions 242 and 243 on TM6 of opsin.
Comparison of These Data with Those of Other Studies and Possible Implications
Gurevich and Benovic have proposed the presence of at least one “activation recognition” domain each within the segment of residues 16–145 and between residues 145 and 191 of arrestin.66 Because receptor activation exposes the TM5–TM6 cytosolic face, which contains T242 and T243, it is possible that the two sites we have studied, the finger loop (residues 67–79) and the 160 loop (residues 155–165), might be two of the proposed activation recognition domains. Thus, the involvement of the arrestin finger loop is not surprising, given the various lines of evidence pointing to its role in receptor binding.12,15,17,20,21,28,59,61,62 We also note that during revision of this manuscript, a paper appeared with a similar conclusion, based on extensive arrestin mutagenesis and modeling.67
However, our observation that the 160 loop of arrestin makes direct contact with rhodopsin was not anticipated, as it has not previously been strongly implicated in binding. There are, however, several clues suggesting it may be involved, including observations that a peptide corresponding to residues 151–170 of this region inhibits the binding of arrestin to metarhodopsin II,8 and the fact that binding of an anti-Myc antibody to a c-Myc tag inserted into this loop abolishes the binding of arrestin to RP*.9 We also note that even if the binding affinity of the 160 loop is enhanced by the introduction of hydrophobic Trp residues, our results still indicate that this region of arrestin has access to, and can bind, these sites on opsin.
Interestingly, the sequence of the 160 loop falls into a category of polypeptides, termed chameleons, that are thought to have a context-dependent structure, indicating a possible functional role.14,68 Consistent with this, there is considerable structural plasticity for the 160 loop in different crystal isoforms of arrestin, and recent double electron–electron resonance (DEER) EPR studies also find considerable plasticity in the 160 loop upon the binding of rhodopsin.62 Other EPR studies looking at the mobility of a spin-label probe at residue T157 of arrestin found a slight loss of mobility of the probe upon the binding of phosphorylated rhodopsin.12 This is consistent with our result, because although we see some quenching of fluorescence by T157W, it is not very strong, possibly indicating that this site is not juxtaposed at the binding interface.
Possible Models of the Arrestin–Rhodopsin Interaction
Our experiments were designed to map the arrestin–rhodopsin binding interface and use the distance and orientation constraints obtained from TrIQ measurements to model this interaction. However, modeling the arrestin–rhodopsin interaction is challenging because of the extremely flexible nature of the arrestin loops. In fact, the same loop may even adopt a number of different conformations upon binding to rhodopsin.62 Such ambiguity mandates that any model of the arrestin–rhodopsin binding interface based on presently available data will inherently be suggestive at best. With these caveats in mind, and assuming there are no large-scale rearrangements of arrestin or rhodopsin, below we discuss our structural models of the arrestin–rhodopsin binding interface generated from analysis of our current data.
Finger Loop Binding Mode
The most likely mode of binding involves the finger loop docking into the cleft exposed in the rhodopsin cytoplasmic face upon receptor activation and TM6 movement, as shown in Figure 6A. This model is consistent with other data suggesting the importance of this region in arrestin for binding, as well as our current data, in which we see arrestin binding is impaired by Gtα peptide binding (Figure 2b) and mutations in the “hydrophobic patch” (Figure 2c and Supporting Information). Such a binding orientation could also provide favorable contacts between the numerous hydrophobic residues on the tip of the arrestin finger loop (I72, V74, and M75) and the “hydrophobic patch” residues on the inner face of rhodopsin TM5 [L226 and V230 (shown as gray spheres in Figure 6A)]. Interestingly, this docking model also places a number of other hydrophobic residues on the arrestin finger loop (V74, M75, and L77) in direct contact with a string of hydrophobic residues on the inner face of rhodopsin TM6 (A246, V250, and M253), suggesting this region may act as a previously unanticipated “hydrophobic patch” specific for arrestin. We propose this arrangement likely reflects the “high-affinity” arrestin binding mode.
Figure 6.

Possible arrestin–rhodopsin binding models. On rhodopsin (orange), the sites where the fluorophore bimane was attached on TM6 are shown as green spheres, and the sites of the “hydrophobic patch” residues on TM5 are shown as gray spheres. On arrestin (gray), the sites where Trp residues were introduced into the arrestin finger loop are indicated with red spheres and in the 160 loops with blue spheres. The spheres reflect the Cα position sites for rhodopsin and Cβ position sites for arrestin. (a) Model of the arrestin finger loop binding to the cleft in the rhodopsin cytoplasmic face, near the “hydrophobic patch”. Note that with some rearrangement of rhodopsin loops, this docking mode can also accommodate the finger loop adopting an α-helix, as has been proposed from NMR studies,61 as shown in Figure S10 of the Supporting Information. (b) Model of the arrestin 160 loop binding to the cleft in rhodopsin. Interestingly, although binding orientation a or b is consistent with the biochemical data reported here, neither orientation can by itself explain all of the TrIQ data shown in Figures 4 and 5. One possibility is that the TrIQ data reflect heterogeneous binding (binding in which both modes a and b occur simultaneously to different receptors). Alternatively, the TrIQ data can also be interpreted to reflect the binding of arrestin to a rhodopsin dimer. One possibility is shown in panel c, which illustrates that a rhodopsin dimer with a TM1/TM4/H8 interface satisfies the same individual arrestin–rhodopsin orientations shown in panels a and b. Interestingly, it is also possible to satisfy the TrIQ data using a model in which arrestin binds to a rhodopsin dimer with a TM4/TM5 interface, as shown in panel d. Other models involving large structural changes cannot be ruled out. Models for M257Y opsin (PDB entry 4A4M, chain A) and arrestin (PDB entry 4J2Q, chain B) were generated using Chimera. Further details are provided in the text. Space-filling representations of each model are provided as insets in the bottom left corner of each panel.
Docking of the finger loop into the cleft is also consistent with our TrIQ data, as it models the Trp residues introduced on the edge of the arrestin finger loop (Y67W and F79W) to have access to (and thus be able to quench) the bimane probe at position 243 of rhodopsin.
The 160 Loop Binding Mode
Our data are also consistent with an unexpected, alternate binding mode, in which the arrestin 160 loop docks into the cleft. As shown in Figure 6B, such an orientation is sterically allowed and would also satisfy the TrIQ results. We note that the TrIQ data clearly show a number of Trp residues in the 160 loop form static quenching complexes with the bimane probe at site 242 on opsin, indicating direct contact between two molecules. This was initially surprising, because it is hard to imagine a single 160 loop conformation that could make this possible for all of the different Trp residues. However, this anomaly could be explained if the 160 loop adopts more than one conformation upon binding opsin, as has been suggested by recent EPR studies.62 Thus, we stress that the model shown in Figure 6B is only suggestive at present (given the highly variable structure for the 160 loop) and is unlikely to reflect a high-affinity binding mode.
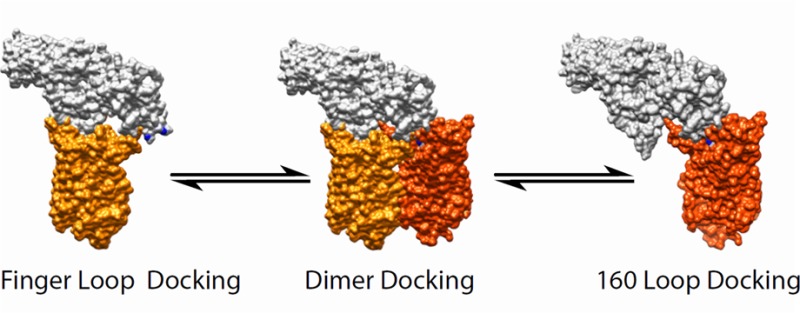
Evidence That Arrestin Uses Heterogeneous Binding Modes and/or Binds to Rhodopsin Dimers
Either binding mode discussed above is plausible and consistent with the data presented here. However, together they present a conundrum, as neither model allows for an orientation of arrestin in which the Trp residues on the finger loop and 160 loop can both access and quench the bimane probes at positions 242 and 243 on opsin at the same time.
In either model, one set of the Trp quenchers is too far away (almost 30 Å apart) to be optimal for the TrIQ results we see here. More importantly, the orientations involved do not allow equal access for both sets of Trp residues to quench the bimane probes. Although arrestin has been proposed to undergo a significant conformational rearrangement, none of the changes indicated by recent structural data produce movements large enough to allow both the finger loop and 160 loop in arrestin to be in the proximity,4,62,69,70 as would be required for our data. Thus, our TrIQ data cannot be readily explained by the exclusive use of only one of the monomeric binding models presented above.
There are several intriguing possible interpretations of our data. One is that arrestin binding is heterogeneous; that is, some fraction of the arrestin binds in one orientation and the other in the alternate (for example, panels a and b of Figure 6). In other words, there is a mixed population of receptors in which some arrestin is bound with the finger loop placed inside the cleft in rhodopsin, near the base of TM6, and others with arrestins having the 160 loop positioned into this cleft. We note there is mounting evidence supporting this possibility.20,21
An alternate possibility is that the data reflect the binding of arrestin to dimers of rhodopsin. We find arrestin can be docked to a rhodopsin dimer in such a way that both sets of TrIQ data can be simultaneously satisfied, if it straddles a dimer in which two rhodopsin molecules are facing each other at their TM1 and TM4 helices, with the H8 forms next to each other in an antiparallel fashion (Figure 6c). With such an arrangement, the probes at positions 242 and 243 on TM6 could simultaneously be quenched by the Trp residues on the arrestin finger loop and the 160 loop. This suggestion is consistent with existing experimental evidence that arrestin can bind to opsin dimers.27 Moreover, multiple techniques (EM, cross-linking, and crystallography) have suggested a TM1/4 dimer is indeed a biologically significant oligomeric state.41,71,72 Intriguingly, we also found that both sets of TrIQ data could be satisfied when arrestin is modeled straddling a rhodopsin dimer with a TM4/5 interface (Figure 6d), an orientation for which there is also significant evidence for both rhodopsin57,73,74 and other GPCRs.75,76
In summary, we provide models consistent with our data in which arrestin binding utilizes the same crevice exposed on rhodopsin activation as does transducin, and we propose that at least part of this binding requires interaction with a “hydrophobic patch” on TM5, and perhaps a second such patch on TM6. Moreover, using the TrIQ approach, we have identified two sites on TM6 of rhodopsin near this crevice that make contact with two sites on arrestin, the finger loop and the 160 loop.
As discussed above, because it is not clear how the two extreme ends of the N-domain separated by almost 30 Å could interact with or lie near the same site on rhodopsin, we propose that our data reflect the fact that arrestin binds in different orientations, binds to rhodopsin dimers, or binds in both possible ways. We currently cannot rule out any of these models, based on the data presented here, nor can we rule out the possibility that some large-scale structural change occurs in both proteins upon binding that would allow the quenching we report here. More work is needed to identify other sites of contact to better triangulate and refine the models, and to test the idea of monomeric versus dimeric interactions.
However, although more sets of interacting pairs between the two proteins will be required to determine the exact mode(s) of arrestin–rhodopsin interaction, our studies described here do provide preliminary structural constraints that can be used to design further experiments and begin modeling the arrestin–rhodopsin interaction. We also note we have identified conditions under which arrestin can bind ligand free M257Y opsin and in so doing form a stable, long-lasting complex (Figure S9 of the Supporting Information). These mutants and conditions may prove to be useful for forming complexes that are stable enough for cocrystallization or electron microscopy studies.
Finally, we are hopeful that our use of TrIQ to map protein–protein interaction sites can be adapted to other systems. With further refinement and calibration, the general approach we outline here (defining sites of contact between proteins by analyzing fluorescence lifetime and steady-state fluorescence quenching data) should prove to be applicable to the analysis of other interacting proteins.
Acknowledgments
We thank the late Dr. Kevin Ridge (University of Texas, Houston, TX) for providing the initial arrestin plasmid and Dr. Ujwal Shinde for thoughtful discussions and suggestions.
Glossary
Abbreviations
- TM
transmembrane helix
- TrIQ
tryptophan-induced quenching
- mBBr
monobromobimane
- GPCR
G protein-coupled receptor
- ROS
rod outer segment
- R
rhodopsin
- R*
light-activated rhodopsin
- RP
phosphorylated rhodopsin
- RP*
light-activated, phosphorylated rhodopsin
- GTP
guanosine triphosphate
- GTPγS
guanosine 5′-O-(3-thiotriphosphate)
- Gt
transducin
- Gtα
transducin Gα subunit
- DOPA
1,2-dioleoyl-sn-glycero-3-phosphate
- DOPS
1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- DM
n-dodecyl β-d-maltoside
- PDB
Protein Data Bank.
Supporting Information Available
Spectra for the retinal trapping experiments, the determination of free label in TrIQ experiments, an SDS–PAGE analysis of the G protein transducin purification, an example of the how a peptide corresponding to the Gtα C-terminal tail inhibits the TrIQ observed upon arrestin binding, alternate models for the arrestin–rhodopsin interaction, including one in which the finger loop of arrestin takes a helical conformation, and fluorescence quenching ratios as well as lifetimes for each Trp-containing arrestin and bimane-labeled opsin pair (tables). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† A.S. and A.M.J.B. contributed equally to the final aspects of this work.
This work was supported by National Institutes of Health Grant R01 EY015436 (D.L.F.) and Training Grants 5T32GM071338 (A.M.J.B.) and 1T32EY023211 and 5T32GM071338-08 (C.T.S.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Luttrell L. M. (2006) Transmembrane signaling by G protein-coupled receptors. Methods Mol. Biol. 332, 3–49. [DOI] [PubMed] [Google Scholar]
- Gurevich V. V.; Gurevich E. V. (2008) Rich tapestry of G protein-coupled receptor signaling and regulatory mechanisms. Mol. Pharmacol. 74, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich V. V.; Hanson S. M.; Song X.; Vishnivetskiy S. A.; Gurevich E. V. (2011) The functional cycle of visual arrestins in photoreceptor cells. Prog. Retinal Eye Res. 30, 405–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich V. V.; Gurevich E. V. (2006) The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol. Ther. 110, 465–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieselbach T.; Irrgang K. D.; Ruppel H. (1994) A segment corresponding to amino acids Val170-Arg182 of bovine arrestin is capable of binding to phosphorylated rhodopsin. Eur. J. Biochem. 226, 87–97. [DOI] [PubMed] [Google Scholar]
- Ohguro H.; Palczewski K.; Walsh K. A.; Johnson R. S. (1994) Topographic study of arrestin using differential chemical modifications and hydrogen/deuterium exchange. Protein Sci. 3, 2428–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith W. C.; McDowell J. H.; Dugger D. R.; Miller R.; Arendt A.; Popp M. P.; Hargrave P. A. (1999) Identification of regions of arrestin that bind to rhodopsin. Biochemistry 38, 2752–2761. [DOI] [PubMed] [Google Scholar]
- Pulvermuller A.; Schroder K.; Fischer T.; Hofmann K. P. (2000) Interactions of metarhodopsin II. Arrestin peptides compete with arrestin and transducin. J. Biol. Chem. 275, 37679–37685. [DOI] [PubMed] [Google Scholar]
- Dinculescu A.; McDowell J. H.; Amici S. A.; Dugger D. R.; Richards N.; Hargrave P. A.; Smith W. C. (2002) Insertional mutagenesis and immunochemical analysis of visual arrestin interaction with rhodopsin. J. Biol. Chem. 277, 11703–11708. [DOI] [PubMed] [Google Scholar]
- Smith W. C.; Dinculescu A.; Peterson J. J.; McDowell J. H. (2004) The surface of visual arrestin that binds to rhodopsin. Mol. Vision 10, 392–398. [PubMed] [Google Scholar]
- Vishnivetskiy S. A.; Hosey M. M.; Benovic J. L.; Gurevich V. V. (2004) Mapping the arrestin-receptor interface. Structural elements responsible for receptor specificity of arrestin proteins. J. Biol. Chem. 279, 1262–1268. [DOI] [PubMed] [Google Scholar]
- Hanson S. M.; Francis D. J.; Vishnivetskiy S. A.; Kolobova E. A.; Hubbell W. L.; Klug C. S.; Gurevich V. V. (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 4900–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy S. A.; Gimenez L. E.; Francis D. J.; Hanson S. M.; Hubbell W. L.; Klug C. S.; Gurevich V. V. (2011) Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J. Biol. Chem. 286, 24288–24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch J. A.; Schubert C.; Gurevich V. V.; Sigler P. B. (1999) The 2.8 Å crystal structure of visual arrestin: A model for arrestin’s regulation. Cell 97, 257–269. [DOI] [PubMed] [Google Scholar]
- Sommer M. E.; Farrens D. L.; McDowell J. H.; Weber L. A.; Smith W. C. (2007) Dynamics of arrestin-rhodopsin interactions: Loop movement is involved in arrestin activation and receptor binding. J. Biol. Chem. 282, 25560–25568. [DOI] [PubMed] [Google Scholar]
- Sommer M. E.; Smith W. C.; Farrens D. L. (2005) Dynamics of arrestin-rhodopsin interactions: Arrestin and retinal release are directly linked events. J. Biol. Chem. 280, 6861–6871. [DOI] [PubMed] [Google Scholar]
- Krupnick J. G.; Gurevich V. V.; Schepers T.; Hamm H. E.; Benovic J. L. (1994) Arrestin-rhodopsin interaction. Multi-site binding delineated by peptide inhibition. J. Biol. Chem. 269, 3226–3232. [PubMed] [Google Scholar]
- Shi W.; Sports C. D.; Raman D.; Shirakawa S.; Osawa S.; Weiss E. R. (1998) Rhodopsin arginine-135 mutants are phosphorylated by rhodopsin kinase and bind arrestin in the absence of 11-cis-retinal. Biochemistry 37, 4869–4874. [DOI] [PubMed] [Google Scholar]
- Raman D.; Osawa S.; Weiss E. R. (1999) Binding of arrestin to cytoplasmic loop mutants of bovine rhodopsin. Biochemistry 38, 5117–5123. [DOI] [PubMed] [Google Scholar]
- Sommer M. E.; Hofmann K. P.; Heck M. (2012) Distinct loops in arrestin differentially regulate ligand binding within the GPCR opsin. Nat. Commun. 3, 995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang T.; Chen Q.; Cho M. K.; Vishnivetskiy S. A.; Iverson T. M.; Gurevich V. V.; Sanders C. R. (2013) Involvement of distinct arrestin-1 elements in binding to different functional forms of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 110, 942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C.; Kusnetzow A. K.; Ernst O. P.; Hofmann K. P.; Hubbell W. L. (2008) High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc. Natl. Acad. Sci. U.S.A. 105, 7439–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C.; Yang K.; Farrens D. L.; Farahbakhsh Z. T.; Khorana H. G.; Hubbell W. L. (1996) Structural features and light-dependent changes in the cytoplasmic interhelical E-F loop region of rhodopsin: A site-directed spin-labeling study. Biochemistry 35, 12470–12478. [DOI] [PubMed] [Google Scholar]
- Hanson S. M.; Van Eps N.; Francis D. J.; Altenbach C.; Vishnivetskiy S. A.; Arshavsky V. Y.; Klug C. S.; Hubbell W. L.; Gurevich V. V. (2007) Structure and function of the visual arrestin oligomer. EMBO J. 26, 1726–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson S. M.; Vishnivetskiy S. A.; Hubbell W. L.; Gurevich V. V. (2008) Opposing effects of inositol hexakisphosphate on rod arrestin and arrestin2 self-association. Biochemistry 47, 1070–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham T. D.; Farrens D. L. (1999) Conformational changes in rhodopsin. Movement of helix f detected by site-specific chemical labeling and fluorescence spectroscopy. J. Biol. Chem. 274, 1683–1690. [DOI] [PubMed] [Google Scholar]
- Sommer M. E.; Hofmann K. P.; Heck M. (2011) Arrestin-rhodopsin binding stoichiometry in isolated rod outer segment membranes depends on the percentage of activated receptors. J. Biol. Chem. 286, 7359–7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer M. E.; Smith W. C.; Farrens D. L. (2006) Dynamics of arrestin-rhodopsin interactions: Acidic phospholipids enable binding of arrestin to purified rhodopsin in detergent. J. Biol. Chem. 281, 9407–9417. [DOI] [PubMed] [Google Scholar]
- Hubbell W. L.; Altenbach C.; Hubbell C. M.; Khorana H. G. (2003) Rhodopsin structure, dynamics, and activation: A perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv. Protein Chem. 63, 243–290. [DOI] [PubMed] [Google Scholar]
- Farrens D. L.; Altenbach C.; Yang K.; Hubbell W. L.; Khorana H. G. (1996) Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274, 768–770. [DOI] [PubMed] [Google Scholar]
- Farrens D. L. (2010) What site-directed labeling studies tell us about the mechanism of rhodopsin activation and G-protein binding. Photochem. Photobiol. Sci. 9, 1466–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz J. M.; Farrens D. L. (2004) Rhodopsin activation exposes a key hydrophobic binding site for the transducin α-subunit C terminus. J. Biol. Chem. 279, 29767–29773. [DOI] [PubMed] [Google Scholar]
- Sheikh S. P.; Zvyaga T. A.; Lichtarge O.; Sakmar T. P.; Bourne H. R. (1996) Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature 383, 347–350. [DOI] [PubMed] [Google Scholar]
- Acharya S.; Saad Y.; Karnik S. S. (1997) Transducin-α C-terminal peptide binding site consists of C-D and E-F loops of rhodopsin. J. Biol. Chem. 272, 6519–6524. [DOI] [PubMed] [Google Scholar]
- Van Eps N.; Anderson L. L.; Kisselev O. G.; Baranski T. J.; Hubbell W. L.; Marshall G. R. (2010) Electron paramagnetic resonance studies of functionally active, nitroxide spin-labeled peptide analogues of the C-terminus of a G-protein α subunit. Biochemistry 49, 6877–6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H. W.; Kim Y. J.; Park J. H.; Morizumi T.; Pai E. F.; Krauss N.; Hofmann K. P.; Scheerer P.; Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655. [DOI] [PubMed] [Google Scholar]
- Scheerer P.; Park J. H.; Hildebrand P. W.; Kim Y. J.; Krauss N.; Choe H. W.; Hofmann K. P.; Ernst O. P. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502. [DOI] [PubMed] [Google Scholar]
- Mansoor S. E.; Dewitt M. A.; Farrens D. L. (2010) Distance mapping in proteins using fluorescence spectroscopy: The tryptophan-induced quenching (TrIQ) method. Biochemistry 49, 9722–9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standfuss J.; Xie G.; Edwards P. C.; Burghammer M.; Oprian D. D.; Schertler G. F. (2007) Crystal structure of a thermally stable rhodopsin mutant. J. Mol. Biol. 372, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M.; Smith S. O.; Sakmar T. P. (1998) Constitutive activation of opsin by mutation of methionine 257 on transmembrane helix 6. Biochemistry 37, 8253–8261. [DOI] [PubMed] [Google Scholar]
- Deupi X.; Edwards P.; Singhal A.; Nickle B.; Oprian D.; Schertler G.; Standfuss J. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl. Acad. Sci. U.S.A. 109, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G.; Gross A. K.; Oprian D. D. (2003) An opsin mutant with increased thermal stability. Biochemistry 42, 1995–2001. [DOI] [PubMed] [Google Scholar]
- Gurevich V. V.; Dion S. B.; Onorato J. J.; Ptasienski J.; Kim C. M.; Sterne-Marr R.; Hosey M. M.; Benovic J. L. (1995) Arrestin interactions with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, β2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 270, 720–731. [DOI] [PubMed] [Google Scholar]
- Molday R. S.; MacKenzie D. (1983) Monoclonal antibodies to rhodopsin: Characterization, cross-reactivity, and application as structural probes. Biochemistry 22, 653–660. [DOI] [PubMed] [Google Scholar]
- Molday R. S.; MacKenzie D. (1985) Inhibition of monoclonal antibody binding and proteolysis by light-induced phosphorylation of rhodopsin. Biochemistry 24, 776–781. [DOI] [PubMed] [Google Scholar]
- Papermaster D. S. (1982) Preparation of retinal rod outer segments. Methods Enzymol. 81, 48–52. [DOI] [PubMed] [Google Scholar]
- Janz J. M.; Farrens D. L. (2004) Rhodopsin activation exposes a key hydrophobic binding site for the transducin α-subunit C terminus. J. Biol. Chem. 279, 29767–29773. [DOI] [PubMed] [Google Scholar]
- Resek J. F.; Farahbakhsh Z. T.; Hubbell W. L.; Khorana H. G. (1993) Formation of the meta II photointermediate is accompanied by conformational changes in the cytoplasmic surface of rhodopsin. Biochemistry 32, 12025–12032. [DOI] [PubMed] [Google Scholar]
- Resek J. F.; Farrens D.; Khorana H. G. (1994) Structure and function in rhodopsin: Covalent crosslinking of the rhodopsin (metarhodopsin II)-transducin complex—the rhodopsin cytoplasmic face links to the transducin α subunit. Proc. Natl. Acad. Sci. U.S.A. 91, 7643–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Mao X.; Abdulaev N. G.; Ngo T.; Liu W.; Ridge K. D. (2012) One-step purification of a functional, constitutively activated form of visual arrestin. Protein Expression Purif. 82, 55–60. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H.; Sinha A.; DeWitt M.; Farrens D. L. (2010) Monomeric rhodopsin is the minimal functional unit required for arrestin binding. J. Mol. Biol. 399, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd ed., Springer, Berlin. [Google Scholar]
- Matsuda T.; Fukada Y. (2000) Functional analysis of farnesylation and methylation of transducin. Methods Enzymol. 316, 465–481. [DOI] [PubMed] [Google Scholar]
- Goc A.; Angel T. E.; Jastrzebska B.; Wang B.; Wintrode P. L.; Palczewski K. (2008) Different properties of the native and reconstituted heterotrimeric G protein transducin. Biochemistry 47, 12409–12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya A. I.; Thaker T. M.; Preininger A. M.; Iverson T. M.; Hamm H. E. (2011) Coupling efficiency of rhodopsin and transducin in bicelles. Biochemistry 50, 3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoor S. E.; Palczewski K.; Farrens D. L. (2006) Rhodopsin self-associates in asolectin liposomes. Proc. Natl. Acad. Sci. U.S.A. 103, 3060–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D.; Liang Y.; Filipek S.; Saperstein D. A.; Engel A.; Palczewski K. (2003) Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature 421, 127–128. [DOI] [PubMed] [Google Scholar]
- Yang K.; Farrens D. L.; Hubbell W. L.; Khorana H. G. (1996) Structure and function in rhodopsin. Single cysteine substitution mutants in the cytoplasmic interhelical E-F loop region show position-specific effects in transducin activation. Biochemistry 35, 12464–12469. [DOI] [PubMed] [Google Scholar]
- Sommer M. E.; Farrens D. L. (2006) Arrestin can act as a regulator of rhodopsin photochemistry. Vision Res. 46, 4532–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexiev U.; Farrens D. L. (2014) Fluorescence spectroscopy of rhodopsins: Insights and approaches. Biochim. Biophys. Acta 1837, 694–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein S. E.; Pulvermuller A.; Hartmann R.; Granzin J.; Stoldt M.; Henklein P.; Ernst O. P.; Heck M.; Willbold D.; Koenig B. W. (2009) Helix formation in arrestin accompanies recognition of photoactivated rhodopsin. Biochemistry 48, 10733–10742. [DOI] [PubMed] [Google Scholar]
- Kim M.; Vishnivetskiy S. A.; Van Eps N.; Alexander N. S.; Cleghorn W. M.; Zhan X.; Hanson S. M.; Morizumi T.; Ernst O. P.; Meiler J.; Gurevich V. V.; Hubbell W. L. (2012) Conformation of receptor-bound visual arrestin. Proc. Natl. Acad. Sci. U.S.A. 109, 18407–18412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupnick J. G.; Gurevich V. V.; Benovic J. L. (1997) Mechanism of quenching of phototransduction. Binding competition between arrestin and transducin for phosphorhodopsin. J. Biol. Chem. 272, 18125–18131. [DOI] [PubMed] [Google Scholar]
- Knierim B.; Hofmann K. P.; Ernst O. P.; Hubbell W. L. (2007) Sequence of late molecular events in the activation of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 104, 20290–20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoor S. E.; Farrens D. L. (2004) High-throughput protein structural analysis using site-directed fluorescence labeling and the bimane derivative (2-pyridyl)dithiobimane. Biochemistry 43, 9426–9438. [DOI] [PubMed] [Google Scholar]
- Gurevich V. V.; Benovic J. L. (1993) Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J. Biol. Chem. 268, 11628–11638. [PubMed] [Google Scholar]
- Ostermaier M. K.; Peterhans C.; Jaussi R.; Deupi X.; Standfuss J. (2014) Functional map of arrestin-1 at single amino acid resolution. Proc. Natl. Acad. Sci. U.S.A. 111, 1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzin J.; Wilden U.; Choe H. W.; Labahn J.; Krafft B.; Buldt G. (1998) X-ray crystal structure of arrestin from bovine rod outer segments. Nature 391, 918–921. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy S. A.; Hirsch J. A.; Velez M. G.; Gurevich Y. V.; Gurevich V. V. (2002) Transition of arrestin into the active receptor-binding state requires an extended interdomain hinge. J. Biol. Chem. 277, 43961–43967. [DOI] [PubMed] [Google Scholar]
- Shukla A. K.; Manglik A.; Kruse A. C.; Xiao K.; Reis R. I.; Tseng W. C.; Staus D. P.; Hilger D.; Uysal S.; Huang L. Y.; Paduch M.; Tripathi-Shukla P.; Koide A.; Koide S.; Weis W. I.; Kossiakoff A. A.; Kobilka B. K.; Lefkowitz R. J. (2013) Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht J. J.; Mielke T.; Vogel R.; Villa C.; Schertler G. F. (2004) Electron crystallography reveals the structure of metarhodopsin I. EMBO J. 23, 3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepp A. M.; Periole X.; Marrink S. J.; Sakmar T. P.; Huber T. (2012) Rhodopsin forms a dimer with cytoplasmic helix 8 contacts in native membranes. Biochemistry 51, 1819–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota P.; Reeves P. J.; Rajbhandary U. L.; Khorana H. G. (2006) Opsin is present as dimers in COS1 cells: Identification of amino acids at the dimeric interface. Proc. Natl. Acad. Sci. U.S.A. 103, 3054–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Fotiadis D.; Filipek S.; Saperstein D. A.; Palczewski K.; Engel A. (2003) Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J. Biol. Chem. 278, 21655–21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baneres J. L.; Parello J. (2003) Structure-based analysis of GPCR function: Evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J. Mol. Biol. 329, 815–829. [DOI] [PubMed] [Google Scholar]
- Johnston J. M.; Wang H.; Provasi D.; Filizola M. (2012) Assessing the relative stability of dimer interfaces in g protein-coupled receptors. PLoS Comput. Biol. 8, e1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.