

Abstract
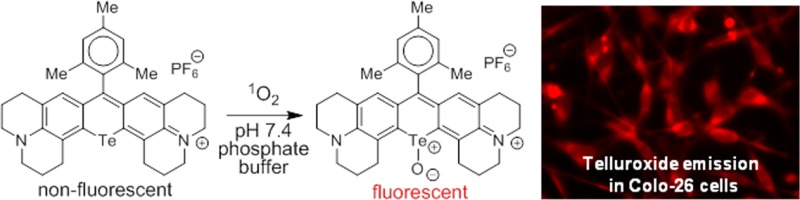
Analogues of Texas red incorporating the heavy chalcogens S, Se, and Te atoms in the xanthylium core were prepared from the addition of aryl Grignard reagents to appropriate chalcogenoxanthone precursors. The xanthones were prepared via directed metalation of amide precursors, addition of dichalcogenide electrophiles, and electrophilic cyclization of the resulting chalcogenides with phosphorus oxychloride and triethylamine. The Texas red analogues incorporate two fused julolidine rings containing the rhodamine nitrogen atoms. Analogues containing two “half-julolidine” groups (a trimethyltetrahydroquinoline) and one julolidine and one “half-julolidine” were also prepared. The photophysics of the Texas red analogues were examined. The S-analogues were highly fluorescent, the Se-analogues generated single oxygen (1O2) efficiently upon irradiation, and the Te-analogues were easily oxidized to rhodamines with the telluroxide oxidation state. The tellurorhodamine telluroxides absorb at wavelengths ≥690 nm and emit with fluorescence maxima >720 nm. A mesityl-substituted tellurorhodamine derivative localized in the mitochondria of Colo-26 cells (a murine colon carcinoma cell line) and was oxidized in vitro to the fluorescent telluroxide.
Introduction
The two fused julolidine rings of the rhodamine Texas red (1, Chart 1) lock the N atoms into conjugation with the rhodamine xanthylium core leading to longer wavelengths of absorption. Compound 1 and related structures have found numerous applications, primarily based on their wavelength and efficiency of fluorescence and their flat, rigid nature, which has assisted in binding to biopolymers.1−12 Analogues of 1 with even longer wavelengths of absorbance and emission have been of interest.12−14
Chart 1.

We have prepared heavy-atom analogues of the tetramethylrosamines/rhodamines (TMR-E, Chart 1)15−17 and analogues incorporating one julolidine group18 and, more recently, one “half-julolidine” group (a trimethyltetrahydroquinoline, Chart 1) in the xanthylium core.19 The introduction of a single fused julolidine or half-julolidine group gives a small increase in absorption maxima (λmax), but larger shifts in λmax are realized by replacing the oxygen atom of the xanthylium core with the heavier chalcogen atoms S, Se, and Te. As the chalcogen atoms increase in size, the resulting rhodamines have decreasing quantum yields of fluorescence (ΦFL) and increasing quantum yields for the generation of triplets and singlet oxygen [Φ(1O2)].15−18 The synthesis of Texas red derivatives incorporating the heavier chalcogen atoms S, Se, and Te should give rhodamine analogues with values of λmax > 600 nm with control of relative yields for fluorescence and triplet production for different applications.
A second observation regarding analogues of TMR-Te is the facile oxidation to the telluroxide oxidation state. The rhodamine telluroxides absorb at much longer wavelengths (>60 nm longer) than the reduced forms. Furthermore, the rhodamine telluroxides are highly fluorescent, while the reduced tellurorhodamine/rosamines are nonfluorescent.20,21 Tellurium-containing derivatives of 1 should be readily oxidized to telluroxides with absorption and emission wavelengths in the near-infrared.
Herein, we describe the synthesis of S-, Se-, and Te-containing analogues of 1 and related compounds via the chalcogenoxanthone precursors 2–4 (Chart 2). The Texas red analogues 5–7 with a 9-phenyl substituent were then prepared (Chart 2). The S-containing analogues were highly fluorescent, the Se-containing analogues were an order of magnitude less fluorescent and generated singlet oxygen (1O2) efficiently upon irradiation, and the Te-containing analogues were easily oxidized to the telluroxide oxidation state and absorb at wavelengths >690 nm and emit with fluorescence maxima >720 nm. Preliminary studies also indicate that tellurorhodamines localize in the mitochondria of cells and that in vitro oxidation of a tellurorhodamine and emission from the resulting telluroxide can be observed in cells in culture.
Chart 2.

Results and Discussion
Synthesis
Our synthetic approach is analyzed in Scheme 1. The key to the successful synthesis of TMR-related molecules or derivatives incorporating a julolidine or half-julolidine was access to the corresponding chalcogenoxanthones as precursors to the rhodamine analogues.18,19,22−24 The synthesis of chalcogenoxanthones is similarly key to this work. Texas red analogues such as 5–7 can be prepared via the addition of PhMgBr (or other Grignard or organolithium reagents) to chalcogenoxanthones 2–4 followed by treatment with acid. Chalcogenoxanthones 2–4 can be prepared via electrophilic cyclization of the diaryl chalcogenide intermediates A (Scheme 1) under modified Friedel–Crafts conditions. Structures related to A have shown a propensity to cyclize para to the amino substituent under these conditions.19,22−24 Intermediates A can be prepared via the addition of a diaryl dichalcogenide B bearing a m-amino substituent to an anion C prepared by amide-directed metalation.19,22−24
Scheme 1.

The specific dichalcogenide precursors B were prepared as shown in Scheme 2. 3-Bromoaniline (8) was converted to 8-bromojulolidine (9) using a modification of the literature procedure used to prepare other 8-substituted julolidines.25 Alkylation of 8 with excess 1-bromo-3-chloropropane at 140 °C in the presence of Na2CO3 gave dialkylation of 8 in essentially quantitative yield. The unpurified dialkylation product was isolated by extraction, dried, and then heated at 160 °C in anhydrous DMF for 48 h to give 9(26) in 68% isolated yield overall. Formation of the Grignard reagent from 9 with Mg turnings in THF proceeded only when the concentration of 9 in THF was at least 2.0 M. At lower concentrations, the Grignard reagent either did not form or formed sluggishly. Elemental S, Se, or Te was then added to the Grignard reagent from 9 to give the corresponding julolidyl dichalcogenides 10 following air oxidation.
Scheme 2.

The half-julolidyl analogues 13 were prepared via allylation of commercially available 3-bromo-N-methylaniline with 1-chloro-3-methylbut-2-ene in DMF at 95 °C to give aniline 11 in 95% yield (Scheme 2). These conditions are more strenuous than the literature conditions25 used for the allylation of N-methylaniline (1-chloro-3-methylbut-2-ene and K2CO3 in CH3CN at 40 °C) and were necessary to give product formation. Cyclization of 11 with concentrated H2SO4 gave tetrahydroquinoline 12.27 The Grignard reagent from 12 reacted with elemental S, Se, or Te to give dichalcogenides 13 following air oxidation.
Chalcogenoxanthones 2–4 were prepared as shown in Scheme 3. Julolidine-9-carboxamide 14(22) was treated with s-BuLi and TMEDA to give directed ortho-lithiation. Treating the resulting anion with a diaryl dichalcogenide 10 gave the unsymmetrical diaryl chalcogenides 15, which were cyclized to xanthones 2 with POCl3 and Et3N in CH3CN.23 Similarly, tetrahydroquinoline carboxamide 16(19) was treated with s-BuLi and TMEDA followed by a diaryl dichalcogenide 13 to give the unsymmetrical diaryl chalcogenides 17, which were cyclized to xanthones 3 with POCl3 and Et3N in CH3CN.23 The unsymmetrical xanthones 4 were prepared from carboxamide 14 and diaryl dichalcogenides 13 via the intermediacy of diaryl chalcogenides 18. The cyclization of 18 with POCl3 and Et3N in CH3CN23 gave xanthones 4.
Scheme 3.

Alternatively, xanthones 4 were prepared from directed metalation of 16 followed by treating the resulting anion with a dichalcogenide 10-E. This approach gave poorer yields than the procedure shown in Scheme 3.
Compounds 2–4 were converted to the Texas red analogues 5–7 (Scheme 2) with PhMgBr in THF followed by work up with aqueous HPF6 (Table 1). Other 9-substituents should be readily introduced using other Grignard or organolithium reagents.15,16,18−22
Table 1. Synthetic Yields, Absorption Maxima (λmax), and Molar Extinction Coefficients (ε) in CH3OH, Fluorescence Emission Maxima (λFL) and Quantum Yields for Fluorescence (ΦFL) in CH3OH, Quantum Yields for the Generation of Singlet Oxygen [Φ(1O2)] in CH3OH for 5-E–7-E, and Values of λmax, ε, and Φ(1O2) for 19-Se and 20-Se.
| compd | % yield | λmax (nm) | ε (M–1 cm–1) | λFL (nm) | ΦFLa | Φ(1O2)a |
|---|---|---|---|---|---|---|
| TMR-Ob | 550 | 8.66 × 104 | 575 | 0.84 | 0.08 | |
| TMR-Sb | 571 | 6.26 × 104 | 599 | 0.44 | 0.21 | |
| TMR-Seb | 581 | 4.4 × 104 | 608 | 0.009 | 0.87 | |
| TMR-Te | 597c | 8.1 × 104c | <0.005d | 0.43d | ||
| 1 | 587e | 8.5 × 104e | 615e | |||
| 5-S | 91 | 594 | 1.24 × 105 | 621 | 0.42 | <0.05 |
| 5-Se | 86 | 604 | 1.35 × 105 | 631 | 0.06 | 0.68 |
| 5-Te | 92 | 617 | 1.44 × 105 | 642 | 0.002 | 0.53 |
| 6-S | 92 | 579 | 1.04 × 105 | 606 | 0.53 | <0.05 |
| 6-Se | 78 | 590 | 1.36 × 105 | 617 | 0.03 | 0.79 |
| 6-Te | 75 | 607 | 1.21 × 105 | 632 | 0.002 | 0.47 |
| 7-S | 90 | 587 | 1.20 × 105 | 614 | 0.47 | <0.05 |
| 7-Se | 75 | 597 | 1.22 × 105 | 624 | 0.02 | 0.81 |
| 7-Te | 89 | 610 | 1.27 × 105 | 637 | 0.003 | 0.40 |
| 19-Sef | 588 | 1.26 × 105 | 0.85 | |||
| 20-Se | 74 | 585 | 1.35 × 105 | 0.88 |
It should be noted that the directed metalation of 16 gave only one regioisomer following the addition of the dichalcogenide electrophile. No product resulting from electrophilic attack at the carbon next to the gem-dimethyl substituents was observed. The electrophilic cyclization of intermediates 17 gave only one product–acylation at the para-position relative to the amino substituent. The synthetic routes to chalcogenoxanthones 2 and 4 could only lead to one regioisomer in the directed metalation reaction and, for 2, only one product in the electrophilic cyclization.
Photophysical Properties
Texas red (1) has an absorbance maximum, λmax, of 587 nm in MeOH with a molar extinction coefficient, ε, of 8.5 × 104 M–1 cm–1 (Table 1).1 The Texas red analogues 5 incorporating the heavier chalcogen analogues have values of λmax at longer wavelengths (594–617 nm, Table 1). For comparison purposes, values of λmax and ε in MeOH for the TMR-E analogues (E = O–Se16 and Te15) are also compiled in Table 1. The heavy-chalcogen analogues 5 of the Texas reds are all red-shifted 20–23 nm relative to their corresponding TMR-E derivative, and all have values of ε ≥ 1.24 × 105 M–1 cm–1. Compounds 6 with two half-julolidine groups are red-shifted 8–9 nm relative to the corresponding TMR-E analogues, while the red shift of the unsymmetrical dyes 7 is intermediate at 13–16 nm.
Steady-state fluorescence spectra for 5–7 were acquired with excitation at 532 nm.16 The emission maxima (λFL) and quantum yields for fluorescence (ΦFL) for TMR-E (E = O–Se16 and Te21) and dyes 5–7 are compiled in Table 1. Values of ΦFL for the S-containing dyes 5-S, 6-S, and 7-S (ΦF = 0.42–0.53, Table 1) are comparable to ΦFL for TMR-S (ΦF = 0.44).16 Selenium-containing dyes 5-Se, 6-Se, and 7-Se have values of ΦFL roughly an order of magnitude smaller than their corresponding S-analogues (ΦFL = 0.02–0.06, Table 1), and all are more emissive than TMR-Se (ΦF = 0.009).16 Fluorescence from all of the Te-containing analogues (TMR-Te, 5-Te, 6-Te, and 7-Te) is weak (ΦFL < 0.005, Table 1).
Quantum yields for the generation of 1O2 [Φ(1O2)] by 5–7 were evaluated using steady-state 1O2 luminescence with TMR-Se as a reference [Φ(1O2) = 0.87].16 In Figure 1, the smoothed luminescence spectra of 1O2 produced by the sensitization of 5-Se, 5-Te, 6-Se, 6-Te, 7-Se, and 7-Te are compared to the luminescence spectrum of 1O2 produced by the sensitization of TMR-Se. Steady-state 1O2 luminescence emission for the S-containing dyes 5-S, 6-S, and 7-S was difficult to extract from the background and the values of Φ(1O2) for the S-containing compounds are considered to be low [Φ(1O2) < 0.05]. Values of Φ(1O2) are compiled in Table 1 for TMR-E (E = O–Se16 and Te23) derivatives and dyes 5–7.
Figure 1.
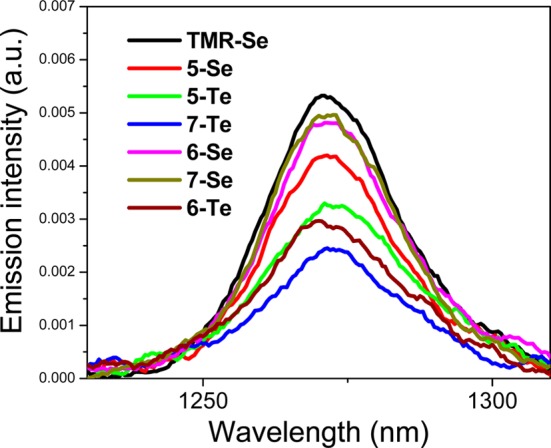
Luminescence of 1O2 sensitized with TMR-Se, 5-Se, 5-Te, 6-Se, 6-Te, 7-Se, and 7-Te through excitation at 532 nm (MeOH solutions). Concentrations of all solutions were adjusted to match OD = 0.10 at 532 nm.
The Se-containing analogues TMR-Se, 5-Se, 6-Se, and 7-Se were found to generate 1O2 more efficiently [Φ(1O2) = 0.68–0.87] than their corresponding Te-containing analogues TMR-Te, 5-Te, 6-Te, and 7-Te [Φ(1O2) = 0.40–0.53]. A possible reason for this can be that as the rate of S1 → T1 intersystem crossing increases, the heavy atom simultaneously increases the rate of T1 → S0 nonradiative transition, causing a decrease in the triplet lifetime that, correspondingly, may decrease the efficiency of the sensitization of singlet oxygen.28,29 The observed intensity of 1O2 luminescence could be also affected by other possible interactions of 1O2, which are different for the Texas red analogues with Se and Te atoms (e.g., possible formation of exciplexes between 1O2 and dye molecules).
In examining the trends observed on the impact of amine structure on Φ(1O2) within the Se-containing dyes, incorporation of the julolidyl fragment lowers Φ(1O2) more than incorporation of the half-julolidyl fragment. Values for 5-Se, 6-Se, and 7-Se were compared to 19-Se(18) incorporating one julolidyl fragment and 20-Se incorporating one-half-julolidyl fragment.
Compound 20-Se was prepared as shown in Scheme 4. Lithiation of amide 16 with s-BuLi/TMEDA followed by the addition of diselenide 21(24) gave diaryl selenide 22 in 46% isolated yield as the only observed product. Cyclization of 22 with POCl3/Et3N in CH3CN23 gave selenoxanthone 23 in 96% yield. The addition of PhMgBr to 23 in THF followed by work-up with aqueous HPF6 gave selenorhodamine 20-Se in 74% isolated yield. The luminescence spectrum of 1O2 produced by sensitization with 20-Se was compared to the luminescence spectrum of 1O2 produced by sensitization of TMR-Se to give Φ(1O2) of 0.88 (Table 1). The value of Φ(1O2) for 19-Se was previously determined to be 0.85.18
Scheme 4.

The selenorhodamine TMR-Se has λmax of 581 nm in MeOH, while values of λmax for 19-Se and 20-Se were 588 and 585 nm, respectively. The impact of the incorporation of a single julolidyl or half-julolidyl moiety in the chromophore is less than half the change observed in λmax for the incorporation of two. The impact of the incorporation of a single julolidyl or half-julolidyl moiety on Φ(1O2) is even smaller. Values of Φ(1O2) for TMR-Se (0.87), 19-Se (0.85), and 20-Se (0.88) are essentially identical within the 3% error of the experiments. When both amino groups are constrained in julolidyl or half-julolidyl fragments, values of Φ(1O2) are lower for 5-Se (0.68) and 6-Se (0.76) relative to TMR-Se, 19-Se, or 20-Se.
Oxidation of Tellurorhodamines to Tellurorhodamine Telluroxides
Earlier studies with chalcogenopyrylium dyes show that rates of reaction of 1O2 with the chalcogen atom in the dye chromophore is Te ≫ S > Se.30 Our recent studies show a rapid oxidation of several tellurorhodamines with 1O2 to give telluroxides 24–26 (Chart 3).20 We examined the oxidations of 5-Te and 6-Te to the corresponding telluroxides in two ways. Photooxidation of 8 × 10–6 M 5-Te in air-saturated 50% aqueous MeOH containing 0.1 M CF3CO2H with 375–800 nm light from a filtered tungsten lamp (50 mW cm–2) gave loss of the chromophore for 5-Te (λmax 617 nm) and appearance of a new chromophore at 716 nm, which was transiently stable. The oxidation was more conveniently carried out using H2O2 as an oxidant as shown in Figure 2. Texas red analogue 5-Te was oxidized with 3 × 10–3 M H2O2 to give presumably the corresponding telluroxide with λmax of 716 nm (ε = 1.1 × 105 M–1 cm–1 assuming 100% conversion of 5-Te to telluroxide) with an isosbestic point at 641 nm. The 716 nm chromophore was lost upon standing under the conditions of reaction over a 2-h time period. Attempts to prepare the telluroxide on a milligram scale resulted decomposition to many products.20
Chart 3.

Figure 2.

Oxidation of 8 × 10–6 M 5-Te with 3 × 10–3 M H2O2 in 50% aqueous MeOH containing 0.1 M CF3CO2H. After an initial scan at 0 s, subsequent scans were taken at 960, 1920, 2400, and 3360 s.
In our earlier work, tellurorhodamine telluroxides 25 and 26 (Chart 3) substituted with 9-aryl groups with ortho-methyl functionality displayed increased hydrolytic stability relative to telluroxide 24 with a 9-phenyl substituent.20 To this end, 9-mesityltellurorhodamine 27 was prepared in 83% yield by the addition of the Grignard reagent from 2-bromomesitylene to telluroxanthone 2-Te at ambient temperature in THF followed by work-up with aqueous HPF6 (Scheme 5). Tellurorhodamine 27 absorbed light with a λmax of 617 nm and ε of 1.65 × 105 M–1 cm–1 in MeOH (Table 2). Tellurorhodamine 27 was oxidized to telluroxide 28 with 3 × 10–3 M H2O2 in either pH 7.4 phosphate buffer or in MeOH containing 0.1 M CF3CO2H. Mesityl-substituted telluroxide 28 was stable with no loss of the 704 nm chromophore (Table 2) after several hours at ambient temperature.
Scheme 5.

Table 2. Absorption Maxima (λmax), Molar Extinction Coefficients (ε), Fluorescence Maxima (λFL), and Fluorescence Quantum Yields (ΦFL) for Tellurorhodamine 27 and 29 and Tellurorhodamine Telluroxides 28 and 30.
| compd | λmax (nm) | ε (M–1 cm–1) | λFL (nm) | ΦFL |
|---|---|---|---|---|
| 27a | 617 | 1.65 × 105 | <0.005 | |
| 28b | 704 | 1.35 × 105 | 740 | 0.16 |
| 29a | 606 | 9.66 × 104 | <0.005 | |
| 30b | 692 | 1.65 × 105 | 720 | 0.16 |
In MeOH.
In CH3OH containing 0.1 M CF3CO2H.
Similarly, the addition of the Grignard reagent from 2-bromomesitylene to telluroxanthone 3-Te at ambient temperature in THF followed by work-up with aqueous HPF6 gave tellurorhodamine 29 in 56% isolated yield (Scheme 5). Tellurorhodamine 29 absorbed light with a λmax of 606 nm and an ε of 9.66 × 104 M–1 cm–1 in MeOH (Table 2). Tellurorhodamine 29 was oxidized with 3 × 10–3 M H2O2 in either pH 7.4 phosphate buffer or in MeOH containing 0.1 M CF3CO2H to give telluroxide 30 with a λmax of 692 nm (Table 2). Telluroxide 30 was less stable than telluroxide 28 and was lost over several hours at ambient temperature in both solvent systems.
On a preparative scale, both 27 and 29 were oxidized in 0.1 M CF3CO2D in CD3OD with H2O2 (0.18 M) to allow the acquisition of NMR spectra. For both compounds, 1H NMR spectra were acquired immediately after the addition of H2O2 and showed complete oxidation of the starting tellurorosamine to telluroxides 28 and 30. For 28, no further changes were observed over a 1.5-h period. For 30, loss of telluroxide signals and the appearance of new products were observed over a 1.5-h period.
Coupling patterns and chemical shifts are similar in the 27/28 and 29/30 pairs, and 1H NMR spectra for both pairs in 0.1 M CF3CO2D in CD3OD are compiled in the Supporting Information. Both tellurorosamine dyes 27 and 29 gave 13C NMR spectra with the expected 19 (11 aromatic and 8 aliphatic) signals. The 13C NMR spectra of telluroxides 28 and 30 were not as well-defined (Supporting Information). All eight different aliphatic carbons were observed for both compounds, but only 8 aromatic signals for 28 and 10 aromatic signals for 30 were observed. Acquisition of data was limited by the stability of the oxidized product. At higher concentrations of 28/30, the rate of loss of 30 was accelerated suggesting second-order processes for the loss of 30. Solvent addition/elimination to the telluroxide functionality might also lead to line broadening on near-by carbons.
Both 28 and 30 were highly emissive. The absorption and emission spectra of telluroxides 28 and 30 in MeOH containing 0.1 M CF3CO2H are shown in Figure 3. Values of ΦF for 28 and 30 were both 0.16 (Table 2) using Rhodamine 700 as a known fluorescence standard with ΦF = 0.38.31
Figure 3.

Absorption and fluorescence spectra for (a) tellurorhodamine telluroxide 28 and (b) tellurorhodamine telluroxide 30 in MeOH with 0.1 M CF3CO2H.
Biological Studies: Cellular Oxidation of 27
In our early work examining telluropyrylium dyes as photosensitizers for the photodynamic therapy (PDT) of cancer, we noted mitochondrial fluorescence from the telluroxide oxidation state following irradiation of dye-treated U-251 MG glioblastoma cells.32,33 Oxidation of the telluropyrylium dyes via self-sensitized generation of singlet oxygen gave telluroxide analogues that absorbed and fluoresced at shorter wavelengths than the reduced form. More recent studies with telluride/telluroxide20,34 and selenide/selenoxide35 pairs examined fluorescence from the chromophore substituted with telluroxide or selenoxide functionality as a probe for redox cycles in living cells. In the case of telluride/telluroxide pairs based on the rhodamine core, the telluroxide absorbs and fluoresces at wavelengths longer than those of the reduced form.19,20
In order to examine the viability of tellurorhodamine 27/telluroxide 28 as a redox pair indicator in cells, Colo-26 cells (a murine colon carcinoma cell line) were incubated first with 0.5 μM MitoTracker Green (MTG) for 10 min and then 0.2 μM tellurorhodamine 27 for an additional 5 min (15 min total for MTG and 5 min total for 27). The cells were washed prior to imaging on a fluorescence microscope. The cells were brought to focus using a halogen lamp, which was used at the end of the experiment to generate the transmission microscopy image shown in Figure 4a. Irradiating the cells with 620 nm light should oxidize nonfluorescent tellurorhodamine 27 (Figure 4b, time = 0 s for 620 nm irradiation) via 27-sensitized generation of 1O2 to give fluorescent telluroxide 28 (Figure 4c, time = 15 s of irradiation with 620 ± 30 nm light) in the cell. Excitation of the MTG (470 ± 20 nm light) is shown in Figure 4d. Figure 4e and f shows the overlays of emission from telluroxide 28 and the MTG. The images of Figure 4 clearly indicate that the reduced tellurorhodamine dye 27 is in the Colo-26 cells and that brief irradiation with 620 nm light gives photooxidation of 27 to fluorescent telluroxide 28 in Figure 4c. The overlay of MTG emission and telluroxide 28 emission in Figure 4f indicates that tellurorhodamine 28 is at least partially targeted to the mitochondria.
Figure 4.
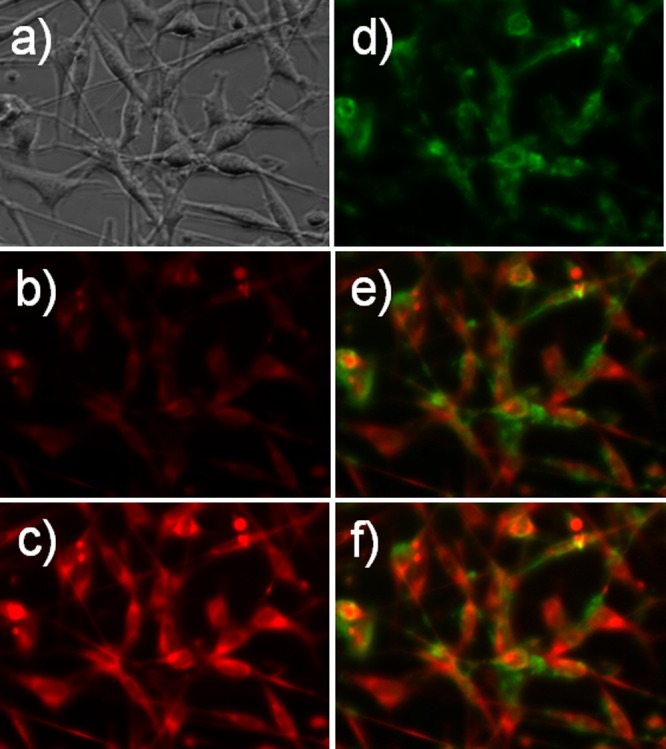
(a) Transmission microscopy image of Colo-26 cells treated with MTG and tellurorhodamine 27. (b) Fluorescence microscopy image of Colo-26 cells before irradiation with 620 ± 30 nm light (t = 0 s). (c) Fluorescence microscopy image of Colo-26 cells from Panel b after 15 s of irradiation with 620 ± 30 nm light (t = 15 s). (d) Fluorescence microscopy image of MTG fluorescence in Colo-26 cells (excitation with 470 ± 20 nm light). (e) Overlay of images from panels b and d. (f) Overlay of images from panels c and d.
Conclusions
We have successfully prepared heavy-chalcogen analogues of Texas red (1) incorporating S, Se, and Te atoms in the xanthylium core. The heavy-chalcogen analogues absorb at longer wavelengths than 1. The excited state of S-containing analogue 5-S is highly emissive (ΦFL = 0.42), while the excited state of the Se- and Te-containing derivatives gives intersystem crossing to the triplet with efficient generation of 1O2 [Φ(1O2) = 0.68 for 5-Se and 0.53 for 5-Te]. We have also prepared derivatives 6-E and 7-E incorporating fused trimethyltetrahydroquinoline (half-julolidine) in the xanthylium core. These derivatives give values of λmax that are intermediate between the TMR-E and 5-E derivatives (Table 1), but their photophysical properties are quite similar to those for 5-E.
Incorporating a single julolidine or a single half-julolidine into the xanthylium core has little impact on the photophysical properties. Singlet oxygen yields for TMR-Se, 19-Se, and 20-Se are essentially identical, while values of λmax are between 581 and 587 nm (Table 1).
The tellurium analogues 5-Te and 6-Te are easily oxidized with 1O2 or H2O2 presumably to the corresponding telluroxides, but the telluroxide derivatives were hydrolytically unstable. Texas red analogues 27 and 29 bearing a 9-mesityl substituent are oxidized to telluroxides 28 and 30, respectively, which are hydrolytically more stable than the oxidation products of 5-Te and 6-Te. Both 28 and 30 are highly fluorescent (ΦF = 0.16 for both). Tellurorhodamine 27 is oxidized to telluroxide 28 by the self-sensitized generation of 1O2 in cells.
The heavy-chalcogen Texas red analogues can be applied to several biological problems. The mesityl-substituted tellurorhodamine 27 and its corresponding telluroxide 28 represent a redox-related, nonfluorescent/fluorescent pair of molecules that can be observed in cells. Fluorescence from telluroxide 28 or related structures (such as 30) in vitro might allow the tracking of cellular redox processes.20 The Se-containing analogues 5-Se, 6-Se, and 7-Se as well as related structures have potential as photosensitizers for the photodynamic therapy (PDT) of malignant cells17,18,36,37 and as photosensitizers for the photodynamic purging of blood-borne viral and bacterial pathogens.38−40 For applications in PDT, the Se-containing analogues 5-Se, 6-Se, and 7-Se absorb light wavelengths >600 nm, where light penetration of tissue is greatest.40
These versatile chromophores can also be applied to problems related to solar energy and solar fuels as has been done with analogues of the TMR-E dyes. Appropriate substitution in the 9-position of the chromophores would allow entry into longer-wavelength-absorbing photosensitizers for dye-sensitized solar cells41,42 and longer-wavelength-absorbing photosensitizers for the solar reduction of protons to hydrogen.43 In the latter systems, control of excited state lifetime (singlet vs triplet) is important, and the suite of Texas red analogues allows control of both wavelengths of absorption and singlet–triplet yields.
Experimental Section
Preparation of 8-Bromo-1,2,3,5,6,7-hexahydropyrido[3,2,1-ij]quinoline (9)25,26
3-Bromoaniline (8, 12.7 mL, 116 mmol, 1.0 equiv), sodium carbonate (49.0 g, 465 mmol), and 1-bromo-3-chloropropane (173 mL, 1.74 mol) were heated at 140 °C for 48 h. After cooling to room temperature, water (300 mL) was added, and products were extracted with CH2Cl2 (3 × 200 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated. Excess 1-bromo-3-chloropropane was then removed at 45 °C at 1 Torr. The crude dialkylated product was then dissolved in 15 mL of DMF and heated at 160 °C for 48 h. After cooling to room temperature, 1 M NaOH (250 mL) was added. The resulting mixture was extracted with diethyl ether (3 × 250 mL). The combined extracts were dried over anhydrous MgSO4, filtered, and concentrated. The crude product was recrystallized from MeOH to give 20.0 g (68.2% overall) of 9 as a white solid, mp 31–32 °C: 1H NMR (500 MHz, CD2Cl2) δ 6.73 (d, 1 H, J = 7.5 Hz), 6.63 (d, 1 H, J = 8.5 Hz), 3.18–3.07 (m, 4 H), 2.75 (t, 2 H, J = 6.0 Hz), 2.68 (t, 2 H, J = 6.5 Hz), 2.01–1.90 (m, 4 H); 13C NMR (75.5 MHz, CDCl3) δ 144.5, 127.7, 122.9, 120.6, 120.5, 119.4, 50.0, 49.6, 28.5, 27.6, 21.9, 21.8; HRMS (ESI, HRDFMagSec) m/z 252.0381 (calcd for C12H14N79Br + H+: 252.0382).
Preparation of Bisjulolidyl Disulfide 10-S
Compound 9 (2.77 g, 11.0 mmol) and ground Mg turnings (294 mg, 12.1 mmol) in 5.5 mL of THF were heated at reflux for 3 h. The reaction mixture was cooled to room temperature, and elemental S (388 mg, 12.1 mmol) and an additional 5.5 mL of THF were added. The resulting mixture was stirred at room temperature for 3 h before adding 10 mL of 1.0 M HCl (10 mL) and water (100 mL). The reaction mixture was stirred overnight under air, and the products were then extracted with ether (3 × 250 mL). The combined extracts were dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified via column chromatography on SiO2 eluted with 0.5:9.5 EtOAc/hexanes (Rf = 0.5) to give 1.35 g (60.3%) of 10-S as a yellow solid, mp 107–108 °C: 1H NMR (500 MHz, CD2Cl2) δ 6.77 (d, 2 H, J = 7.5 Hz), 6.70 (d, 2 H, J = 8.0 Hz), 3.16–3.06 (m, 8 H), 2.83 (t, 4 H, J = 6.0 Hz), 2.70 (t, 4 H, J = 6.0 Hz), 2.00–1.88 (m, 8 H); 13C NMR (75.5 MHz, CDCl3) δ 143.2, 133.0, 126.9, 120.8, 120.4, 116.3, 50.1, 49.5, 27.6, 25.1, 21.8, 21.6; HRMS (ESI, HRDFMagSec) m/z 408.1690 (calcd for C24H28N2S2+: 408.1688).
Preparation of Bisjulolidyl Diselenide 10-Se
The procedure described for the preparation of 10-S was followed using 9 (5.00 g, 19.8 mmol), Mg turnings (530 mg, 12.1 mmol), and elemental Se (1.72 mg, 21.8 mmol) in a total of 20 mL of THF. Work-up as described and purification via chromatography on SiO2 (0.5:9.5 EtOAc/hexanes) gave 2.84 g (57.0%) of 10-Se as an orange solid, mp 126–128 °C: 1H NMR (400 MHz, CDCl3) δ 6.95 (d, 2 H, J = 7.6 Hz), 6.69 (d, 2 H, J = 8.0 Hz), 3.15–3.07 (m, 8 H), 2.83 (t, 4 H, J = 6.4 Hz), 2.73 (t, 4 H, J = 6.4 Hz), 2.01–1.90 (m, 8 H); 13C NMR (75.5 MHz, CDCl3) δ 143.3, 128.8, 127.2, 121.9, 121.5, 120.5, 50.2, 49.7, 27.7, 27.6, 22.1, 21.9; HRMS (ESI, HRDFMagSec) m/z 505.0649 (calcd for C24H28N280Se2 + H+: 505.0656).
Preparation of Bisjulolidyl Ditelluride 10-Te
The procedure described for the preparation of 10-S was followed using 9 (2.00 g, 7.93 mmol), Mg turnings (212 mg, 8.73 mmol), and elemental Te (1.11 g, 8.73 mmol) in a total of 8 mL of THF. Work-up as described and purification via recrystallization from CH2Cl2/hexanes gave 1.70 g (71.4%) of 10-Te as an orange solid, mp 138–139 °C: 1H NMR (500 MHz, CDCl3) δ 7.18 (d, 2 H, J = 8.0 Hz), 6.59 (d, 2 H, J = 8.0 Hz), 3.15–3.06 (m, 8 H), 2.82 (t, 4 H, J = 6.5 Hz), 2.78 (t, 4 H, J = 6.5 Hz), 2.10–1.90 (m, 8 H); 13C NMR (75.5 MHz, CDCl3) δ 143.5, 128.1, 127.5, 124.6, 122.8, 109.5, 50.4, 50.2, 33.5, 28.1, 23.0, 22.4; HRMS (EI, HRDFMagSec) m/z 604.0354 (calcd for C24H28N2130Te2 + H+: 604.0350).
Preparation of 3-Bromo-N-methyl-N-(3-methylbut-2-enyl)aniline (11)
1-Chloro-3-methylbut-2-ene (5.87 mL, 49.5 mmol, 1.5 equiv) was slowly added to a stirred solution of potassium carbonate (6.84 g, 49.5 mmol, 1.5 equiv) and commercially available 3-bromo-N-methylaniline (6.45 g, 33.0 mmol, 1.0 equiv) in DMF (30 mL). The resulting solution was heated to 95 °C for 16 h before it was allowed to cool to ambient temperature. Water (30 mL) was added, and the mixture was extracted with diethyl ether (3 × 100 mL). The combined organic fractions were washed with water (4 × 100 mL), dried over anhydrous magnesium sulfate, vacuum filtered, and concentrated in vacuo. The crude products were purified via column chromatography on SiO2 eluted with 30:70 CH2Cl2/hexanes (Rf = 0.60) to give 8.00 g (95.3%) of 11 as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.05 (t, 1 H, J = 8.0 Hz), 6.83 (t, 1 H, J = 2.0 Hz), 6.79 (d, 1 H, J = 8.0 Hz), 6.59 (dd, 1 H, J = 2.0, 8.0 Hz), 5.16 (m, 1 H), 3.87 (d, 2 H, J = 6.5 Hz), 2.88 (s, 3 H), 1.73 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) δ 150.8, 135.1, 130.2, 123.4, 120.1, 118.8, 115.3, 111.1, 50.2, 37.8, 25.7, 17.9; IR (film on NaCl) νmax 2969, 2913, 1953, 1555, 1494, 1450 cm–1; HRMS (ESI, HRDFMagSec) m/z 253.0459 (calcd for C12H16N79Br+: 253.0461).
Preparation of 7-Bromo-1,4,4-trimethyl-1,2,3,4-tetrahydroquinoline (12)27
Concentrated sulfuric acid (23.6 mL) was added dropwise to aniline 11 (7.87 g, 31.0 mmol, 1.0 equiv) at 0 °C. The resulting mixture was warmed to ambient temperature and was stirred for 1 h before it was transferred via pipet into water at 0 °C. Sodium hydroxide pellets were added until the solution was basic to litmus. The mixture was extracted with ether (4 × 100 mL), and the combined organic fractions were dried over anhydrous magnesium sulfate, filtered, and concentrated. The crude product was recrystallized from hot methanol to give 6.26 g (79.5%) of 12 as a brown solid, mp 55–57 °C: 1H NMR (500 MHz, CDCl3) δ 7.02 (d, 1 H, J = 8.0 Hz), 6.73 (dd, 1 H, J = 2.0, 8.0 Hz), 6.66 (d, 1 H, J = 2.0 Hz), 3.25 (t, 2 H, J = 6.0 Hz), 2.89 (s, 3 H), 1.73 (t, 2 H, J = 6.0 Hz), 1.25 (s, 6 H); 13C NMR (75.5 MHz, CD2Cl2) δ 147.1, 130.8, 127.4, 120.7, 118.5, 113.4, 47.8, 39.3, 37.1, 32.1, 30.6; HRMS (EI, HRDFMagSec) m/z 253.0456 (calcd for C12H16N79Br+: 253.0461).
Preparation of Diaryl Sulfide 13-S
Freshly ground magnesium turnings (522 mg, 21.5 mmol, 1.1 equiv) were suspended in a stirred solution of 12 (5.00 g, 19.6 mmol, 1.0 equiv) in THF (30 mL). A crystal of elemental iodine was added, and the resulting solution was heated at reflux for 3 h, then cooled to ambient temperature, and elemental sulfur (628 mg, 19.6 mmol, 1.0 equiv) was added. The resulting solution was stirred at ambient temperature for 3 h, and then a solution of 1 M aqueous HCl (30 mL) was added. The mixture was allowed to stir overnight under air before it was extracted with diethyl ether (3 × 100 mL), and the combined organic fractions were dried over anhydrous magnesium sulfate, filtered, and concentrated. The crude products were purified via column chromatography on SiO2 eluted with 3:7 CH2Cl2/hexanes (Rf = 0.5), to give 2.81 g (70.0%) of 13-S as a pale yellow solid, mp 66–68 °C: 1H NMR (500 MHz, CDCl3) δ 7.08 (d, 2 H, J = 8.0 Hz), 6.81 (dd, 2 H, J = 1.5, 8.0 Hz), 6.72 (d, 2 H, J = 1.5 Hz), 3.21 (t, 4 H, J = 6.0 Hz), 2.84 (s, 6 H), 1.72 (t, 4 H, J = 6.0 Hz), 1.24 (s, 12 H); 13C NMR (75.5 MHz, CDCl3) δ 145.6, 135.6, 130.7, 126.1, 115.6, 110.2, 47.5, 39.1, 37.0, 31.8, 30.7; HRMS (EI, HRDFMagSec) m/z 412.2008 (calcd for C24H32N2S2+: 412.2001).
Preparation of Diaryl Diselenide 13-Se
The procedure described for the preparation of 13-S was followed using 12 (2.00 g, 7.87 mmol), Mg turnings (211 mg, 8.66 mmol), and elemental Se (1.72 g, 21.8 mmol) in a total of 16 mL of THF. Work-up as described followed by purification via column chromatography on SiO2 eluted with 2:8 CH2Cl2/hexanes (Rf = 0.4) and recrystallization from hexanes gave 1.33 g (66.8%) of 13-Se as an orange solid, mp 76–78 °C: 1H NMR (500 MHz, CDCl3) δ 7.05 (d, 2 H, J = 8.0 Hz), 6.92 (dd, 2 H, J = 1.5, 7.5 Hz), 6.82 (d, 2 H, J = 2.0 Hz), 3.20 (t, 4 H, J = 6.0 Hz), 2.82 (s, 6 H), 1.72 (t, 4 H, J = 6.0 Hz), 1.24 (s, 12 H); 13C NMR (75.5 MHz, CDCl3) δ 145.4, 130.8, 129.0, 125.9, 118.9, 113.7, 47.2, 38.8, 36.7, 31.6, 30.8; HRMS (EI, HRDFMagSec) m/z 508.0895 (calcd for C24H32N280Se2+: 508.0896).
Preparation of Diaryl Ditelluride 13-Te
The procedure described for the preparation of 13-S was followed using 12 (2.50 g, 9.84 mmol), Mg turnings (263 mg, 10.8 mmol), and elemental Te (1.72 g, 21.8 mmol) in a total of 20 mL of THF. Workup as described followed by purification via column chromatography on SiO2 eluted with 2:8 CH2Cl2/hexanes (Rf = 0.4) and recrystallization from hexanes gave 2.64 g (88.9%) of 13-Te as a dark red solid, mp 85–87 °C: 1H NMR (500 MHz, CDCl3) δ 7.08 (dd, 2 H, J = 1.5, 7.5 Hz), 6.98 (d, 2 H, J = 1.5 Hz), 6.94 (d, 2 H, J = 8.0 Hz), 3.19 (t, 4 H, J = 6.0 Hz), 2.81 (s, 6 H), 1.72 (t, 4 H, J = 6.0 Hz), 1.24 (s, 12 H); 13C NMR (75.5 MHz, CDCl3) δ 145.6, 131.4, 126.1, 124.8, 119.9, 105.9, 47.4, 39.0, 36.9, 31.7, 30.7; HRMS (ESI, HRDFMagSec) m/z 609.0758 (calcd for C24H32N2130Te2 + H+: 609.0763).
Preparation of Diaryl Sulfide 15-S
sec-Butyllithium (1.14 M in cyclohexane, 3.39 mL, 3.87 mmol) was added dropwise to a stirred solution of TMEDA (576 μL, 3.87 mmol) and amide 1417 (1.00 g, 3.52 mmol) in THF (70 mL) at −78 °C. A solution of 10-S (1.58 g, 3.87 mmol) in THF (70 mL) at −78 °C was added immediately. The resulting mixture was stirred at −78 °C for 3 h and then 15 h at room temperature. A solution of saturated NH4Cl (25 mL) was added, and the products were extracted with CH2Cl2 (3 × 100 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified via chromatography on SiO2 (1:9 Et2O/DCM, Rf = 0.5) to give 765 mg (44.5%) of 15-S as a yellow oil: 1H NMR (500 MHz, CD2Cl2) δ 6.76 (s, 1 H), 6.60 (d, 1 H, J = 8.0 Hz), 6.01 (d, 1 H, J = 8.0 Hz), 3.78–3.68 (m, 1 H), 3.65–3.54 (m, 1 H), 3.34–3.20 (m, 4 H), 3.18–3.05 (m, 6 H), 2.74–2.62 (m, 8 H), 2.13–1.93 (m, 6 H), 1.92–1.83 (m, 2 H), 1.72–1.53 (m, 4 H), 1.47–1.32 (m, 2 H); 13C NMR (75.5 MHz, CD2Cl2) δ 168.9, 143.8, 143.5, 134.3, 131.3, 126.9, 126.0, 125.2, 124.8, 122.9, 118.5, 118.2, 113.8, 50.5, 50.1, 49.8, 49.7, 48.1, 42.2, 28.1, 27.8, 26.3, 26.1, 25.8, 25.3, 24.9, 22.5, 22.4, 21.9, 21.8; IR (film on NaCl) 2934, 2854, 1630, 1587, 1488, 1438, 1305, 1202 cm–1; HRMS (EI, HRDFMagSec) m/z 487.2652 (calcd for C30H37ON3S+: 487.2658).
Preparation of Diaryl Selenide 15-Se
sec-Butyllithium (1.03 M in cyclohexane, 1.88 mL, 1.93 mmol), TMEDA (288 μL, 1.93 mmol), amide 14(17) (500 mg, 1.76 mmol, 1.0 equiv), and 10-Se (972 mg, 1.93 mmol) in THF (2 × 35 mL) were treated as described for the preparation of 15-S. Following chromatography on SiO2 (1:9 Et2O/DCM, Rf = 0.5), 15-Se was isolated in 450 mg (47.9%) yield as a yellow oil: 1H NMR (500 MHz, CD2Cl2) δ 6.73 (s, 1 H), 6.54 (d, 1 H, J = 8.0 Hz), 6.09 (d, 1 H, J = 8.5 Hz), 3.76–3.68 (m, 1 H), 3.59–3.51 (m, 1 H), 3.29–3.07 (m, 10 H), 2.86–2.71 (m, 6 H), 2.70–2.62 (m, 2 H), 2.08–1.91 (m, 6 H), 1.86 (quintet, 2 H, J = 6.5 Hz), 1.69–1.52 (m, 4 H), 1.42–1.30 (m, 2 H); 13C NMR (75.5 MHz, CD2Cl2) δ 169.8, 143.9, 143.8, 131.9, 131.4, 127.3, 126.1, 124.8, 123.4, 122.9, 119.8, 119.3, 116.1, 54.2, 50.5, 50.2, 49.9, 48.3, 42.3, 28.6, 28.2, 27.8, 27.0, 26.5, 25.8, 25.0, 22.5, 22.4, 22.3, 22.0; IR (film on NaCl) 2934, 2854, 1627, 1596, 1502, 1461, 1437, 1390, 1316, 1304, 1262, 1211 cm–1; HRMS (ESI, HRDFMagSec) m/z 536.2184 (calcd for C30H37ON380Se + H+: 536.2175).
Preparation of Diaryl Telluride 15-Te
sec-Butyllithium (1.08 M in cyclohexane, 3.58 mL, 3.52 mmol), TMEDA (576 μL, 3.87 mmol), amide 14(17) (1.00 g, 3.52 mmol, 1.0 equiv), and 10-Te (2.32 g, 3.87 mmol) in THF (2 × 70 mL) were treated as described for the preparation of 15-S. Following chromatography on SiO2 (1:9 Et2O/DCM, Rf = 0.5), 15-Te was isolated in 763 mg (37.2%) yield as a yellow oil: 1H NMR (500 MHz, CD2Cl2) δ 6.75 (s, 1 H), 6.51 (d, 1 H, J = 8.0 Hz), 6.37 (d, 1 H, J = 8.0 Hz), 3.80–3.55 (m, 2 H), 3.40–3.26 (m, 2 H), 3.23 (t, 2 H, J = 6.0 Hz), 3.18–3.05 (m, 6 H), 3.00–2.76 (m, 4 H), 2.74–2.55 (m, 4 H), 2.07–1.92 (m, 6 H), 1.89–1.80 (m, 2 H), 1.75–1.51 (m, 4 H), 1.4–1.38 (m, 2H); 13C NMR (75.5 MHz, CD2Cl2) δ 171.3, 143.9, 143.1, 134.3, 127.8, 127.5, 124.5, 123.1, 122.8, 121.5, 120.4, 118.4, 114.2, 50.4, 50.1, 50.0, 49.9, 48.4, 42.5, 33.8, 31.0, 28.1, 27.8, 26.6, 25.7, 24.9, 22.7, 22.4, 22.3, 22.0; IR (film on NaCl) 2934, 2852, 1623, 1582, 1486, 1436, 1388, 1328, 1305, 1202 cm–1; HRMS (ESI, HRDFMagSec) m/z 586.2073 (calcd for C30H37ON3130Te + H+: 586.2072).
Preparation of Diaryl Sulfide 17-S
sec-Butyllithium (0.94 M in 92:8 cyclohexane/hexanes, 4.09 mL, 3.84 mmol), TMEDA (572 μL, 3.84 mmol), amide 16(21) (1.00 g, 3.49 mmol), and 13-S (1.58 g, 3.84 mmol) in THF (2 × 70 mL) were treated as described for the preparation of 15-S. Following chromatography on SiO2 (1:9 EtOAc/CH2Cl2, Rf = 0.6), 17-S was isolated in 1.06 g (61.6%) yield as a yellow solid, mp 83–85 °C: 1H NMR (500 MHz, CDCl3) δ 7.07 (d, 1 H, J = 8.0 Hz), 7.00 (s, 1 H), 6.68–6.62 (m, 2 H), 6.43 (s, 1 H), 3.66 (br s, 2 H), 3.26–3.04 (m, 6 H), 2.84 (s, 3 H), 2.73 (s, 3 H), 1.76–1.66 (m, 4 H), 1.63–1.44 (m, 6 H), 1.24 (s, 12 H); 13C NMR (MHz, CDCl3) δ 168.8, 145.1, 132.3, 131.1, 130.0, 129.5, 125.6, 125.2, 123.7, 119.2, 113.7, 112.9, 47.5 (br), 47.0, 46.8, 42.1 (br), 38.7, 38.4, 36.6, 36.2, 31.3, 31.2, 30.3, 30.1, 25.4 (br), 24.1; IR (film on NaCl) 2931, 2852, 1630, 1593, 1501, 1467, 1434 cm–1; HRMS (ESI, HRDFMagSec) m/z 492.3035 (calcd for C30H41N3OS + H+: 492.3043).
Preparation of Diaryl Selenide 17-Se
sec-Butyllithium (1.19 M in 92:8 cyclohexane/hexanes, 3.23 mL, 3.84 mmol), TMEDA (572 μL, 3.84 mmol), amide 16(21) (1.00 g, 3.49 mmol), and 13-Se (1.94 g, 3.84 mmol) in THF (2 × 55 mL) were treated as described for the preparation of 15-S. Following chromatography on SiO2 (1:9 Et2O/CH2Cl2, Rf = 0.6), 17-Se was isolated in 1.04 g (55.3%) yield as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.07 (d, 1 H, J = 7.5 Hz), 6.99 (s, 1 H), 6.86–6.78 (m, 2 H), 6.43 (s, 1 H), 3.44 (br s, 4 H), 3.18 (quintet, 4 H, J = 6.0 Hz), 2.83 (s, 3 H), 2.65 (s, 3 H), 1.71 (t, 2 H, J = 6.0 Hz), 1.68 (t, 2 H, J = 6.0 Hz), 1.64–1.57 (m, 2 H), 1.57–1.48 (m, 4 H), 1.23 (s, 6 H), 1.21 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) 169.6, 145.3, 130.7, 128.9, 128.6, 127.4, 125.8, 124.8, 123.8, 122.1, 116.7, 113.7, 47.1, 46.9, 45.0 (br), 38.7, 38.2, 36.6, 36.3, 31.4, 31.2, 30.3, 30.1, 25.6, 24.2; IR (film on NaCl) 2930, 1627, 1593, 1499, 1467, 1430 cm–1; HRMS (ESI, HRDFMagSec) m/z 562.2330 (calcd for C30H41N3O80Se + Na+: 562.2307).
Preparation of Diaryl Telluride 17-Te
sec-Butyllithium (1.19 M in 92:8 cyclohexane/hexanes, 3.23 mL, 2.88 mmol), TMEDA (429 μL, 2.88 mmol), amide 16(21) (750 mg, 2.62 mmol), and 13-Te (1.74 g, 2.88 mmol) in THF (55 + 40 mL) were treated as described for the preparation of 15-S. Following chromatography on SiO2 (1:19 EtOAc/DCM, Rf = 0.7), 17-Te was isolated in 764 mg (49.6%) yield as a yellow solid, mp 182–184 °C: 1H NMR (500 MHz, CDCl3) δ 7.23 (d × d, 1 H, J = 1.0, 7.5 Hz), 7.18 (d, 1 H, J = 1.0 Hz), 7.10–7.04 (m, 2 H), 6.40 (s, 1 H), 3.60–3.53 (m, 4 H), 3.21 (t, 2 H, J = 6.0 Hz), 3.17 (t, 2 H, J = 6.0 Hz), 1.72–1.65 (m, 4 H), 1.65–1.59 (m, 4 H), 1.26 (s, 6 H), 1.21 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) δ 171.5, 145.9, 145.5, 131.5, 128.7, 127.7, 126.1, 125.1, 123.5, 123.4, 119.8, 115.7, 114.2, 47.2, 46.9, 46.4 (br), 38.9, 37.9, 36.7, 36.2, 31.6, 31.1, 30.4, 30.1, 25.9, 24.4; IR (film on NaCl) 2931, 2852, 1591 cm–1; HRMS (ESI, HRDFMagSec) m/z 590.2380 (calcd for C30H41N3O130Te + H+: 590.2385).
Preparation of Diaryl Sulfide 18-S
sec-Butyllithium (1.00 M in 92:8 cyclohexane/hexanes, 3.87 mL, 3.87 mmol), TMEDA (577 μL, 3.87 mmol), amide 14(17) (1.00 g, 3.52 mmol), and 13-S (1.60 g, 3.87 mmol) in THF (2 × 60 mL) were treated as described for the preparation of 15-S. Following chromatography on SiO2 (1:9 Et2O/CH2Cl2, Rf = 0.5), 18-S was isolated in 992 mg (55.4%) yield as a white solid, mp 141–143 °C: 1H NMR (500 MHz, CDCl3) δ 6.94 (d, 1 H, J = 8.0 Hz), 6.69 (s, 1 H), 6.44 (s, 1 H), 6.30 (d, 1 H, J = 8.0 Hz), 3.72–3.65 (m, 1 H), 3.63–3.55 (m, 1 H), 3.22–3.05 (m, 7 H), 3.04–2.89 (m, 2 H), 2.80 (s, 3 H), 2.78–2.68 (m, 3 H), 2.02–1.90 (m, 2 H), 1.87 (quintet, 2 H, J = 6.0 Hz), 1.70 (t, 2 H, J = 6.0 Hz), 1.62–1.44 (m, 4 H), 1.32–1.23 (m, 2 H), 1.20 (s, 3 H), 1.19 (s, 3 H); 13C NMR (75.5 MHz, CDCl3) δ 169.0, 145.1, 143.2, 134.8, 130.3, 128.2, 125.7, 125.6, 125.2, 124.1, 122.1, 114.6, 109.4, 49.5, 49.1, 47.6, 47.1, 41.7, 38.8, 36.8, 31.2, 30.5, 30.4, 27.4, 25.6, 25.4, 25.0, 24.1, 21.3, 21.2; IR (film on NaCl) 2934, 2853, 1630, 1590, 1552, 1500, 1438 cm–1; HRMS (ESI, HRDFMagSec) m/z 490.2878 (calcd for C30H39N3OS + H+: 490.2887).
Preparation of Diarylselenide 18-Se
sec-Butyllithium (1.23 M in 92:8 cyclohexane/hexanes, 3.15 mL, 3.87 mmol), TMEDA (577 μL, 3.87 mmol), amide 14(17) (1.00 g, 3.52 mmol), and 13-Se (1.96 g, 3.87 mmol) in THF (2 × 40 mL) were treated as described for the preparation of 15-S. Following chromatography on SiO2 eluted with 2:9 EtOAc/CH2Cl2, (Rf = 0.5), 18-Se was isolated in 1.21 g (64.0%) yield as a red solid, mp 55–57 °C: 1H NMR (500 MHz, CDCl3) δ 6.93 (d, 1 H, J = 8.0 Hz), 6.68 (s, 1 H), 6.54 (d, 1 H, J = 1.5 Hz), 6.43 (dd, 1 H, J = 1.5, 8.0 Hz), 3.78–3.66 (m, 1 H), 3.62–3.51 (m, 1 H), 3.22–3.12 (m, 5 H), 3.11–3.05 (m, 2 H), 3.04–2.97 (m, 1 H), 2.89–1.89 (m, 2 H), 2.76 (s, 3 H), 2.72–2.68 (m, 2 H), 2.00–1.89 (m, 2 H), 1.82 (m, 2 H), 1.70 (m, 2 H), 1.65–1.43 (m, 4 H), 1.34–1.21 (m, 2 H), 1.20 (s, 3 H), 1.19 (s, 3 H); 13C NMR (75.5 MHz, CDCl3) δ 170.2, 145.7, 143.4, 131.2, 130.4, 129.3, 126.2, 126.0, 124.3, 123.9, 122.4, 117.5, 112.1, 49.9, 49.6, 48.0, 47.6, 42.1, 39.2, 37.2, 31.7, 30.9, 30.8, 28.5, 27.8, 25.9, 25.4, 24.5, 21.9, 21.6; IR (film on NaCl) 2933, 2853, 1628, 1587, 1499 cm–1; HRMS (ESI, HRDFMagSec) m/z 537.2254 (calcd for C30H39N3O80Se+: 537.2253).
Preparation of Diaryltelluride 18-Te
sec-Butyllithium (0.94 M in 92:8 cyclohexane/hexanes, 1.23 mL, 1.16 mmol), TMEDA (173 μL, 1.16 mmol), amide 14(17) (300 mg, 1.05 mmol), and 13-Te (700 mg, 1.16 mmol) in THF (2 × 40 mL) were treated as described for the preparation of 15-S. Following chromatography on SiO2 eluted with 1:9 EtOAc/CH2Cl2, (Rf = 0.5), 18-Te was isolated in 233 mg (37.9%) yield as a light brown solid, mp 81–83 °C: 1H NMR (500 MHz, CDCl3) δ 6.90 (d, 1 H, J = 8.0 Hz), 6.72 (s, 1 H), 6.70–6.63 (m, 2 H), 3.66 (br s, 2 H), 3.32–3.10 (m, 7 H), 3.07 (t, 2 H, J = 5.5 Hz), 2.99–2.87 (m, 1 H), 2.79 (s, 3 H), 2.74 (t, 2 H, J = 6.5 Hz), 1.93 (quintet, 2 H, J = 6.0 Hz), 1.83 (quintet, 2 H, J = 6.0 Hz), 1.71 (t, 2 H, J = 5.5 Hz), 1.66–1.50 (m, 4 H), 1.42–1.33 (m, 2 H), 1.21 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) δ 171.7, 145.7, 142.7, 133.6, 130.2, 127.3, 126.3, 123.9, 123.4, 117.7, 114.0, 113.6, 49.7, 49.6, 48.2 (br), 47.4, 42.3 (br), 39.0, 37.0, 33.7, 31.6, 30.7, 27.7, 26.1 (br), 25.2 (br), 24.4, 22.2, 21.4; IR (film on NaCl) 2932, 2852, 1623, 1582, 1546, 1497, 1434 cm–1; HRMS (ESI, HRDFMagSec) m/z 588.2231 (calcd for C30H39N3O130Te+: 588.2228).
Preparation of Bisjulolidyl Thioxanthone 2-S
Phosphorus oxychloride (1.73 mL, 18.6 mmol) was added dropwise to a solution of triethylamine (2.59 mL, 18.6 mmol) and 15-S (755 mg, 1.55 mmol) in anhydrous CH3CN (50 mL). The resulting mixture was heated at reflux for 12 h and was then cooled to 0 °C. A solution of 1 M NaOH (10 mL) was added slowly, and the resulting mixture was poured into a stirred solution of cold 1 M NaOH (200 mL). The resulting mixture was stirred for 6 h, and products were then extracted with CH2Cl2 (3 × 250 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated. The resulting solid was recrystallized from CH2Cl2/hexanes to give 511 mg (82.0%) of 2-S as a brown solid, mp >250 °C: 1H NMR (300 MHz, CD2Cl2) δ 7.95 (s, 2 H), 3.41–3.10 (m, 8 H), 2.95–2.65 (m, 8 H), 2.14–1.82 (m, 8 H); 13C NMR (75.5 MHz, CD2Cl2) δ 178.1, 145.8, 134.8, 127.7, 120.7, 118.1, 113.5, 50.6, 49.7, 28.2, 24.5, 21.9, 21.6; IR (film on NaCl) 2932, 1590, 1421, 1331, 1302 cm–1; HRMS [ESI, high-resolution, double focusing magnetic sector (HRDFMagSec)] m/z 403.1845 (calcd for C25H26O1N2S1 + H+: 403.1839).
Preparation of Bisjulolidyl Selenoxanthone 2-Se
Phosphorus oxychloride (1.46 mL, 15.7 mmol), triethylamine (2.19 mL, 15.7 mmol), and 15-Se (700 mg, 1.31 mmol) in anhydrous CH3CN (30 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 410 mg (70.0%) of 2-Se as a brown solid, mp >250 °C: 1H NMR (500 MHz, CDCl3) δ 8.17 (s, 2 H), 3.31–3.24 (m, 8 H), 2.86 (t, 4 H, J = 6.5 Hz), 2.79 (t, 4 H, J = 6.5 Hz), 2.08 (quintet, 4 H, J = 6.5 Hz), 1.98 (quintet, 4 H, J = 6.5 Hz); 13C NMR (125 MHz, CDCl3) δ 180.3, 145.5, 133.6, 129.3, 120.2, 119.3, 114.6, 50.2, 49.4, 27.6, 25.5, 21.5, 21.3; IR (film on NaCl) 2927, 2834, 1586, 1569, 1540, 1416, 1329, 1301, 1204 cm–1; HRMS (ESI, HRDFMagSec) m/z 451.1289 (calcd for C25H26ON280Se + H+: 451.1283).
Preparation of Bisjulolidyl Telluroxanthone 2-Te
Phosphorus oxychloride (1.44 mL, 15.5 mmol), triethylamine (2.16 mL, 15.5 mmol), and 15-Te (753 mg, 1.29 mmol) in anhydrous CH3CN (30 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 380 mg (59.1%) of 2-Te as a brown solid, mp >250 °C: 1H NMR (500 MHz, CD2Cl2) δ 8.13 (s, 2 H), 3.51–3.23 (m, 8 H), 2.85 (t, 4 H, J = 6.5 Hz), 2.70 (t, 4 H, J = 6.5 Hz), 2.07 (quintet, 4 H, J = 6.5 Hz), 1.97 (quintet, 4 H, J = 6.5 Hz); 13C NMR (125 MHz, CDCl3) δ 184.2, 146.1, 131.4, 122.8, 122.3, 121.3, 118.6, 50.5, 49.9, 30.0, 28.1, 22.2, 21.9; IR (film on NaCl) 2940, 2836, 1583, 1556, 1535, 1504, 1406, 1321, 1292, 1204 cm–1; HRMS (ESI, HRDFMagSec) m/z 501.1172 (calcd for C25H26ON2130Te + H+: 501.1180).
Preparation of Bishalf-julolidyl Thioxanthone 3-S
Phosphorus oxychloride (2.36 mL, 25.3 mmol), triethylamine (3.53, 25.3 mmol), and 17-S (1.04 g, 2.11 mmol) in anhydrous CH3CN (25 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 708 mg (82.5%) of 3-S as a brown solid, mp >250 °C: 1H NMR (500 MHz, CDCl3) δ 8.39 (s, 2 H), 6.44 (s, 2 H), 3.38 (t, 4 H J = 6.0 Hz), 3.01 (s, 6 H), 1.76 (t, 4 H, J = 6.0 Hz), 1.35 (s, 12 H); 13C NMR (75.5 MHz, CDCl3) δ 177.5, 147.3, 136.7, 130.3, 125.9, 118.2, 103.4, 47.1, 38.7, 35.9, 31.7, 29.7; IR (film, NaCl) 2957, 1594, 1574, 1520, 1434 cm–1; HRMS (ESI, HRDFMagSec) m/z 407.2149 (calcd for C25H30N2OS + H+: 407.2152).
Preparation of Bishalf-julolidyl Selenoxanthone 3-Se
Phosphorus oxychloride (0.849 mL, 9.11 mmol), triethylamine (1.27 mL, 9.11 mmol), and 17-Se (409 mg, 0.759 mmol) in anhydrous CH3CN (15 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 250 mg (72.5%) of 3-Se as a brown solid, mp >250 °C: 1H NMR (500 MHz, CDCl3) δ 8.47 (s, 2 H), 6.53 (s, 2 H), 3.38 (t, 4 H, J = 6.0 Hz), 3.00 (s, 6 H), 1.75 (t, 4 H, J = 6.0 Hz), 1.34 (s, 12 H); 13C NMR (75.5 MHz, CDCl3) δ 179.5, 147.3, 134.1, 130.5, 127.5, 119.7, 106.2, 47.2, 38.8, 36.0, 31.8, 29.8; IR (film on NaCl) 2955, 1590, 1562, 1517 cm–1; HRMS (ESI, HRDFMagSec) m/z 455.1599 (calcd for C25H30N2O80Se + H+: 455.1596).
Preparation of Bishalf-julolidyl Telluroxanthone 3-Te
Phosphorus oxychloride (1.29 mL, 13.8 mmol), triethylamine (1.92 mL, 13.8 mmol), and 17-Te (677 mg, 1.15 mmol) in anhydrous CH3CN (25 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 507 mg (87.4%) of 3-Te as a brown solid, mp >250 °C: 1H NMR (500 MHz, CDCl3) δ 8.57 (s, 2 H), 6.64 (s, 2 H), 3.36 (t, 4 H, J = 6.0 Hz), 2.98 (s, 6 H), 1.75 (t, 4 H, J = 6.0 Hz), 1.34 (s, 12 H); 13C NMR (75.5 MHz, CDCl3) δ 183.2, 147.1, 131.0, 129.3, 122.6, 118.9, 112.3, 47.2, 38.7, 36.0, 31.7, 29.8; IR (film on NaCl) 2924, 1585 cm–1; HRMS (ESI, HRDFMagSec) m/z 505.1500 (calcd for C25H30N2O130Te + H+: 505.1493).
Preparation of Julolidyl/Half-julolidyl Thioxanthone 4-S
Phosphorus oxychloride (1.72 mL, 18.4 mmol), triethylamine (2.56 mL, 11.0 mmol), and 18-S (750 mg, 1.53 mmol) in anhydrous CH3CN (30 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 538 mg (86.9%) of 4-S as a brown solid, mp >250 °C: 1H NMR (500 MHz, CDCl3) δ 8.37 (s, 1 H), 8.10 (s, 1 H), 6.50 (s, 1 H), 3.37 (t, 2 H, J = 6.0 Hz), 3.28–3.21 (m, 4 H), 3.00 (s, 3 H), 2.85 (t, 2 H, J = 6.5 Hz), 2.75 (t, 2 H, J = 6.5 Hz), 2.05 (quintet, 2 H, J = 6.5 Hz), 1.96 (quintet, 2 H, J = 6.5 Hz), 1.75 (t, 2 H, J = 6.0 Hz), 1.35 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) δ 177.5, 147.1, 144.9, 136.3, 134.6, 130.3, 127.4, 125.5, 119.5, 117.8, 117.7, 112.2, 103.7, 49.7, 48.9, 46.9, 38.5, 35.7, 31.6, 29.5, 27.4, 23.7, 21.1, 20.7; IR (film on NaCl) 2931, 1592, 1573, 1513, 1421 cm–1; HRMS (ESI, HRDFMagSec) m/z 404.1917 (calcd for C25H28N2OS+: 404.1917).
Preparation of Julolidyl/Half-julolidyl Selenoxanthone 4-Se
Phosphorus oxychloride (2.46 mL, 26.4 mmol), triethylamine (3.68 mL, 26.4 mmol), and 18-Se (1.18 g, 2.20 mmol) in anhydrous CH3CN (40 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 610 mg (61.4%) of 4-Se as a brown solid, mp >250 °C: 1H NMR (500 MHz, CDCl3) δ 8.43 (s, 1 H), 8.14 (s, 1 H), 6.55 (s, 1 H), 3.31 (t, 2 H, J = 6.0 Hz), 3.22–3.13 (m, 4 H), 2.94 (s, 3 H), 2.80 (t, 2 H, J = 6.0 Hz), 2.63 (t, 2 H, J = 6.0 Hz), 2.00 (quintet, 2 H, J = 6.0 Hz), 1.91 (quintet, 2 H, J = 6.0 Hz), 1.70 (t, 2 H, J = 6.0 Hz), 1.32 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) δ 179.8, 147.3, 145.4, 134.1, 133.9, 130.7, 129.4, 127.3, 119.9, 119.5, 119.4, 114.4, 106.3, 50.0, 49.2, 47.2, 38.8, 36.0, 31.8, 29.7, 27.5, 25.5, 21.3, 21.2; IR (film on NaCl) 2930, 1589, 1569, 1514 cm–1; HRMS (ESI, HRDFMagSec) m/z 453.1432 (calcd for C25H28N2O80Se + H+: 453.1440).
Preparation of Julolidyl/Half-julolidyl Telluroxanthone 4-Te
Phosphorus oxychloride (306 μL, 3.28 mmol), triethylamine (457 μL, 3.28 mmol), and 18-Te (160 mg, 0.273 mmol) in anhydrous CH3CN (20 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 83.0 mg (60.6%) of 4-Te as a brown solid, mp >250 °C: 1H NMR (300 MHz, CD2Cl2) δ 8.47 (s, 1 H), 8.17 (s, 1 H), 6.74 (s, 1 H), 3.32 (t, 2 H, J = 6.0 Hz), 3.26–3.14 (m, 4 H), 2.95 (s, 3 H), 2.83 (t, 2 H, J = 6.3 Hz), 2.53 (t, 2 H, J = 6.3 Hz), 2.06–1.86 (m, 4 H), 1.72 (t, 2 H, J = 6.0 Hz), 1.34 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) δ 183.6, 147.6, 146.0, 131.7, 131.6, 129.0, 122.9, 122.8, 122.7, 120.9, 119.5, 118.6, 112.5, 50.3, 49.8, 47.6, 39.1, 36.5, 32.1, 30.1, 30.0, 27.9, 22.0, 21.8; IR (film on NaCl) 2928, 1583, 1560, 1406, 1304, 1280, 1261, 1207 cm–1; HRMS (ESI, HRDFMagSec) m/z 503.1329 (calcd for C25H28ON2130Te + H+: 503.1337).
Preparation of Bisjulolidyl Thiorhodamine 5-S
Phenylmagnesium bromide (1 M in THF, 1.24 mL, 1.24 mmol) was added dropwise to a stirred solution of 2-S (100 mg, 0.248 mmol) in THF (10 mL), and the resulting mixture was heated at reflux for 16 h and then cooled to ambient temperature. The reaction was quenched by the addition of glacial acetic acid (2 mL) and the resulting mixture was poured into 200 mL of aqueous 10% HPF6. After stirring for 12 h, the precipitate was collected via filtration and washed with water (30 mL) and diethyl ether (4 × 25 mL) to give 138 mg (91.4%) of 5-S as a green solid, mp >250 °C: 1H NMR (500 MHz, CD2Cl2) δ 7.63–7.56 (m, 3 H), 7.30–7.23 (m, 2 H), 6.95 (s, 2 H), 3.54–3.44 (m, 8 H), 2.94 (t, 4 H, J = 6.0 Hz), 2.65 (t, 4 H, J = 6.0 Hz), 2.16 (quintet, 4 H, J = 6.0 Hz), 1.95 (quintet, 4 H, J = 6.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 157.3, 148.5, 138.8, 137.0, 132.8, 129.8, 129.2, 128.8, 125.1, 118.6, 114.1, 51.5, 50.5, 28.2, 24.4, 20.8, 20.4; λmax (MeOH) 594 nm (ε = 1.24 × 10–5 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 463.2203 (calcd for C31H31N2S+: 463.2202).
Preparation of Bisjulolidyl Selenorhodamine 5-Se
Phenylmagnesium bromide (1 M in THF, 2.23 mL, 2.23 mmol) and 2-Se (100 mg, 0.223 mmol) in THF (10 mL) were treated as described for the preparation of 5-S to give 125 mg (85.6%) of 5-Se as a blue solid, mp >250 °C: 1H NMR (500 MHz, CDCl3) δ 7.62–7.54 (m, 3 H), 7.28–7.20 (m, 2 H), 6.98 (s, 2 H), 3.56–3.40 (m, 8 H), 2.86 (t, 4 H, J = 6.0 Hz), 2.62 (t, 4 H, J = 6.0 Hz), 2.18 (quintet, 4 H, J = 6.0 Hz), 1.94 (quintet, 4 H, J = 6.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 159.1, 148.3, 141.0, 138.4, 135.1, 129.8, 129.0, 128.7, 124.5, 119.4, 116.5, 51.5, 50.6, 28.1, 26.2, 20.8, 20.6; λmax (MeOH) 604 nm (ε = 1.35 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 511.1649 (calcd for C31H31N280Se+: 511.1647).
Preparation of Bisjulolidyl Tellurorhodamine 5-Te
Phenylmagnesium bromide (1 M in THF, 1.00 mL, 1.00 mmol) and 2-Te (100 mg, 0.201 mmol) in THF (10 mL) were treated as described for the preparation of 5-S to give 130 mg (92.3%) of 5-Te as a purple solid, mp >250 °C: 1H NMR (500 MHz, CD2Cl2) δ 7.60–7.51 (m, 3 H), 7.25–7.19 (m, 2 H), 7.09 (s, 2 H), 3.47 (t, 4 H, J = 6.0 Hz), 3.43 (t, 4 H, J = 6.0 Hz), 2.73 (t, 4 H, J = 6.5 Hz), 2.59 (t, 4 H, J = 6.0 Hz), 2.20 (quintet, 4 H, J = 6.0 Hz), 1.93 (quintet, 4 H, J = 6.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 162.2, 147.7, 140.6, 138.1, 135.6, 129.7, 128.6, 128.5, 124.2, 121.5, 121.0, 51.4, 50.7, 30.5, 28.0, 21.0, 20.8; λmax (MeOH) 615 nm (ε = 1.44 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 561.1540 (calcd for C31H31N2130Te+: 561.1544).
Preparation of Bishalf-julolidyl Thiorhodamine 6-S
Phenylmagnesium bromide (1 M in THF, 2.46 mL, 2.46 mmol) and 3-S (100 mg, 0.246 mmol) in THF (10 mL) were treated as described for the preparation of 5-S to give 124 mg (82.1%) of 6-S as a green solid, mp 252–254 °C: 1H NMR (500 MHz, CD2Cl2) δ 7.68–7.61 (m, 3 H), 7.36–7.30 (m, 2 H), 7.29 (s, 2 H), 7.00 (s, 2 H), 3.58 (t, 4 H, J = 6.0 Hz), 3.24 (s, 6 H), 1.75 (t, 4 H, J = 6.0 Hz), 1.09 (s, 12 H); 13C NMR (75.5 MHz, CD2Cl2) δ 158.7, 150.5, 142.9, 136.2, 135.1, 130.6, 129.7, 129.6, 128.9, 119.4, 104.6, 48.5, 40.0, 34.6, 32.2, 28.6; λmax (MeOH) 579 nm (ε = 1.04 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 467.2514 (calcd for C31H35N2S+: 467.2515).
Preparation of Bishalf-julolidyl Selenorhodamine 6-Se
Phenylmagnesium bromide (1 M in THF, 1.10 mL, 1.10 mmol) and 3-Se (100 mg, 0.22 mmol) in THF (10 mL) were treated as described for the preparation of 5-S to give 113 mg (77.9%) of 6-Se as a green solid, mp 246–248 °C: 1H NMR (500 MHz, CD2Cl2) δ 7.65–7.59 (m, 3 H), 7.33–7.27 (m, 4 H), 7.17 (s, 2 H), 3.55 (t, 4 H, J = 6.0 Hz), 3.23 (s, 6 H), 1.74 (t, 4 H, J = 6.0 Hz), 1.05 (s, 12 H); 13C NMR (75.5 MHz, CDCl3) δ 159.6, 149.6, 144.5, 137.5, 134.1, 132.1, 129.0, 128.5, 127.1, 119.8, 107.9, 48.3, 39.9, 34.5, 31.8, 28.6; λmax (MeOH) 590 nm (ε = 1.36 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 515.1960 (calcd for C31H35N280Se+: 515.1960).
Preparation of Bishalf-julolidyl Tellurorhodamine 6-Te
Phenylmagnesium bromide (1 M in THF, 1.98 mL, 1.98 mmol) and 3-Te (100 mg, 0.198 mmol) in THF (10 mL) were treated as described for the preparation of 5-S to give 105 mg (75.0%) of 6-Te as a green solid, mp 250–252 °C: 1H NMR (500 MHz, CDCl3) δ 7.61–7.55 (m, 3 H), 7.48 (s, 2 H), 7.35 (s, 2 H), 7.25–7.21 (m, 2 H), 3.49 (t, 4 H, J = 6.0 Hz), 3.24 (s, 6 H), 1.71 (t, 4 H, J = 6.0 Hz), 1.00 (s, 12 H); 13C NMR (75.5 MHz, CD2Cl2) δ 163.5, 149.2, 140.0, 136.0, 135.5, 134.2, 129.3, 129.0, 128.7, 122.1, 114.8, 48.6, 39.9, 34.8, 32.0, 28.6; λmax (MeOH) 607 nm (ε = 1.21 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 565.1857 (calcd for C31H35N2130Te+: 565.1857).
Preparation of Julolidyl/Half-julolidyl Thiorhodamine 7-S
Phenylmagnesium bromide (1 M in THF, 2.47 mL, 2.47 mmol) and 4-S (100 mg, 0.247 mmol) in THF (10 mL) were treated as described for the preparation of 5-S to give 136 mg (90.1%) of 7-S as a purple solid, mp 249–251 °C: 1H NMR (500 MHz, CD2Cl2) δ 7.66–7.58 (m, 3 H), 7.33–7.27 (m, 2 H), 7.19 (s, 1 H), 7.06 (s, 1 H), 7.03 (s, 1 H), 3.57 (t, 2 H, J = 6.5 Hz), 3.54–3.48 (m, 4 H), 3.22 (s, 3 H), 2.88 (t, 2 H, J = 6.5 Hz), 2.67 (t, 2 H, J = 6.5 Hz), 2.17 (quintet, 2 H, J = 6.0 Hz), 1.96 (quintet, 2 H, J = 6.0 Hz), 1.74 (t, 2 H, J = 6.5 Hz), 1.07 (s, 6 H); 13C NMR (75.5 MHz, CD2Cl2) δ 157.8, 150.0, 148.9, 141.9, 139.6, 136.6, 134.8, 133.2, 130.2, 129.7, 129.4, 128.8, 125.4, 119.1, 118.8, 113.9, 104.8, 51.5, 50.6, 48.4, 39.8, 34.7, 32.1, 28.6, 28.1, 24.3, 20.7, 20.2; λmax (MeOH) 587 nm (ε = 1.20 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 465.2358 (calcd for C31H33N2S+: 465.2359).
Preparation of Julolidyl/Half-julolidyl Selenorhodamine 7-Se
Phenylmagnesium bromide (1 M in THF, 1.11 mL, 1.11 mmol) and 4-Se (100 mg, 0.222 mmol) in THF (10 mL) were treated as described for the preparation of 5-S to give 110 mg (75.3%) of 7-Se as a purple solid, mp 237–239 °C: 1H NMR (500 MHz, CDCl3) δ 7.65–7.54 (m, 3 H), 7.30–7.23 (m, 2 H), 7.19 (s, 1 H), 7.18 (s, 1 H), 7.10 (s, 1 H), 3.56–3.44 (m, 6 H), 3.21 (s, 3 H), 2.78 (t, 2 H, J = 6.0 Hz), 2.64 (t, 2 H, J = 6.0 Hz), 2.18 (quintet, 2 H, J = 6.0 Hz), 1.95 (quintet, 2 H, J = 6.0 Hz), 1.73 (t, 2 H, J = 6.0 Hz), 1.03 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) 158.7, 149.1, 148.1, 142.6, 141.6, 137.6, 134.9, 133.8, 131.7, 129.0, 128.8, 128.4, 124.3, 119.2, 119.1, 116.2, 107.4, 51.1, 50.2, 48.0, 39.5, 34.4, 31.7, 28.6, 27.6, 25.7, 20.2, 19.9; λmax (MeOH) 597 nm (ε = 1.22 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 513.1803 (calcd for C31H33N280Se+: 513.1803).
Preparation of Julolidyl/Half-julolidyl Tellurorhodamine 7-Te
Phenylmagnesium bromide (1 M in THF, 1.00 mL, 1.00 mmol) and 4-Te (100 mg, 0.200 mmol) in THF (10 mL) were treated as described for the preparation of 5-S to give 125 mg (88.7%) of 7-Te as a purple solid, mp >250 °C: 1H NMR (500 MHz, CD2Cl2) δ 7.62–7.54 (m, 3 H), 7.41 (s, 1 H), 7.28 (s, 1 H), 7.27–7.23 (m, 2 H), 7.22 (s, 1 H), 3.53–3.46 (m, 4 H), 3.44 (t, 2 H, J = 6.0 Hz), 3.19 (s, 3 H), 2.67–2.58 (m, 4 H), 2.24–2.17 (m, 2 H), 1.98–1.90 (m, 2 H), 1.71 (t, 2 H, J = 6.0 Hz), 0.99 (s, 6 H); 13C NMR (75.5 MHz, CDCl3) δ 162.9, 148.8, 148.3, 140.3, 138.7, 136.8, 135.1, 135.0, 134.1, 129.6, 128.8, 128.6, 124.5, 122.0, 121.7, 121.6, 114.3, 51.6, 50.9, 48.5, 39.8, 34.9, 32.0, 30.8, 28.7, 27.9, 20.9, 20.7; λmax (MeOH) 610 nm (ε = 1.27 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 563.1697 (calcd for C31H33N2130Te+: 563.1700).
Preparation of Diaryl Selenide 22
sec-Butyllithium (1.00 M in cyclohexane, 0.96 mL, 0.96 mmol), TMEDA (144 μL, 0.964 mmol), amide 16(19) (0.250 g, 0.873 mmol), and diselenide 21(22) (0.730 g, 1.83 mmol) in THF (2 × 30 mL) were treated as described for the preparation of 15-S. The crude product was purified via column chromatography (SiO2, 4:6 EtOAc/hexanes) to yield 194 mg (45.9%) of 22 as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.13 (t, 1 H, J = 8.1 Hz), 7.04–6.95 (m, 2 H), 6.90 (d, 1 H, J = 8.1 Hz), 6.64 (dd, 1 H, J = 2.4, 8.1 Hz), 6.48 (s, 1 H), 3.62–3.36 (m, 4 H), 3.19 (t, 2 H, J = 6.0 Hz), 2.91 (s, 6 H), 2.67 (s, 3 H), 1.71 (t, 2 H, J = 6.0 Hz), 1.65 (m, 2 H), 1.58 (m, 4 H), 1.23 (s, 6 H); 13C NMR (300 MHz, CDCl3) δ 169.8, 150.7, 145.6, 130.7, 129.4, 129.2, 128.3, 125.2, 124.1, 122.2, 118.1, 114.0, 111.5, 47.1, 45.0 (br), 40.2, 38.5, 36.4, 31.5, 30.3, 25.8, 24.4; IR (film on NaCl) 2933, 2852, 1625, 1590, 1557, 1533, 1493, 1467, 1430, 1347, 1317, 1260 cm–1; HRMS (ESI, HRDFMagSec) m/z 486.2018 (calcd for C26H35N3O80Se + H+: 486.2018).
Preparation of Selenoxanthone 23
Phosphorus oxychloride (0.380 mL, 4.08 mmol), triethylamine (0.567 mL, 4.07 mmol), and selenide 22 (165 mg, 0.339 mmol) in anhydrous CH3CN (15 mL) were treated as described for the preparation of 2-S. The crude product was recrystallized from CH2Cl2/hexanes to give 130 mg (95.7%) of 23 as a yellow solid, mp 200–203 °C: 1H NMR (500 MHz, CDCl3) δ 8.94 (d, 1 H, J = 9.0 Hz), 8.46 (s, 1 H), 6.76 (dd, 1 H, J = 3.0, 9.0 Hz), 6.67 (d, 1 H, J = 3.0 Hz), 6.54 (s, 1 H), 3.39 (t, 2 H, J = 6.0 Hz), 3.07 (s, 6 H), 3.02 (s, 3 H), 1.76 (t, 2 H, J = 6.0 Hz), 1.35 (s, 6 H); 13C NMR (500 MHz, CDCl3) δ 179.8, 151.6, 147.7, 136.4, 134.4, 132.2, 130.9, 127.8, 120.5, 119.9, 111.3, 107.9, 106.4, 47.5, 40.0, 39.0, 36.2, 32.0, 29.9; IR (film on NaCl) 1590, 1314, 1287 cm–1; HRMS (ESI, HRDFMagSec) m/z 401.1126 (calcd for C21H24N2O80Se + H+: 401.1127).
Preparation of Half-julolidyl Selenorhodamine 20-Se
Phenylmagnesium bromide (1.0 M in THF, 2.50 mL, 2.50 mmol) and selenoxanthone 23 (200 mg, 0.501 mmol) in THF (20 mL) were treated as described for the preparation of 5-S to give 225 mg (74.3%) of 20-Se as a green solid, mp 215–218 °C: 1H NMR (500 MHz, CD2Cl2) δ 7.66–7.59 (m, 2 H), 7.45 (d, 1 H, J = 9.0 Hz), 7.36 (s, 1 H) 7.32–7.27 (m, 3 H), 7.23 (d, 1 H, J = 2.5 Hz), 7.19 (s, 1 H), 6.82 (dd, 1 H, J = 2.5, 9.5 Hz), 3.57 (t, 2 H, J = 6.5 Hz), 3.26 (s, 3 H), 3.23 (s, 6 H), 1.75 (t, 2 H, J = 6.5 Hz), 1.05 (s, 6 H); 13C NMR (500 MHz, CD2Cl2) δ 161.3, 153.0, 150.7, 145.4, 145.1, 138.4, 137.6, 135.0, 133.1, 129.5, 128.9, 120.5, 120.0, 114.7, 108.9, 108.3, 48.9, 40.7, 40.3, 34.6, 32.1, 28.5; λmax (CH3OH) 585 nm (ε = 1.35 × 105 M–1 cm–1); IR (film on NaCl) 1591, 1509, 1448, 1387, 1356, 1330, 1252, 1211 cm–1; HRMS (ESI, HRDFMagSec) m/z 461.1490 (calcd for C27H29N2Se+: 461.1497).
Preparation of Tellurorhodamine 27
2-Bromomesitylene (469 μL, 3.07 mmol) was dissolved in dry THF (1.5 mL) in a round-bottomed flask. Magnesium (82.0 mg, 3.37 mmol) was added and the solution stirred at room temperature until most of the magnesium was consumed. The resulting solution of Grignard reagent was added via cannula to a stirred suspension of 2-Te (100 mg, 0.204 mmol) in dry THF (10 mL). The resulting solution was heated at reflux for 24 h, cooled to ambient temperature, and poured into 200 mL of an ∼10% aqueous HPF6 solution. The resulting precipitate was collected via filtration after 12 h of stirring and was washed with water (100 mL) and diethyl ether (100 mL) to give 127 mg (83.0%) of 27 as a dark purple solid, mp >250 °C. 1H NMR (500 MHz, CD2Cl2) δ 7.10 (s, 2 H), 7.03 (s, 2 H), 3.47 (t, 4 H, J = 6.0 Hz), 3.43 (t, 4 H, J = 6.0 Hz), 2.74 (t, 4 H, J = 6.5 Hz), 2.61 (t, 4 H, J = 6.5 Hz), 2.42 (s, 3 H), 2.21 (quintet, 4 H, J = 6.0 Hz), 1.93 (quintet, 4 H, J = 6.5 Hz), 1.80 (s, 6 H); 13C NMR (75.5 MHz, CD2Cl2) δ 162.4, 147.9, 138.5, 136.6, 136.0, 135.9, 135.4, 128.7, 125.1, 120.9, 120.8, 51.4, 50.7, 30.5, 28.0, 21.2, 21.0, 20.8, 19.7; λmax (MeOH) 617 nm (ε = 1.65 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 603.2013 (calcd for C34H37N2130Te+: 603.2012).
Oxidation of Tellurorhodamine 27 to Tellurorhodamine Telluroxide 28
Tellurorhodamine 27 (1.5 mg, 0.20 μmol) was dissolved in 0.60 mL of a solution of 0.1 M CF3CO2D in CD3OD in a 5 mm NMR tube. The 1H NMR spectrum of 27 in this solvent system was acquired: δ 7.09 (s, 2 H), 7.07 (s, 2 H), 3.54–3.48 (m, 4 H), 3.47–3.43 (m, 4 H), 2.82–2.74 (m, 4 H), 2.64–2.56 (m, 4 H), 2.42 (s, 3 H), 2.23–2.15 (m, 4 H), 1.98–1.88 (m, 4 H), 1.79 (s, 6 H). To this solution was added 12.5 μL of an 8.8 M solution of H2O2 (0.11 mmol, 0.18 M final concentration). The 1H NMR spectrum of the resulting solution was recorded within 3 min of addition of the H2O2, and oxidation was complete at this point: 1H NMR (500 MHz, CD3OD 0.1 M CF3C(O)OD) δ 7.05 (s, 2 H), 6.81 (s, 2 H), 3.75–3.67 (m, 4 H), 3.66–3.60 (m, 4 H), 3.46–3.36 (m, 4 H), 2.58–2.49 (m, 4 H), 2.39 (s, 3 H), 2.29–2.17 (m, 4 H), 2.00–1.86 (m, 4 H), 1.90 (s, 6 H); 13C NMR (500 MHz, 0.1 M CF3C(O)OD in CD3OD) δ 151.1, 139.3, 137.2, 136.2, 135.4, 130.1, 128.5, 126.0, 52.0, 51.4, 30.4, 27.3, 20.8, 20.2, 20.1, 18.6; λmax (CH3OH) 704 nm (ε = 1.35 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 619.1963 (calcd for C34H37N2O130Te+: 619.1963).
Preparation of Tellurorhodamine 29
2-Bromomesitylene (461 μL, 2.98 mmol) was added to a stirred suspension of ground magnesium turnings (79.7 mg, 3.28 mmol) in THF (3 mL). The resulting solution was stirred at ambient temperature for 3 h before it was transferred via cannula to a stirred solution of telluroxanthone 3-Te (150 mg, 0.298 mmol) at room temperature in THF (5 mL). The mixture was heated at reflux overnight, then cooled to ambient temperature before glacial acetic acid (2 mL) was added. The resulting solution was poured into cold, stirring 10% aqueous HPF6 (200 mL), and the resulting mixture was stirred for 12 h. The precipitate was collected via filtration and washed with water (50 mL) and hexanes (75 mL), yielding 125 mg (55.8%) of 29 as a blue solid, mp 166–168 °C: 1H NMR (500 MHz, CD2Cl2) δ 7.40 (s, 2 H), 7.39 (s, 2 H), 7.08 (s, 2 H), 3.50 (t, 4 H, J = 6.0 Hz), 3.20 (s, 6 H), 2.44 (s, 3 H), 1.82 (s, 6 H), 1.73 (t, 4 H, J = 6.0 Hz), 1.01 (s, 12 H); 13C NMR (75.5 MHz, CD2Cl2) δ 164.1, 149.5, 138.9, 135.9, 135.8, 135.6, 135.1, 133.7, 128.7, 121.5, 114.9, 48.5, 39.9, 34.8, 31.9, 28.8, 21.2, 19.5; λmax (CH3OH) 606 nm (ε = 9.66 × 104 M–1 cm–1); HRMS m/z 607.2329 (ESI, HRDFMagSec) (C34H41N2130Te+: 607.2326).
Oxidation of Tellurorhodamine 29 to Tellurorhodamine Telluroxide 30
Tellurorhodamine 29 (1.5 mg, 0.20 μmol) was dissolved in 0.60 mL of a solution of 0.1 M CF3CO2D in CD3OD in a 5 mm NMR tube. The 1H NMR spectrum of 29 in this solvent system was acquired: δ 7.73 (s, 2 H), 7.43 (s, 2 H), 7.16 (s, 2 H), 3.54 (t, 4 H, J = 6.0 Hz), 3.21 (s, 4 H), 2.42 (s, 3 H), 1.82 (s, 6 H), 1.82 (t, 4 H, J = 6.0 Hz), 1.00 (s, 6 H). To this solution was added 12.5 μL of an 8.8 M solution of H2O2 (0.11 mmol, 0.18 M final concentration). The 1H NMR spectrum of the resulting solution was recorded within 3 min of addition of the H2O2, and oxidation was complete at this point: 1H NMR (500 MHz, CD3OD 0.1 M CF3C(O)OD) δ 8.00 (s, 2 H), 7.19 (s, 2 H), 7.15 (s, 2 H), 3.76 (t, 4 H, J = 6.0 Hz), 3.48 (s, 4 H), 2.42 (s, 3 H), 1.93 (s, 6 H), 1.77 (t, 4 H, J = 6.0 Hz), 1.00 (s, 6 H); 13C NMR (500 MHz, 0.1 M CF3C(O)OD in CD3OD) δ 153.4, 140.8, 137.0, 136.8, 135.7, 129.8, 129.4, 123.7, 50.2, 40.8, 35.3, 32.6, 32.3, 28.9, 21.2, 19.6; λmax (CH3OH) 692 nm (ε = 1.65 × 105 M–1 cm–1); HRMS (ESI, HRDFMagSec) m/z 623.2279 (calcd for C34H41N2O130Te+: 623.2276).
Determination of Singlet Oxygen Yields from Singlet Oxygen Phosphorescence Spectroscopy
Generation of singlet oxygen (1O2) was assessed by its phosphorescence peak at 1270 nm. A spectrometer equipped with a near-infrared photodetector was used for acquisition of the emission spectra in NIR spectral range. A diode-pumped solid-state laser at 532 nm was the excitation source. The emission signal was collected at 90° relative to the exciting laser beam with the use of a 950 nm long-pass filter to attenuate the scattered light and fluorescence from the samples. The samples (methanol solutions of the compounds in quarts cuvettes) were placed in front of the spectrometer entrance slit.
Fluorescence Experiments
Measurements of fluorescence quantum yield were performed on a spectrofluorometer using either fluorescent dye Rhodamine 6G with known ΦFL = 0.9344 in MeOH for 5-E–7-E or fluorescent dye LD 700 perchlorate (Rhodamine 700) as a reference with known ΦFL = 0.3831 in MeOH for 27 and 28.
Cell Experiments
Colo-26, a murine colon carcinoma cell line, was maintained in RPMI 1640 supplemented with 10% fetal calf serum (FCS) and antibiotics at 37 °C, and 5% CO2. Colo-26 cells were harvested, counted, and seeded at a density of 1.5 × 105 cells per well in 0.5 mL of RPMI supplemented medium the night before the assay in a 24-well plate. The day of the assay, the medium was removed from a single well at a time, and 0.5 mL of HBSS was added to the cells. MitoTracker Green (0.25 μL of stock solution and 0.5 μM final concentration) was added to the well, and the plate was incubated for 10 min. Tellurorhodamine 27 (0.2 μM) was added, and the plate was incubated for an additional 5 min (total incubation = 15 min). The HBSS containing tellurorhodamine 27 and MitoTracker Green was removed, and 0.5 mL of fresh HBSS was added.
A camera-equipped fluorescence microscope was used in the cell imaging experiments. Tellurorhodamine 27 and/or telluroxide 28 were excited with 620 ± 30 nm light, and MitoTracker Green was excited with 470 ± 20 nm light. ImageJ software was used to color the images and to give an overlay of images.
Acknowledgments
This research was supported in part by the NIH (GM-94367) and the National Science Foundation (CHE-1151379).
Supporting Information Available
General experimental procedures and 1H and 13C NMR spectra for 2-E–7-E, 9, 10-E, 11, 12, 13-E, 15-E, 17-E, 18-E, 20-E, 22, 23, and 27–30. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ M.W.K. and G.A.S. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Titus J. A.; Haugland R.; Sharrow S. O.; Segal D. M. J. Immunol. Methods 1982, 50, 193–204. [DOI] [PubMed] [Google Scholar]
- Wang X.; He F.; Li L.; Wang H.; Yan R.; Li L. ACS Appl. Mater. Interfaces 2013, 5, 5700–5708. [DOI] [PubMed] [Google Scholar]
- Stepanyants N.; Zhang H.; Lobovkina T.; Dommersnes P.; Jeffries G. D. M.; Jesorka A.; Orwar O. J. Soft Mater. 2013, 9, 5155–5159. [Google Scholar]
- Yuan J.; Hao C.; Chen M.; Berini P.; Zou S. Langmuir 2013, 29, 221–227. [DOI] [PubMed] [Google Scholar]
- Cherevatskaya M.; Neumann M.; Fueldner S.; Harlander C.; Kuemmel S.; Dankesreiter S.; Pfitzner A.; Zeitler K.; Koenig B. Angew. Chem., Int. Ed. 2012, 51, 4062–4066. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Schmidt T.; Mahrous M.; Guieu V.; Perrier S.; Ravelet C.; Peyrin E. Anal. Chim. Acta 2011, 707, 191–196. [DOI] [PubMed] [Google Scholar]
- Taldone T.; Gomes-DaGama E. M.; Zong H.; Sen Si.; Alpaugh M. L.; Zatorska D.; Alonso-Sabadell R.; Guzman M. L.; Chiosis G. Bioorg. Med. Chem. Lett. 2011, 21, 5347–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y.; Tataurov A. V.; Owczarzy R. Biopolymers 2011, 95, 472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monson C. F.; Pace H. P.; Liu C.; Cremer P. S. Anal. Chem. 2011, 83, 2090–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moula G.; Aroca R. F. Anal. Chem. 2011, 83, 284–288. [DOI] [PubMed] [Google Scholar]
- Richardson J. A.; Gerowska M.; Shelbourne M.; French D.; Brown T. ChemBioChem 2010, 11, 2530–2533. [DOI] [PubMed] [Google Scholar]
- Mottram L. F.; Forbes S.; Ackley B. D.; Peterson B. R. Beilstein J. Org. Chem. 2012, 8, 2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmakov K.; Wurm C. A.; Hennig R.; Rapp E.; Jakobs S.; Belov V. N.; Hell S. W. Chem.—Eur. J. 2012, 18, 12986–12998. [DOI] [PubMed] [Google Scholar]
- Kolmakov K.; Belov V. N.; Bierwagen J.; Ringemann C.; Iler V. M.; Eggeling C.; Hell S. W. Chem.—Eur. J. 2010, 16, 158–166. [DOI] [PubMed] [Google Scholar]
- Calitree B.; Donnelly D. J.; Holt J. J.; Gannon M. K.; Nygren C. L.; Sukumaran D. K.; Autschbach J.; Detty M. R. Organometallics 2007, 26, 6248–6257. [Google Scholar]
- Ohulchanskyy T. Y.; Donnelly D. J.; Detty M. R.; Prasad P. N. J. Phys. Chem. B 2004, 108, 8668–8672. [Google Scholar]
- Detty M. R.; Prasad P. N.; Donnelly D. J.; Ohulchanskyy T.; Gibson S. L.; Hilf R. Bioorg. Med. Chem. 2004, 12, 2537–2544. [DOI] [PubMed] [Google Scholar]
- Holt J. J.; Gannon M. K.; Tombline G.; McCarty T. A.; Page P. M.; Bright F. V.; Detty M. R. Biorg. Med. Chem. 2006, 14, 8635–8643. [DOI] [PubMed] [Google Scholar]
- Orchard A.; Schamerhorn G. A.; Calitree B. D.; Sawada G. A.; Loo T. W.; Bartlett M. C.; Clarke D. M.; Detty M. R. Bioorg. Med. Chem. 2012, 20, 4290–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryman M. W.; Schamerhorn G. A.; Yung K.; Sathyamoorthy B.; Sukumaran D. K.; Ohulchanskyy T. Y.; Benedict J. B.; Detty M. R. Organometallics 2013, 32, 4321–4333. [Google Scholar]
- Koide Y.; Kawaguchi M.; Urano Y.; Hanaoka K.; Komatsu T.; Abo M.; Terai T.; Nagano T. Chem. Commun. 2012, 48, 3091–3093. [DOI] [PubMed] [Google Scholar]
- Holt J. J.; Calitree B. D.; Vincek J.; Gannon M. K. II; Detty M. R. J. Org. Chem. 2007, 72, 2690–2693. [DOI] [PubMed] [Google Scholar]
- Del Valle D. J.; Donnelly D. J.; Holt J. J.; Detty M. R. Organometallics 2005, 24, 3807–3810. [Google Scholar]
- Brennan N. K.; Donnelly D. J.; Detty M. R. J. Org. Chem. 2003, 68, 3344–3347. [DOI] [PubMed] [Google Scholar]
- Katayama H.; Abe E.; Kaneko K. J. Heterocycl. Chem. 1982, 19, 925–926. [Google Scholar]
- Rosevear J.; Wilkshire J. F. K. Aust. J. Chem. 1977, 30, 1561–1572. [Google Scholar]
- Klaus M.; Mohr P.; Weiss E. European Patent EP350846, Jan 17, 1990.
- Rubio N.; Prat P.; Bou N.; Borrell J. I.; Teixidó J.; Villanueva Á.; Juarranz Á.; Cañete M.; Stockert J. C.; Nonell S. New J. Chem. 2005, 29, 378–384. [Google Scholar]
- Azenha E. G.; Serra A. C.; Pineiro M.; Pereira M. M.; de Melo J. S.; Arnaut L. G.; Formosinho S. J.; Gonsalves A. M. d’A. R. Chem. Phys. 2002, 280, 177–190. [Google Scholar]
- Merkel P. B.; Detty M. R. J. Am. Chem. Soc. 1990, 112, 3845–3855. [Google Scholar]
- Sauer M.; Han K.-T.; Müller R.; Nord S.; Schulz A.; Seeger S.; Wolfrum J.; Arden-Jacob J.; Deltau G.; Marx N. J.; Zander C.; Drexhage K. J. Fluoresc. 1995, 5, 247–261. [DOI] [PubMed] [Google Scholar]
- Detty M. R.; Merkel P. B.; Hilf R.; Gibson S. L.; Powers S. K. J. Med. Chem. 1990, 33, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Powers S. K.; Walstad D. L.; Brown J. T.; Detty M. R.; Watkins P. J. J. Neuro-Oncol. 1989, 7, 179–188. [DOI] [PubMed] [Google Scholar]
- Yu F.; Li P.; Wang B.; Han K. J. Am. Chem. Soc. 2013, 135, 7674–7680. [DOI] [PubMed] [Google Scholar]
- B. Wang B.; Li P.; Yu F.; Song P.; Sun X.; Yang S.; Lou Z.; Han K. Chem. Commun. 2013, 49, 1014–1016. [DOI] [PubMed] [Google Scholar]
- Gibson S. L.; Hilf R.; Donnelly D. J.; Detty M. R. Bioorg. Med. Chem. 2004, 12, 4625–4631. [DOI] [PubMed] [Google Scholar]
- Gibson S. L.; Holt J. J.; Ye M.; Donnelly D. J.; Ohulchanskyy T. Y.; You Y.; Detty M. R. Biorg. Med. Chem. 2005, 13, 6394–6403. [DOI] [PubMed] [Google Scholar]
- Wagner S. J.; Skripchenko A.; Donnelly D. J.; Ramaswamy K.; Detty M. R. Biorg. Med. Chem. 2005, 13, 5927–5935. [DOI] [PubMed] [Google Scholar]
- Wagner S. J.; Skripchenko A.; Thompson-Montgomery D.; Awatefe H.; Donnelly D. J.; Detty M. R. Photochem. Photobiol. 2006, 76, 514–517.12462646 [Google Scholar]
- Detty M. R.; Gibson S. L.; Wagner S. J. Med. Chem. 2004, 47, 3897–3915. [DOI] [PubMed] [Google Scholar]
- Mann J. R.; Gannon M. K.; Fitzgibbons T. C.; Detty M. R.; Watson D. F. J. Phys. Chem. C 2008, 112, 13057–13061. [Google Scholar]
- Mulhern K. R.; Orchard A.; Watson D. F.; Detty M. R. Langmuir 2012, 28, 7071–7082. [DOI] [PubMed] [Google Scholar]
- McCormick T. M.; Calitree B. D.; Orchard A.; Kraut N. D.; Bright F. V.; Detty M. R.; Eisenberg R. J. Am. Chem. Soc. 2010, 132, 15480–15483. [DOI] [PubMed] [Google Scholar]
- Magde D. R.; Wong R.; Seybold P. G. Photochem. Photobiol. 2002, 75, 327–334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.