

Abstract
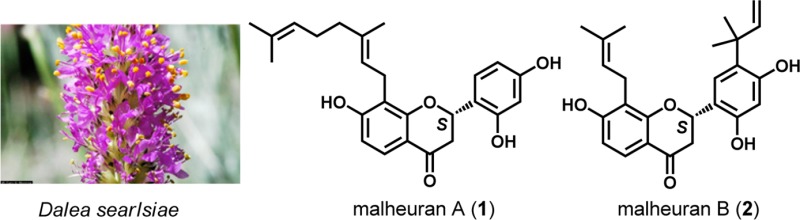
Continued interest in the chemistry of Dalea spp. led to investigation of Dalea searlsiae, a plant native to areas of the western United States. Methanol extractions of D. searlsiae roots and subsequent chromatographic fractionation afforded the new prenylated and geranylated flavanones malheurans A–D (1–4) and known flavanones (5 and 6). Known rotenoids (7 and 8) and isoflavones (9 and 10) were isolated from aerial portions. Structure determination of pure compounds was accomplished primarily by extensive 1D- and 2D-NMR spectroscopy. The absolute configurations of compounds 1–5, 7, and 8 were assigned using electronic circular dichroism spectroscopy. Antimicrobial bioassays revealed significant activity concentrated in the plant roots. Compounds 1–6 exhibited MICs of 2–8 μg/mL against Streptococcus mutans, Bacillus cereus, and oxacillin-sensitive and -resistant Staphylococcus aureus. Aerial metabolites 7–10 were inactive against these organisms, but the presence of 7 and 8 prompted investigation of the antiinsectan activity of D. searlsiae metabolites toward the major crop pest Spodoptera frugiperda (fall armyworm). While compounds 1–10 all caused significant reductions in larval growth rates, associated mortality (33–66%) was highest with flavanone 4 and rotenoids 7 and 8. These findings suggest a differential allocation of antimicrobial and antiinsectan plant resources to root and aerial portions of the plant, respectively.
Dalea searlsiae (A. Gray) Barneby (Searle’s prairie clover; syn. Petalostemon searlsiae A. Gray) (Fabaceae) is an herbaceous perennial plant, native to the Great Basin, Colorado Plateau, and northern Mojave Desert areas of the western United States.1 The plant genus Dalea was named by Linnaeus after Samuel Dale (1659–1739), a English apothecary, physician, and author of a well-regarded materia medica first published in 1693.2 The species was named after a 19-year-old amateur botanist, Fanny Searls, who returned from a trip to southeastern Nevada in 1872 with numerous plant specimens.3 Coincidental to the present study, D. searlsiae is a subject of research on the reintroduction of native flora, via seed distribution, to rangelands of the western U.S.,4,5 as inclusion of D. searlsiae in the diet of cattle may help improve nutrient use and prevent the potentially fatal condition “bloat”.6 An understanding of the chemistry of this plant is therefore of ecological and agricultural relevance.
Dalea spp. are prolific producers of phenolic secondary metabolites including flavonoids, isoflavonoids, chalcones, pterocarpans, stilbenes, and other biosynthetically related chemotypes.7−11 Secondary metabolites of mixed biogenetic origin are common in this genus, with frequent incorporation of isoprene units in the form of prenyl, and occasionally geranyl, side chains. Taxonomically, Dalea are in the Amorpheae tribe of a newly described Dalbergioid clade.12 The more phylogenetically distant genus Glycyrrhiza,13 however, produces similar metabolites that exhibit antibacterial activity against oral pathogens, including Streptococcus mutans.14−16 The prolific and diverse chemical content of Dalea spp. has resulted in the discovery of metabolites exhibiting a wide range of biological activities. No prior work has been done on the ecological roles of these metabolites or their localization within the plants.
Previous work in our laboratory has yielded higher percentages of new compounds from the roots of Dalea spp. as compared to the aerial parts.9 The role of flavonoids in plant roots has been a relatively recent area of interest. Root metabolites may be selectively allocated to defend against a complex variety of soil-borne pathogens, including Gram-positive bacteria, fungi, nematodes, and insect larvae, among others.17 These phenomena are complex, however, and may be dependent on many factors such as herbivory, growth conditions, symbiotic Gram-negative bacteria and other microbial populations, and the relative importance of plant parts (e.g., reproductive versus other tissues). Secondary metabolites allocated to aerial parts, on the other hand, may play a role in defense against herbivorous insects as well as microbes and other threats.18 Ultimately, the localization of metabolites in Dalea, as with other plants of importance, has implications for its use in agriculture or medicine.
An objective that evolved during the course of this study was therefore to assess the antibacterial and antiinsectan activities of root and aerial metabolites using the Gram-positive bacteria Bacillus cereus, oxacillin-resistant and -sensitive Staphylococcus aureus, S. mutans, and the generalist lepidopteran herbivore and major crop pest Spodoptera frugiperda (fall armyworm). All three chosen bacteria have medical relevance. B. cereus, an endemic soil-dwelling bacterium, may serve as a model organism for gauging plant defense in the root environment. B. cereus also has the potential to cause food-borne illness in humans.19 Multidrug-resistant (MDR) strains of Staph. aureus pose a serious health risk and are a primary concern of the U.S. Centers for Disease Control (CDC).20 Methicillin-resistant Staph. aureus (MRSA) infections have become disturbingly common; annual death rates in the United States due to MRSA have recently eclipsed those due to AIDS.21 Originally confined to hospitals, MRSA infections now also occur in the general community. S. aureus itself is a common commensal bacterium present in ∼30% of the population, but MRSA isolates increased in frequency from 36 to 64% from 1992 to 2003.22 MRSA is typically referred to as synonymous with oxacillin-resistant Staph. aureus (ORSA). Staph. aureus susceptibility to these compounds is nearly identical, and oxacillin is widely used as a surrogate for methicillin for in vitro testing.23 A wide variety of chemical classes of antistaphylococcal plant natural products have been reported to date, and, of these, prenylated and geranylated flavanones have been found to be particularly active.24 Another area where antibacterial plant natural products may be of use is for prevention of dental caries. It is widely accepted that S. mutans is one of three species of endogenous oral bacteria that are strongly correlated with the formation of caries.25,26 This occurs through the production of acids by bacterial biofilms that gradually dissolve tooth enamel. Isoflavones of Glycyrrhiza uralensis were recently reported to have moderate growth-inhibitory properties against S. mutans.16 Determination of the mechanism of action of flavonoids toward bacteria, which certainly differs from that of β-lactams, is an area of high interest.27 The discovery of new compounds that exhibit good activity and diverse structure–activity relationships will likely contribute to these efforts.
Larvae of S. frugiperda are a major agricultural pest of crops such as peanut, corn, and most grains and cause annual losses in the hundreds of millions of dollars in the United States.28−30 Damage to corn by this pest is of economic and medical significance, as it can lead to reduced overall yields and plant fungal infections that may be associated with mycotoxin production and food contamination.31 Control of S. frugiperda using commercial pesticides is difficult, and there is growing concern over unintended harm that these pesticides may cause to the environment and human health. The effects of neonicotinoid pesticides such as imidacloprid have been widely publicized and appear closely associated with massive declines in honeybee populations.32,33 Agents used for control of S. frugiperda include pyrethroids such as cyfluthrin and permethrin, which are known to be toxic to beneficial insects, including bees, and many other organisms.34 Malathion has been recommended for control of S. frugiperda, and recent reports associate this organophosphate pesticide with amphibian declines and ADHD in humans.35,36 In addition, valid concern over the development of resistance to certain pesticides by S. frugiperda(37) adds to renewed interest in alternative or supplemental control strategies. One such approach may involve the identification and use of plant metabolites that possess insecticidal properties but pose minimal risks to the environment and human health.
Herein, the structures and associated biological activities of a series of prenylated and geranylated isoflavanones, rotenoids, and other flavonoids are reported, and early observations of their apparent distribution in root and aerial portions of D. searlsiae are discussed.
Results and Discussion
Metabolite Isolation and Identification
Metabolites were isolated from roots and aerial parts of the plant using similar isolation methods. Fractionation of extracts of D. searlsiae by silica gel VLC, Sephadex LH-20, and gradient chromatography over silica gel afforded the four new flavanones malheurans A–D (1–4) and two known prenylated flavanones, 5 and prostratol F (6), from the roots. Aerial parts of the plant yielded the known pterocarpans tephrosin (7) and milletosin (8) and the known isoflavonoids griffonianone E (9) and calopogonium isoflavone A (10). The structures of all 10 compounds were determined by NMR and HRESIMS, and the absolute configurations of compounds 1–5, 7, and 8 were determined by electronic circular dichroism (ECD). Compounds 2 and 3 were analogous to two known metabolites of D. versicolor(10) and D. scandens var. paucifolia(7) that differed only in the presence of hydroxy groups at C-5.

The HRESIMS, 13C NMR (Table 1), and DEPT data for compound 1 indicated a molecular formula of C25H28O5. A pattern characteristic of a flavanone core structure was indicated by HSQC correlations between oxymethine (δH 5.70; Table 1) and methylene (δH 2.94 and 2.72) proton resonances of the C-ring and carbons at δC 76.0 and 44.0. The 13C and DEPT data indicated the presence of 10 nonprotonated carbons, including a ketocarbonyl at δC 191.7 and four oxygenated sp2 carbons between δC 156 and 163. Three hydroxy protons and the multiplicities of the five observed aromatic protons permitted the placement of two substituents each on the flavanone A- and B-rings, while the H-5 o-coupled (8.6 Hz) doublet indicated a 7,8-disubstituted flavanone A-ring. The molecular formula supported the presence of a geranyl group as the fourth aromatic substituent, and its placement at C-8 was confirmed through HMBC correlations from H2-1″ to C-7, C-8, and C-9 and from H-2″ to C-8. The C-2″ double bond was assigned an E-configuration, as the C-3″ methyl resonated at δC 16.4 instead of at δC ∼23, as would be expected for a Z-configuration. The 1H NMR data indicated a B-ring with an ABX spin system, and HMBC correlations from H-6′ to C-2, C-2′, and C-4′ and from H-2 to C-1′, C-2′, and C-6′ verified the substitution pattern of the B-ring and its connectivity to the C-ring. The overall structural connectivity was established by HSQC and HMBC spectroscopic data (Figures S10 and S11, Supporting Information) and by comparison with known compounds.11,38 Thus, in conjunction with ECD data (vide infra), the structure of 1 was assigned as the new compound (2S,E)-8-(3,7-dimethylocta-2,6-dien-1-yl)-7,2′,4′-trihydroxyflavanone and named malheuran A.
Table 1. NMR Data for Geranylated Flavanones 1, 4, and 6 (Acetone-d6).
|
1 |
4 |
6 |
||||
|---|---|---|---|---|---|---|
| position | δCa | δHb (mult.; JHH) | δCa | δHb (mult.; JHH) | δCa | δHb (mult.; JHH) |
| 2 | 76.0 | 5.70 (dd; 13.1, 2.8) | 76.3 | 5.69 (dd; 13.2, 2.7) | 80.5 | 5.44 (dd; 12.9, 2.8) |
| 3a | 44.0 | 2.94 (dd; 16.7, 13.1) | 44.1 | 2.98 (dd; 16.7, 13.2) | 44.7 | 3.00 (dd; 16.7, 12.9) |
| 3b | 2.72 (dd; 16.7, 2.8) | 2.73 (dd; 16.7, 2.7) | 2.70 (dd; 16.7, 2.8) | |||
| 4 | 191.7 | 191.8 | 191.1 | |||
| 5 | 126.4 | 7.60 (d; 8.6) | 126.4 | 7.61 (d; 8.6) | 131.7 | 7.59 (d; 8.6) |
| 6 | 110.5 | 6.63 (d; 8.6) | 110.5c | 6.63 (d; 8.6) | 110.6 | 6.63 (d; 8.6) |
| 7 | 162.6 | 162.3 | 162.4 | |||
| 7-OH | 9.30 (s) | 9.30 (s) | not obsd | |||
| 8 | 116.7 | 116.7 | 115.5 | |||
| 9 | 162.3 | 162.6 | 162.1 | |||
| 10 | 115.5 | 115.5 | 116.7 | |||
| 1′ | 118.5 | 117.5 | 126.4 | |||
| 2′ | 156.2 | 154.2 | 128.9 | 7.42 (d; 8.5) | ||
| 2′-OH | 8.63 (s) | 8.43 (s) | ||||
| 3′ | 103.5 | 6.47 (d; 2.2) | 104.7 | 6.48 (s) | 116.2 | 6.90 (d; 8.5) |
| 4′ | 159.4 | 157.0 | 158.6 | |||
| 4′-OH | 8.38 (s) | 8.11 (s) | not obsd | |||
| 5′ | 107.9 | 6.44 (dd; 8.3, 2.2) | 126.3 | 116.2 | 6.90 (d; 8.5) | |
| 6′ | 128.7 | 7.38 (d; 8.3) | 126.6 | 7.44 (s) | 128.9 | 7.42 (d; 8.5) |
| 1a″ | 22.8 | 3.37 (m) | 22.9 | 3.42 (dd, 13.7, 7.6) | 22.8 | 3.35 (d; 7.1) |
| 1b″ | 3.33 (dd, 13.7, 7.0) | |||||
| 2″ | 123.2 | 5.28 (br t; 6.9) | 123.2 | 5.32 (br t; 7.2) | 123.2 | 5.26 (m) |
| 3″ | 135.5 | 135.5 | 135.5 | |||
| 3″-Me | 16.4 | 1.67 (s) | 16.4 | 1.67 (s) | 16.4 | 1.64 (s) |
| 4″ | 40.6 | 1.93 (m) | 40.7 | 1.93 (m) | 40.6 | 1.93 (m) |
| 5″ | 27.5 | 2.03 (m) | 27.5 | 2.02 (m) | 27.5 | 2.00 (m) |
| 6″ | 125.3 | 5.06 (m) | 125.2 | 5.05 (m) | 125.2 | 5.07 (m) |
| 7″ | 131.7 | 131.6 | 131.7 | |||
| 7″-Me | 17.8 | 1.53 (s) | 17.8 | 1.53 (s) | 17.8 | 1.55 (s) |
| 8″ | 25.9 | 1.59 (s) | 25.9 | 1.58 (s) | 25.9 | 1.60 (s) |
| 2‴ | 40.9 | |||||
| 3‴ | 149.1 | 6.29 (dd; 17.3, 10.6) | ||||
| 4‴ | 110.5c | 5.01 (dd; 17.3, 1.3) | ||||
| 4.95 (dd; 10.6, 1.3) | ||||||
| 1‴ | 27.7 | 1.48 (s) | ||||
| 2‴-Me | ||||||
75 MHz.
400 MHz.
Assignments may be interchanged.
The molecular formula, C25H28O5, of compound 2 was established via its 13C NMR and HRESIMS data. As with 1, HSQC data (Table 2) confirmed a flavanone core structure, with oxymethine (δH 5.71) and methylene (δH 3.00 and 2.74) proton resonances correlating to carbons at δC 76.3 (C-2) and 44.0 (C-3), respectively. Three hydroxy protons and the multiplicities of the four aromatic protons indicated di- and trisubstituted A- and B-rings, respectively. Similar to 1, the A-ring of 2 contained o-coupled (8.6 Hz) doublets for H-5 and H-6. The structure and placement of the C-8 prenyl group and the overall substitution pattern of the A-ring were readily deduced from HMBC correlations. The NMR assignments of H2-3 were based on coupling constants relative to H-2. H-3a and H-3b exhibited HMBC correlations to C-2, C-10, and C-1′ similar to those observed for compound 1. The B-ring protons of 2 resonated as singlets, assignable to p-oriented H-3′ (δH 6.49) and H-6′ (δH 7.44). HMBC correlations from H-6′ to C-2 and to the quaternary C-2‴ (δC 40.8) verified the regiochemistry of this portion of the B-ring and the nature of the C-5′ prenyl group. All three protons characteristic of the vinylic portion of this group (δH 6.29, 5.01, and 4.95) correlated to C-2‴ in the HMBC spectrum. The C-3‴ proton correlated to C-5′, establishing the location of the prenyl group at this carbon. The remaining regiochemistry of the B-ring was assigned via 1D- and 2D-NMR methods (Figures S12–S15, Supporting Information). Following ECD analysis (vide infra), the structure of the new compound 2 was assigned as (2S)-5′-(2-methylbut-3-en-2-yl)-8-(3-methylbut-2-en-1-yl)-7,2′,4′-trihydroxyflavanone and named malheuran B.
Table 2. NMR Data for Prenylated Flavanones 2, 3, and 5 (Acetone-d6).
|
2 |
3 |
5 |
||||
|---|---|---|---|---|---|---|
| position | δCa | δHb (mult.; JHH) | δCa | δHb (mult.; JHH) | δCa | δHb (mult.; JHH) |
| 2 | 76.3 | 5.71 (dd; 13.2, 2.8) | 75.9 | 5.66 (dd; 13.1, 2.7) | 75.8 | 5.67 (dd; 13.0, 2.4) |
| 3a | 44.0 | 3.00 (dd: 16.4, 13.2) | 44.1 | 2.91 (dd; 16.6, 13.1) | 42.9 | 3.12 (17.1, 13.0) |
| 3b | 2.74 (dd; 16.4, 2.8) | 2.67 (dd; 16.6, 2.8) | 2.75 (dd; 17.1, 2.4) | |||
| 4 | 192.0 | 191.5 | 198.2 | |||
| 5 | 126.4 | 7.62 (d; 8.6) | 126.4c | 7.60 (d; 8.6) | 163.1 | |
| 5-OH | 12.52 (s) | |||||
| 6 | 110.4c | 6.63 (d; 8.6) | 110.5 | 6.62 (d; 8.6) | 96.3 | 6.03 (s) |
| 7 | 162.3 | 162.2 | 164.8 | |||
| 7-OH | 9.28 (s) | 9.25 (s) | 9.54 (br s) | |||
| 8 | 116.6 | 116.6 | 108.3 | |||
| 9 | 162.6 | 162.5 | 161.7 | |||
| 10 | 115.4 | 115.5 | 103.4 | |||
| 1′ | 117.5 | 119.1 | 117.1 | |||
| 2′ | 154.1 | 156.6 | 154.3 | |||
| 2′-OH | 8.44 (br s) | 8.44 (s) | ||||
| 2′-OCHH3 | 55.9 | 3.79 (s) | ||||
| 3′ | 104.7 | 6.49 (s) | 101.0 | 6.55 (s) | 104.8 | 6.48 (s) |
| 4′ | 157.0 | 157.3 | 157.2 | |||
| 4′-OH | 8.10 (br s) | 8.19 (s) | 8.12 (s) | |||
| 5′ | 126.3 | 126.6 | 126.4 | |||
| 6′ | 126.6 | 7.44 (s) | 126.5c | 7.50 (s) | 126.7 | 7.40 (s) |
| 1a″ | 22.9 | 3.37 (dd; 13.8, 7.6) | 22.9 | 3.40 (m) | 22.4 | 3.24 (m) |
| 1b″ | 3.28 (dd; 13.8, 7.0) | 3.32 (m) | ||||
| 2″ | 123.2 | 5.30 (m) | 123.3 | 5.28 (m) | 123.8 | 5.25 (br t; 7.0) |
| 3″ | 131.7 | 131.7 | 131.3 | |||
| 3″-Me | 18.1 | 1.65 (s) | 18.1 | 1.64 (s) | 18.0 | 1.62 (s) |
| 4″ | 26.0 | 1.62 (s) | 26.0 | 1.62 (s) | 26.0 | 1.61 (s) |
| 2‴ | 40.8 | 40.9 | 40.9 | |||
| 3‴ | 149.1 | 6.29 (dd; 17.9, 10.7) | 149.0 | 6.29 (dd; 17.6, 10.7) | 149.1 | 6.28 (dd; 17.6, 11.0) |
| 4‴ | 110.5c | 5.01 (d; 17.9) | 110.7 | 5.02 (d; 17.5) | 110.5 | 5.00 (d; 17.6) |
| 4.95 (d; 10.7) | 4.97 (d; 10.7) | 4.95 (d; 11.0) | ||||
| 1‴ | 27.7 | 1.47 (s) | 27.6 | 1.49 (s) | 27.7 | 1.47 (s) |
| 2‴-Me | ||||||
75 MHz.
400 MHz.
Assignments may be interchanged.
The HRESIMS and 13C NMR data of compound 3 indicated a molecular formula of C26H30O5. Spectroscopic data for 3 (Table 2; Figures S16–S19, Supporting Information) were similar to those of 2 with the additional feature of an O-methyl group (δH 3.79, δC 55.9), consistent with the difference in the molecular formula as compared to 2. The placement of the O-methyl and two prenyl groups of 3 was verified by HMBC correlations from O-methyl, methylene, and methine protons to neighboring A- and B-ring carbons, respectively. Remaining structural assignments were made based on analysis of HSQC and HMBC correlations, comparison to the spectroscopic data of 2, and ECD analysis (vide infra). The structure of the new compound was therefore assigned as (2S)-7,4′-dihydroxy-2′-methoxy-5′-(2-methylbut-3-en-2-yl)-8-(3-methylbut-2-en-1-yl)flavanone and named malheuran C (3).
The 1H NMR data for 4 resembled those of 1, except for a six-proton singlet (δH 1.48) and deshielded resonances (δH 6.29, 5.01, and 4.95) with multiplicities and coupling constants (Table 1) characteristic of a vinyl group. The HRESIMS and 13C NMR data of 4 revealed a molecular formula of C30H36O5, consistent with the presence of an additional prenyl group as compared to 1. Two aromatic protons resonating as singlets indicated a p-disubstituted B-ring. The remaining structural assignments were made based on HSQC and HMBC data (Figures S22 and S23, Supporting Information), comparison with 1, and ECD analysis (vide infra). The structure of the new compound was therefore assigned as (2S,E)-8-(3,7-dimethylocta-2,6-dien-1-yl)-5′-(2-methylbut-3-en-2-yl)-7,2′,4′-trihydroxyflavanone and named malheuran D (4).
Spectroscopic data for compound 5 resembled those of 2 with the addition of a C-5 hydrogen-bonded hydroxy proton, as indicated by a 1H NMR resonance at δH 12.52. Comparison of the NMR and ECD data (Figures S24–S27, Supporting Information) with those of 2 and confirmation of the molecular formula C25H28O6 by HRESIMS and 13C NMR data further indicated that 5 is the 5-oxy analogue of compound 2. Compound 5 was therefore identified as the known metabolite (2S)-5′-(2-methylbut-3-en-2-yl)-8-(3-methylbut-2-en-1-yl)-5,7,2′,4′-tetrahydroxyflavanone.7
The absolute configurations for compounds 1–5 were assigned using electronic circular dichroism and NMR data. All five compounds produced similar ECD spectra (Figures S1–S5, Supporting Information) containing high-amplitude positive Cotton effects (CEs) between 320 and 360 nm and high-amplitude negative CEs between 280 and 320 nm. These CEs correspond to the respective n → π* and π → π* electronic transitions of the acetophenone chromophore39,40 and allowed for the assignment of the 2S-configuration for all five compounds. All compounds also gave high-amplitude positive CEs near 240 nm, corresponding to the 1La electronic transition of the aromatic chromophores, consistent with 2S-configuration. This configuration was supported by NMR data, which indicated that the aryl substituent was in the thermodynamically preferred equatorial position. Accordingly, the vicinal JHH coupling constants of ∼13 and ∼2.8 Hz for H-2 (δH ∼5.70) in each compound indicated that this proton was in the axial position, with axial–axial coupling at ∼13 Hz to H-3a (δH 2.9–3.1) and axial–equatorial coupling at ∼2.8 Hz to H-3b (δH 2.67–2.75).40,41 Additionally, in the HMBC spectrum (optimized for 2–3JCH = 8 Hz), H-3a showed correlations to C-2 and C-1′ but not to C-10, while the converse was true of H-3b, which correlates to C-10 but not to C-2 or C-1′. The thermodynamically preferred conformation for the flavanone C-ring leads to P-helicity and a corresponding 2S-configuration based on the modified octant rule applied to aryl ketones as proposed by Snatzke in 1965.42
The 1H and 13C NMR spectra of 6 showed similarities to 1 and 4, with methyl groups at δH 1.55, 1.60, and 1.64 and the characteristic vinylic proton resonances at δH 5.07 and 5.26 indicative of a geranyl substituent. An AA′BB′ spin system at δH 7.42 and 6.90 (J = 8.5 Hz) indicated a p-substituted B-ring. Comparison of the MS and NMR data (Figures S29–S32, Supporting Information) of compound 6 with those of the known compound prostratrol F confirmed their identical gross structures and relative configuration.41 The specific rotation of 6 ([α]20D −40) closely matched the reported value ([α]20D −42) of prostratol F,41 hence establishing the 2S absolute configuration.41 Compound 6 was therefore assigned as the known compound prostratol F, (2S,E)-7,4′-dihydroxy-8-(3,7-dimethylocta-2,6-dien-1-yl)flavanone.
The structures of the known compounds 7–10 were determined by comparison of their MS, NMR, and ECD data (Figures S6, S7, and S33–S48, Supporting Information) with reported values.43−49
Antibacterial Assays
Initial screening in simple disk-diffusion assays (100 μg/disk) revealed zones of inhibition of 11–14 mm for the root extract of D. searlsiae against the tested Gram-positive bacteria, while extracts of the aerial portions of the plant were inactive.
The results of antibacterial broth microdilution assays using the isolated root compounds 1–6 are shown in Table 3. All compounds had moderately potent minimum inhibitory concentration (MIC) values of 2.0–5.3 μg/mL against the cariogenic bacterium S. mutans. These results are consistent with those of previous studies on the antibacterial properties of flavonoids of Glycyrrhiza(16) and suggest potential applications of these metabolites for oral health.
Table 3. Minimum Inhibitory Concentrations (MIC)a and Minimum Bactericidal Concentrations (MBC)a of Compounds 1–6 and Oxacillin (μg/mL) against Gram-Positive Bacteria.
|
Staph. aureus OSSAb |
Staph. aureus ORSAc |
S. mutansd |
B. cereuse |
|||||
|---|---|---|---|---|---|---|---|---|
| compound | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC |
| 1 | 4.3 | 5.3 | 3.7 | 6.0 | 3.3 | 6.0 | 3.7 | 6.5 |
| 2 | 3.4 | 4.3 | 3.7 | 4.7 | 3.0 | 7.3 | 2.7 | 4.7 |
| 3 | 4.6 | 6.0 | 4.3 | 5.7 | 3.7 | 6.3 | 3.0 | 5.7 |
| 4 | 6.1 | 6.7 | 6.5 | 6.7 | 2.0 | 2.7 | 5.0 | 7.3 |
| 5 | 3.1 | 4.3 | 3.4 | 4.7 | 3.0 | 9.0 | 2.3 | 3.3 |
| 6 | 6.8 | 9.0 | 6.4 | 14.0 | 5.3 | 7.3 | 8.0 | 15.0 |
| oxacillin | 0.4 | NAf | 21.3 | NA | 0.5 | NA | 106.7 | NA |
Standard errors for all MICs and MBCs were in the range 0–1.7 except those for MICs of oxacillin against ORSA (5.3) and B. cereus (21.3).
Oxacillin-sensitive Staph. aureus; ATCC 25923.
Oxacillin-resistant Staph. aureus; ATCC 43300.
ATCC 25175.
Wild-type.
Not applicable; oxacillin is bacteriostatic.
All compounds also had MICs of 2.3–8.0 μg/mL against the endemic soil bacterium Bacillus cereus.(19) This supports an ecological view that D. searlsiae may allocate antibacterial resources toward Gram-positive organisms in the root environment. B. cereus is also an opportunistic pathogen, associated with food-borne illness, and is a close relative of B. anthracis, the causative agent of anthrax.19
Compounds 1–6 exhibited antistaphylococcal MIC values of 3.1–6.8 μg/mL, a level of activity (<10 μg/mL) that qualifies as “very interesting” as recently defined by Cushnie and Lamb.50 MICs of 1–6 ranged from 3.1 to 6.8 μg/mL against oxacillin-sensitive Staph. aureus (OSSA) and from 3.4 to 6.5 μg/mL against the resistant strain (ORSA). In contrast, the positive control, oxacillin, was effective against OSSA (0.4 μg/mL) and S. mutans (0.5 μg/mL), but had the expected low activity against ORSA (21.3 μg/mL). The similarity in MICs for 1–6 against both OSSA and ORSA suggests that the antibacterial mechanism of action for these compounds is different than that of oxacillin and related β-lactams.
Previous structure–activity analyses of similar flavonoids associated increased antibacterial activity with oxygenation at C-5,50 although the present data reveal only a slight increase in potency of 5 compared to 1–4 and 6. It may be that an increase in activity due to the presence of lipophilic groups (e.g., geranyl or prenyl), also concluded previously,50 is an overriding factor for the activity of this suite of compounds. The confirmatory measurements of minimum bactericidal concentrations (Table 3), defined as the lowest concentration that results in 99.9% reduction of bacterial density, were only slightly higher than the MICs of all compounds tested, suggesting that these compounds work via a bactericidal mechanism of action. Coupled with their differing mechanism of action from β-lactam agents, the levels of bactericidal activity of 1–6 suggest a potential clinical use against resistant strains of Staph. aureus and various other Gram-positive bacteria. These results also support previous reports of flavonoid activities against β-lactam-resistant Gram-positive bacteria.51
Antiinsectan Assays
The presence of rotenoids 7 and 8 prompted our investigation of the potential antiinsectan properties of D. searlsiae using the generalist herbivore Spodoptera frugiperda (fall armyworm) to evaluate antifeedant and possible larvicidal properties of extracts and isolated flavonoids. Prior reports have demonstrated antifeedant activity of flavonoids toward insects,52 and other natural products have been shown to have such activity specifically toward S. frugiperda.53,54 Initial testing of crude methanolic extracts of the roots and aerial parts of the plant was done at concentrations constituting 0.8% (8 μg/mg disk) and 1.4% (14 μg/mg disk) of a pinto bean diet, respectively. These concentrations were estimated from isolated amounts of the compounds from the original plant material and by taking into account the approximate water content of plants57 that would be encountered by S. frugiperda. After 4 days, the root extract caused a reduction in growth of S. frugiperda larvae of 50–75% (as indicated by comparing weights of survivors), while the extract of the aerial portions caused 75% reduction in growth. These results demonstrate that antiinsectan compounds may be distributed throughout the plant, with slightly higher concentrations in aerial portions, and support the premise that secondary metabolites defend against insect larvae feeding on both roots and shoots.
Further assessment of compounds 1–10 using pinto bean diet disks containing 1% (10 μg/mg disk; 10 000 ppm) of the test compounds was performed, and the reductions in growth and associated mortality relative to controls are shown in Table 4. In all cases, larvae were observed feeding on diet treatments, rather than on agar, as would be seen for antifeedant activity, so effects were likely due primarily to toxicity. As expected, rotenoids 7 and 8 were both highly active, exhibiting greater than 90% reduction in growth, with associated mortality. The flavanone malheuran D (4) was also active, causing 90% growth reduction, with 37% mortality. Notable reductions in growth were observed for all 10 compounds at this test concentration, which was selected for comparisons of relative activity. The three most active compounds were then assayed further at lower concentrations, ranging from 0.0023 to 0.08% (23–831 ppm), approximating those that might be typically encountered by herbivorous Lepidoptera larvae. Growth reduction of >50% relative to controls remained significant (p < 0.0001) for both tephrosin 7 and rotenone (Table 4), while 4 and 8 were less active at the lower test concentrations. The level of effectiveness observed at these concentrations suggests a potential ecological role in plant defense against insect herbivores. The mechanism of action of rotenone toward insects is via inhibition of cellular respiration, specifically within the mitochondrial complex I electron transport system.55 The mechanism of action of nonrotenoid phenolic compounds (e.g., compound 4) toward insects is, for the most part, undetermined.55
Table 4. Activitya of Compounds 1–10 against S. frugiperda First Instar Larvae.
| compound | % reduction in growthb relative to control | % mortalityc | % reduction in growth at lower “natural” concentrationsd relative to control |
|---|---|---|---|
| 1 | 86 | 0 | |
| 2 | 86 | 0 | |
| 3 | 75 | 6 | |
| 4 | 90 | 37 | 6 |
| 5 | 88 | 0 | |
| 6 | 50 | 0 | |
| 7 | 94 | 66 | 56 |
| 8 | 91 | 33 | 18 |
| 9 | 81 | 0 | |
| 10 | 78 | 0 | |
| rotenone | 57 |
A pinto bean diet disk was the sole food source provided. Control disks were treated only with acetone, the solvent used for the test materials. Duration of the assay was 3 days.
Experimental diet disks containing 1% compound (10 000 ppm, or 10 μg/mg disk).
Where mortality occurs, reduction in growth and mortality are typically decreased (but not measured) due to cannibalism of dead insects by survivors.
Test concentrations: 4 = 0.08% (831 ppm), 7 = 0.016% (163 ppm), 8 = 0.0023% (23 ppm), rotenone = 0.016% (163 ppm). No mortality was observed at these concentrations.
Overall, for crude extracts and isolated compounds of D. searlsiae, the apparent larvacidal activities toward S. frugiperda appear to be greater in the aerial portions of the plant, a phenomenon partially attributable to the presence of rotenoids 7 and 8. Conversely, Gram-positive antibacterial metabolites appear to be selectively allocated to root portions of the plant, potentially for defense against soil pathogens.
Experimental Section
General Experimental Procedures
Melting points were measured on a Barnstead International Mel-Temp apparatus, model 12010. Optical rotations were recorded on a PerkinElmer 341 polarimeter (Na lamp, 589 nm); concentrations were reported in g/100 mL. UV spectra were recorded on an HP-Agilent 8453 photodiode array instrument. ECD spectra were obtained on a JASCO J-815 circular dichroism spectrometer using high-purity MeOH as the solvent and a 1 cm path length quartz cuvette. ECD spectra were obtained for each compound at concentrations of 0.01, 0.02, 0.04, 0.06, 0.08, and 1.0 mM, and raw data were smoothed using a binomial smoothing algorithm with 10 passes as provided with the JASCO Spectra Manager Ver. 1.54 software. IR spectra were recorded on a Nicolet Protégé 460 spectrometer. All NMR spectra were obtained on a Bruker Avance 400 MHz system with Topspin 1.3 software. HRESIMS were run on a Waters Q-TOF Premier hybrid mass spectrometer. A Waters Acquity UPLC was used to inject samples in 1:1 MeCN–H2O using flow injection analysis (100 μL/min) with no column. Positive or negative ESIMS was used to generate [M + H]+, [M + Na]+, or [M – H]− ions. TLC plates (EMD Chemicals, Inc.; silica gel 60, F254) were eluted with 9:1 CH2Cl2–MeOH or mixtures of hexanes–EtOAc and visualized with UV (254 nm) and the spray reagent vanillin/H2SO4 (1 g/100 mL w/v) after gentle heating.
Plant Material
Four whole plants of D. searlsiae were received from Dr. Clinton Shock in July 2011. The plants were grown from seed at the Oregon State University, Malheur Experiment Station, Ontario, OR.5 Plant materials were air-dried for approximately 72 h from the time of collection. Roots were washed with water to remove dirt and debris, aerial portions were separated, and material was stored at −20 °C until further use. A voucher specimen was authenticated by Dr. Tom Cottrell, Department of Biological Sciences, Central Washington University, and was deposited in the herbarium of the same department.
Extraction and Isolation
Whole D. searlsiae roots (44 g) were shaved into 3–6 cm slices, soaked for 48 h in MeOH at room temperature, and blended in a Waring blender for 2–3 min with 700 mL of MeOH. The mixture was filtered and the filtrate evaporated under reduced pressure to afford 3.7 g of crude extract. This material was preadsorbed in MeOH solution onto ∼10 g of silica gel, the solvent removed under vacuum, and the resulting powder subjected to VLC over a prepacked column bed (10 × 6 cm; i.d. × h) of TLC-grade (230–400 mesh) silica gel. The column was eluted using a stepwise gradient of solvents beginning with hexanes (1 L) and continuing with mixtures (500 mL each) of EtOAc in hexanes (20, 40, 60, 80, and 100% EtOAc), followed by mixtures of MeOH in CH2Cl2 (2, 5, 8, 10, and 30% MeOH). Fractions 3 and 4 from this column were preadsorbed (∼3 g of silica gel; MeOH), dried, and rechromatographed on a smaller scale (3.5 × 7 cm; i.d. × h), eluting with 300 mL of 100% hexanes, followed by 300 mL each of 10, 20, 30, 35, 40, 75, and 100% EtOAc in hexanes. Fractions 4–7 of this column were combined with fractions 5 and 6 from the first VLC column to afford an enriched flavonoid fraction (1.67 g).
The enriched flavonoid fraction was further purified by Sephadex LH-20 (Sigma) CC (2.5 × 60 cm) eluting with 1 L of 3:1:1 hexanes–toluene–MeOH, followed by 1 L of 100% MeOH at a flow rate of 0.3–0.5 mL/min with ∼8 mL per fraction tube. Materials of similar composition as determined by TLC were pooled to give 46 fractions. Fractions 27 and 28 were combined (76 mg) and further purified over silica gel (2.5 × 8 cm; 60–100 mesh, ∼20 mL/min) using a continuous linear gradient of EtOAc (0–40%) in hexanes. Fractions eluting with 36–40% EtOAc afforded compound 1 (malheuran A; 18 mg), and fractions eluting with 30–34% EtOAc afforded (2S)-5′-(2-methylbut-3-en-2-yl)-8-(3-methylbut-2-en-1-yl)-5,7,2′,4′-tetrahydroxyflavanone (5; 13 mg). Fraction 24 (245 mg) from the Sephadex LH-20 column was further purified in a similar manner to that used for 27 and 28, using a continuous linear gradient of EtOAc in hexanes (0–50%; 2.5 × 11 cm column). Fractions eluting with 31–35% EtOAc yielded compound 2 (malheuran B; 129 mg). Fraction 6 (36 mg) from the Sephadex LH-20 column was further purified over silica gel in two successive stages using gradients of 0–30% EtOAc in hexanes (1.5 × 8 cm column), followed by 0–4% MeOH in CH2Cl2 (1.5 × 7 cm column) to afford compound 3 (malheuran C; 17 mg). Fractions 8 and 9 from the Sephadex LH-20 column were combined (153 mg) and further purified over silica gel in two successive stages using gradients of 0–5% MeOH in CH2Cl2 (2.5 × 10 cm column), followed by 0–25% EtOAc in hexanes (1.5 × 7 cm column) to yield prostratrol F (6; 23 mg). Fraction 10 (96 mg) from the Sephadex LH-20 column was purified over silica gel using 0–5% MeOH in CH2Cl2 (2.5 × 10 cm column) to yield compound 4 (malheuran D; 69 mg).
Aerial portions of D. searlsiae (leaves, stems, and flowers; 196 g) were blended in a Waring blender for 2–3 min with 1.2 L of MeOH. The mixture was filtered, and the filtrate was evaporated under reduced pressure to afford 27.4 g of crude extract. This material was fractionated by VLC over silica gel (10 × 6 cm; i.d. × h), using methods and eluting solvents similar to those used for VLC of the crude root extract. Material from fractions 3 and 4, eluted with 40–60% EtOAc in hexanes from this VLC column, were combined (1.5 g) and fractionated by Sephadex LH-20 chromatography as described above to yield 37 fractions following combinations based on TLC analyses. Fraction 16 from this column yielded (−)-tephrosin (7; 40 mg). Fractions 18–21 from the Sephadex LH-20 column were combined (54 mg) and further purified over silica gel (60–100 mesh) in successive stages with step gradients of MeOH in CH2Cl2 (0–1%; 1.5 × 13 cm column) and 100% CH2Cl2 (1.5 × 5 cm column) to yield (−)-milletosin (8; 8 mg). Fractions 10 and 11 from the Sephadex LH-20 column were combined (72 mg) and further purified over silica gel with continuous linear gradient conditions and solvents identical to those used for 1 above, to afford griffonianone E (9; 18 mg) and calopogonium isoflavone A (10; 12 mg).
Malheuran A (1):
off-white, amorphous solid; [α]20D −66 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) 203 (4.64), 220 (sh) (4.42), 236 (sh) (4.22), 286 (4.14), 315 (sh) (3.79) nm; ECD (c 0.0016, MeOH) λ (θ) 204 (−4.70 × 104), 223 (4.15 × 104), 247 (−3.77 × 103), 303 (−2.54 × 104), 334 (1.34 × 104) nm; IR (film on KBr) νmax 3342 (br OH), 2966, 2920, 1655, 1585, 1520, 1440, 1284, 1167, 1114, 974 cm–1; 1H and 13C NMR data, see Table 1; HMBC correlations (acetone-d6) H-2 → C-4, 1′, 2′, 6′; H-3a→ C-2, 4, 1′; H-3b → C-4, 10; H-5 → C-4, 7, 8*; H-6 → C-7, 10; OH-7 → C-6, 7, 8; OH-2′ → C-1′, 2′, 3′; H-3′ → C-1′, 2′, 4′, 5′; OH-4′ → C-3′, 4′, 5′; H-5′ → C-1′, 3′; H-6′ → C-2, 2′, 3′*, 4′; H2-1″ → C-7, 8, 9, 2″, 3″; H-2″ → C-8, 1″, CH3-3″, 4″; CH3-3″→ C-2″, 3″, 4″; H2-4″ → C-2″, 3″, CH3-3″, 5″; H2-5″ → C-3″, 4″, 6″, 7″; H-6″ → C-4″, 8″, CH3-7″; CH3-7″→ C-6″, 7″, 8″; H3-8″ → C-6″, 7″, CH3-7″ (* indicates weak four-bond correlation); HRESIMS found m/z 407.1863 [M – H]−, calcd for C25H27O5 407.1859.
Malheuran B (2):
yellow, amorphous solid; [α]20D −90 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) 203 (4.65), 220 (sh) (4.43), 235 (sh) (4.24), 287 (4.16), 314 (sh) (3.10) nm; ECD (c 0.0016, MeOH) λ (θ) 208 (−3.66 × 104), 228 (2.01 × 104), 250 (−1.91 × 103), 305 (−2.41 × 104), 335 (1.19 × 104) nm; IR (film on KBr) νmax 3405 (br OH), 2967, 2916, 1655, 1586, 1504, 1437, 1384, 1287, 1213 cm–1; 1H and 13C NMR data, see Table 2; HMBC correlations (acetone-d6) H-2 → C-3, 4, 1′, 2′, 6′; H-3a → C-2, 4, 1′; H-3b → C-4, 10; H-5 → C-4, 7, 8*, 9; H-6 → C-7, 8, 10; H-3′ → C-2*,1′, 2′, 4′, 5′, 2‴*; H-6′ → C-2, 1′, 2′, 3′*, 4′, 2‴; H2-1″ → C-7, 8, 9, 2″, 3″; H-2″ → C-8, 1″, CH3-3″, 4″; CH3-3″ → C-2″, 3″, 4″; H3-4″ → C-2″, 3″, CH3-3″; H3-1‴ → C-5′, 2‴, CH3-2‴, 3‴; CH3-2‴ → C-5′, 1‴, 2‴, 3‴; H-3‴ → C-5′, 1‴, 2‴, CH3-2‴; H2-4‴ → C-5′*, 1‴*, 2‴, CH3-2‴ (*indicates weak four-bond correlation); HRESIMS found m/z 407.1865 [M – H]−, calcd for C25H27O5 407.1858.
Malheuran C (3):
yellow oil; [α]20D −100 (c 0.03, CHCl3); UV (MeOH) λmax (log ε) 204 (4.66), 222 (sh) (4.40), 235 (sh) (4.26), 286 (4.14), 316 (sh) (3.71) nm; ECD (c 0.0017, MeOH) λ (θ) 209 (−4.96 × 104), 230 (1.94 × 104), 246 (−2.66 × 103), 303 (−2.65 × 104), 334 (1.20 × 104) nm; IR (film on KBr) νmax 3467 (br OH), 2965, 2925, 1653, 1586, 1506, 1443, 1383, 1286, 1207 cm–1; 1H and 13C NMR data, see Table 2; HMBC correlations (acetone-d6) H-2 → C-3, 4, 9, 1′, 2′, 6′; H-3α → C-2, 4, 1′; H-3β → C-4, 10; H-5 → C-4, 8*, 9; H-6 → C-7, 8, 10; OH-7 → C-6, 7, 8; OCH3-2′ → C-2′, 3′*; H-3′ → C-2*,1′, 2′, 4′, 5′, 2‴*; OH-4′ → C-3′, 4′, 5′; H-6′ → C-2, 1′, 2′, 3′*, 4′, 2‴; H2-1″ → C-7, 8, 2″, 3″, CH3-3″*; H-2″ → C-8, 1″, 4″, CH3-3″; H3-4″ → C-2″, 3″, CH3-3″; CH3-3″ → C-2″, 3″, 4″; H-1‴ → C-5′, 2‴, CH3-2‴, 3‴; CH3-2‴ → C-5′, 1‴, 2‴, 3‴ (* indicates weak four-bond correlation); HRESIMS found m/z 423.2172 [M + H]+, calcd for C26H31O5 423.2171.
Malheuran D (4):
orange oil; [α]20D −76 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) 204 (4.75), 221 (sh) (4.50), 236 (sh) (4.32), 287 (4.24), 316 (sh) (3.88) nm; ECD (c 0.0019, MeOH) λ (θ) 207 (−5.90 × 104), 227 (3.36 × 104), 248 (−3.69 × 103), 304 (−3.48 × 104), 335 (1.65 × 104) nm; IR (film on KBr) νmax 3394 (br OH), 2967, 2925, 1655, 1586, 1505, 1437, 1383, 1288, 1213 cm–1; 1H and 13C NMR data, see Table 1; HMBC correlations (acetone-d6) H-2 → C-3, 4, 9, 1′, 2′, 6′; H-3a → C-2, 4, 1′; H-3b → C-4, 10; H-5 → C-4, 6, 7, 8*; H-6 → C-7, 8, 10, 1″*; OH-7 → C-8; OH-2′ → C-1′, 2′, 3′; H-3′ → C-2*, 1′, 2′, 4′, 5′, 2‴*; OH-4′ → C-3′, 4′, 5′; H-6′ → C-2, 1′, 2′, 3′*, 4′, 2‴; H2-1″ → C-7, 8, 9, 2″, 3″; H-2″ → C-8, 1″, 4″, 5″; CH3-3″ → C-2″, 3″, 4″; H2-4″ → C-2″, 3″, 5″, 6″; H2-5″ → C-3″, 4″, 6″, 7″; H-6″ → C-7″, CH3-7″, 8″; CH3-7″ → C-6″, 7″, 8″; H3-8″ → C-6″, 7″, CH3-7″; CH3-1‴ → C-5′, 2‴, CH3-2‴, 3‴, 4‴*; CH3-2‴ → C-5′, 1‴, 2‴, 3‴, 4‴*; H-3‴ → C-5′, 1‴, 2‴, CH3-2‴; H2-4‴ → C-5′*, 1‴*, 2‴, CH3-2‴*, 3‴ (* indicates weak four-bond correlation); HRESIMS found m/z 477.2638 [M + H]+, calcd for C30H37O5 477.2641.
(2S)-5′-(2-Methylbut-3-en-2-yl)-8-(3-methylbut-2-en-1-yl)-5,7,2′,4′-tetrahydroxyflavanone (5):
orange oil; [α]20D −50 (c 0.1, CHCl3) (lit. [α]20D −79.1);7 UV (MeOH) λmax (log ε) 203 (4.68), 230 (sh) (4.29), 291 (4.21), 338 (sh) (3.54) nm; ECD (c 0.0017, MeOH) λ (θ) 210 (−2.83 × 104), 230 (1.11 × 104), 254 (648), 294 (−2.49 × 104), 335 (3.91 × 103) nm; IR (film on KBr) νmax 3412 (br OH), 2967, 2927, 1635, 1604, 1504, 1432, 1384, 1300, 1169, 1076 cm–1; 1H and 13C NMR data, see Table 2; HMBC correlations (acetone-d6) H-2 → C-4, 1′, 2′, 6′; H-3a → C-2, 4, 1′; H-3b → C-4, 10; OH-5 → C-5, 6, 10; H-6 → C-4*, 5, 7, 8, 10, 1″; OH-7 → C-8; OH-2′ → C-1′, 2′, 3′; H-3′ → C-2*,1′, 2′, 4′, 5′, 2‴*; OH-4′ → C-3′, 4′, 5′; H-6′ → C-2, 1′, 2′, 3′*, 4′, 2‴; H2-1″ → C-7, 8, 9, 2″, 3″; H-2″ → C-1″, CH3-3‴, 4″; H3-4″ → C-2″, 3″, CH3-3‴; CH3-3″ → C-2″, 3″, 4″; H3-1‴ → C-5′, 2‴, CH3-2‴, 3‴; CH3-2‴ → C-5′, 1‴, 2‴, 3‴; H-3‴ → C-5′, 1‴, 2‴, CH3-2‴; H2-4‴ → C-2‴; 3‴ (* indicates weak four-bond correlation); HRESIMS found m/z 423.1809 [M – H]−, calcd for C25H27O6 423.1808.
Prostratol F (6):
off-white solid; mp 55–64 °C; [α]20D −40 (c 0.03, CHCl3) (lit. [α]20D −42);41 UV (MeOH) λmax (log ε) 202 (4.47), 219 (sh) (4.36), 239 (sh) (4.16), 283 (4.04), 315 (sh) (3.77) nm; IR (film on KBr) νmax 3325 (br OH), 2923, 2854, 1655, 1597, 1518, 1440, 1384, 1335, 1283, 1218 cm–1; 1H and 13C NMR data (Table 1) are consistent with reported data;41 COSY correlations (acetone-d6) H-2 → H-3a, 3b; H-3a → H-2, 3b; H-3b → H-2, 3a; H-5 → H-6; H-6 → H-5; H-2′/6′ → H-3′/5′; H-3′/5′ → H-2′/6′; H2-1″ → H-2″, CH3-3″*, H2-4″*; H-2″ → H2-1″, CH3-3″*, H2-4″*; CH3-3″ → H2-1″*, H-2″*, H2-4″*; H2-4″ → H2-1″*, H-2″*, CH3-3″*, H2-5″; H2-5″ → H2-4″, H-6″, CH3-7″*, H3-8″*; H-6″ → H2-5″, CH3-7″*, H3-8″*; CH3-7″ → H2-5″*, H-6″*; H3-8″ → H2-5″*, H-6″* (* indicates long-range correlation); HRESIMS found m/z 393.2060 [M + H]+, calcd for C25H29O4 393.2066.
(−)-Tephrosin (7):
yellow, microcrystalline solid; mp 84–96 °C; [α]20D −100 (c 0.1, CHCl3) (lit. [α]20D −100);43 UV (MeOH) λmax (log ε) 203 (4.69), 237 (4.47), 251 (4.47), 271 (4.51), 300 (4.17), 319 (sh) (4.14) nm; ECD (c 0.0016, MeOH) λ (θ) 204 (2.52 × 104), 218 (1.04 × 104), 236 (−4.01 × 104), 275 (1.30 × 104), 329 (−1.14 × 104) nm; IR (film on KBr) νmax 3475 (br OH), 2980, 2930, 1674, 1638, 1598, 1578, 1511, 1443, 1331, 1272, 1112 cm–1; 1H and 13C NMR data (acetone-d6, 400 MHz) were consistent with reported values44 and further supported with HSQC and HMBC data (Figures 35 and 36, Supporting Information); HRESIMS found m/z 433.1269 [M + Na]+, calcd for C23H22O7Na 433.1263.
(−)-Milletosin (8):
off-white, microcrystalline solid; mp 81–90 °C; [α]20D −132 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) 202 (4.49), 228 (sh) (4.26), 241 (4.31), 250 (4.30), 271 (4.35), 305 (4.07) nm; ECD (c 0.0016, MeOH) λ (θ); 203 (2.33 × 104), 219 (1.64 × 104), 241 (−1.94 × 104), 275 (1.37 × 104), 300 (4.08 × 103), 329 (−2.40 × 104) nm; IR (film on KBr) νmax 3446 (br OH), 2975, 2926, 1674, 1635, 1599, 1578, 1482, 1331, 1273, 1211, 1166 cm–1; 1H and 13C NMR data (acetone-d6, 400 MHz) were consistent with reported values45 and further supported with HSQC and HMBC data (Figures 39 and 40, Supporting Information); HRESIMS found m/z 395.1136 [M + H]+, calcd for C22H19O7 395.1131.
Griffonianone E (9):
off-white, microcrystalline solid; mp 125–133 °C (lit. 130–131 °C);46 UV (MeOH) λmax (log ε) 205 (4.69), 221 (sh) (4.57), 245 (4.53), 252 (4.52), 303 (4.38) nm; IR (film on KBr) νmax 3447 (br OH), 2918, 1643, 1617, 1601, 1485, 1425, 1268, 1193 cm–1; 1H and 13C NMR data (acetone-d6, 400 MHz) were consistent with reported values46 and further supported with HSQC and HMBC data (Figures 43 and 44, Supporting Information); HRESIMS found m/z 395.1491 [M + H]+, calcd for C23H23O6 395.1495.
Calopogonium isoflavone A (10):
off-white, microcrystalline solid; mp 134–141 °C (lit. 138–141 °C);47 UV (MeOH) λmax (log ε) 202 (4.57), 216 (sh) (4.45), 241 (sh) (4.60), 261 (4.78), 311 (4.03), 321 (sh) (4.03) nm; IR (film on KBr) νmax 3447 (br OH), 3073, 2927, 1629, 1591, 1512, 1440, 1395, 1246, 1211, 1178, 1113 cm–1; 1H and 13C NMR data (acetone-d6, 400 MHz) were consistent with reported values48 and further supported with HSQC and HMBC data (Figures 47 and 48, Supporting Information); HRESIMS found m/z 335.1284 [M + H]+, calcd for C21H19O4 335.1283.
Antibacterial Susceptibility Testing
Two strains of Staphylococcus aureus, oxacillin-sensitive Staph. aureus ATCC 25923 (OSSA) and oxacillin-resistant Staph. aureus ATCC 43300 (ORSA), Bacillus cereus (environmental isolate), and Streptococcus mutans ATCC 25175 were used for these studies. Stock cultures of all bacteria were maintained at 35 °C under aerobic conditions on nutrient broth/agar, and B. cereus and S. mutans were additionally grown on brain-heart infusion broth/agar. B. cereus was isolated from the environment and identified using standard microscopy and staining techniques as a Gram-positive rod that exhibited beta-hemolysis on sheep’s blood agar. Three colonies from agar plates were transferred to broth and grown for 24 h followed by a broth-to-broth transfer using a calibrated loop (5 μL) and growing for 24 h.
Biologically active extracts and pure compounds were initially identified using disk-diffusion bioassay-guided fractionation. Paper disks (6 mm) were permeated with test materials in MeOH (100 μg/disk) and placed on Mueller-Hinton agar plates containing bacterial inoculum. Following incubation (37 °C; 3 days), the diameters (mm) of the resulting zones of inhibition were recorded.
Broth microdilution was performed according to antimicrobial susceptibility testing standards56 (NCCLS) to determine MICs of pure compounds against OSSA, ORSA, B. cereus, and S. mutans. Dilutions of compounds were made at concentrations incrementally decreasing by 1 μg/mL in the appropriate broth with 2% DMSO. Oxacillin was used as a positive control and diluted in broth using a serial 2-fold method. Double-strength test samples were added (75 μL) to sterile 96-well round-bottomed culture plates. A bacterial cell suspension corresponding to 1 × 106 cfu/mL (75 μL) was added to test wells. Control wells with only medium were used to ensure medium sterility. The final concentration of bacteria in the assay wells after dilution with broth and sample was 5 × 105 cfu/mL with 1% DMSO. Culture plates were incubated at 35 °C for 24 h for all strains other than S. mutans, which was incubated for 48 h. At the end of the incubation periods MICs were determined by identifying the drug concentration at which no bacterial growth was visible in the wells. MBCs were determined by transferring 20 μL of broth from wells without visible growth onto either nutrient agar for Staph. aureus and B. cereus or brain-heart infusion agar for S. mutans. Growth was observed at 24 or 48 h, respectively. Averages and standard errors were calculated for MICs and MBCs using measurements of at least three.
Antiinsectan Testing
Laboratory colonies of S. frugiperda were reared on a pinto bean-based diet at 27 ± 1 °C, 50 ± 10% relative humidity, and a 14:10 h light:dark photoperiod. First instar larvae were used in bioassays. Pinto bean-based insect diet disks were prepared by “casting” slabs of diet 1 mm thick, punching out 8 mm diameter disks, and freeze-drying. Disks (∼15 mg each) were stored in a sealed container at 4 °C. For the bioassay, test materials were absorbed into freeze-dried diet disks using acetone at a rate of 25 μL per disk to yield the target concentrations. The disks were placed in a hood for 30 min to evaporate solvent. Petri dishes with tight-fitting lids (Falcon 1006, Becton Dickinson & Co.) containing 5 mL of 3% agar were used as bioassay containers. A 1 cm diameter Teflon disk (Scientific Specialties Service) was added to each plate, and the diet disks with incorporated test materials were placed on top of the Teflon disks. The diet disks were rehydrated with sterile distilled water (25 μL), and 10 first instar larvae were added to each dish. All compounds were tested in duplicate. Each disk provided sufficient material for ad libitum feeding by larvae over the duration of the assay period. The dishes were incubated under the same conditions used in insect rearing except plates were kept in the dark. Dishes were evaluated for dead larvae daily and frozen at the end of the assay period until all survivors of the bioassay could be weighed. Larvae were weighed to the nearest 0.01 mg using an analytical balance (Mettler Instrument Corp.; model AE163). The weights of all surviving larvae on a given plate were averaged. Individual treatment replicates were statistically analyzed, and data were pooled where no statistical differences at p < 0.05 were noted (all cases). Reductions in larval growth were assessed by comparing the average weight of corresponding control larvae to the average weight of larvae on treatment plates, and results are reported as a percentage of the control. Mortality was assessed by calculating the percentage of dead larvae compared to the total larvae found. SAS version 8.0 was used for analysis of variance for weights (Proc GLM) and for chi square analysis of mortality (Proc Freq).
Crude extracts of D. searlsiae root and aerial parts were tested at concentration percentages 1/10 that of the percent of crude extracts resulting from the plant material extracted (e.g., 27.4 g of crude extract from 196 g aerial parts = 14%, tested at 1.4% of diet disks) in order to account for the wet weight of fresh plant material. Crude extracts were tested over 4 days. Initial testing of compounds 1–10 was performed at concentrations of 1% of disk weight (10 μg/mg disk) over 3 days, for relative activity comparisons. Compounds 4, 7, 8, and rotenone were tested at lower doses (see Table 4) corresponding to their probable concentrations, based on isolated amounts, in fresh plant material that contains 50–65% water,57 using the same methods as described above. Average weights and standard errors for these were as follows: control (acetone only) = 0.33 mg ± 0.2; compound 4 = 0.31 mg ± 0.2; compound 7 = 0.14 mg ± 0.1; compound 8 = 0.27 mg ± 0.2; rotenone = 0.14 mg ± 0.2.
Acknowledgments
The authors would like to thank the University of Iowa HRMS Facility, operating under its NIH shared instrument grant (S10-RR023384-01), for discounted services. We gratefully acknowledge the CWU, College of the Sciences Science Talent Expansion Program (STEP), for student financial support and for G.B.’s start-up funding. Special thanks to Prof. C. Shock, OSU, Malheur Experiment Station, for providing plant material and water content measurement, and Dr. W. Massefski, Mass Analytical, for assistance with in-house 2D-NMR experimental setup and troubleshooting. We also thank Dr. D. Rosado and the Department of Chemistry and Biochemistry, Mississippi College, Clinton, MS, for the use of the JASCO J-815 CD spectrometer.
Supporting Information Available
ECD spectra of compounds 1–5, 7, and 8; 1H, 13C, HSQC, and HMBC spectra of compounds 1–5 and 7–10; and 1H, 13C, DEPT, and COSY data of compound 6. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- St. John L.; Tilley D.; Ogle D.; Johnson D.; Bushman S.. Plant Guide for Searls’ Prairie Clover (Dalea searlsiae); USDA-Natural Resources Conservation Service; Plant Materials Center, Aberdeen, ID, and USDA-Agricultural Research Service, Forage and Range Research Laboratory: Logan, UT, 2011; http://www.fs.fed.us/rm/pubs_other/rmrs_2011_stjohn_l001.pdf. [Google Scholar]
- Morris A. D. Proc. R. Soc. Med. 1974, 67, 4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiehm A. Brittonia 1985, 37, 41–43. [Google Scholar]
- Bhattarai K.; Bushman B. S.; Johnson D. A.; Carman J. G. Crop Sci. 2011, 51, 716–727. [Google Scholar]
- Shock C. C.; Feibert E. B. G.; Parris C. A.; Saunders L. D.; Johnson D.; Bushman S.; Shaw N. In Malheur Experiment Station Annual Report, Oregon State University Agricultural Experiment Station; Shock C. C. Ed.; 2011, 2012; pp 136–138; http://cropinfo.net/AnnualReports/2011/ForbEmergenceLegumes2011.php.
- Bushman B. S.; Johnson D. A.. USDA, ARS, The Forage and Range Research Laboratory: Logan, UT, Personal communication.
- Nanayakkara N. P. D.; Burandt C. L. Jr.; Jacob M. R. Planta Med. 2002, 68, 519–522. [DOI] [PubMed] [Google Scholar]
- Peralta M. A.; Ortega M. G.; Agnese A. M.; Cabrera J. L. J. Nat. Prod. 2011, 74, 158–162. [DOI] [PubMed] [Google Scholar]
- Belofsky G.; Kolaczkowski M.; Adams E.; Schreiber J.; Eisenberg V.; Coleman C. M.; Zou Y.; Ferreira D. J. Nat. Prod. 2013, 76, 915–925. [DOI] [PubMed] [Google Scholar]
- Belofsky G.; Percivill D.; Lewis K.; Tegos G. P.; Ekart J. J. Nat. Prod. 2004, 67, 481–484. [DOI] [PubMed] [Google Scholar]
- Belofsky G.; French A. N.; Wallace D. R.; Dodson S. L. J. Nat. Prod. 2004, 67, 26–30. [DOI] [PubMed] [Google Scholar]
- Wojciechowski M. F. In Advances in Legume Systematics, Part 10, Higher Level Systematics; Klitgaard B. B.; Bruneau A., Eds.; Royal Botanical Gardens: Kew, UK, 2003; pp 5–35. [Google Scholar]
- Wojciechowski M. F.; Sanderson M. J.; Steele K. P.; Liston A. In Advances in Legume Systematics, Part 9; Herendeen P. S.; Bruneau A., Eds.; Royal Botanical Gardens: Kew, UK, 2000; pp 277–298. [Google Scholar]
- Mitscher L. A.; Park Y. H.; Clark D.; Beal J. L. J. Nat. Prod. 1980, 43, 259–269. [DOI] [PubMed] [Google Scholar]
- Kinoshita T.; Kajiyama K.; Hiraga Y.; Takahashi K.; Tamura Y.; Mizutani K. Heterocycles 1996, 43, 581–588. [Google Scholar]
- Gafner S.; Bergeron C.; Villinski J. R.; Godejohann M.; Kessler P.; Cardellina J. H. II; Ferreira D.; Feghali K.; Grenier D. J. Nat. Prod. 2011, 74, 2514–2519. [DOI] [PubMed] [Google Scholar]
- Cesco S.; Mimmo T.; Tonon G.; Tomasi N.; Pinton R.; Terzano R.; Neumann G.; Weisskopf L.; Renella G.; Landi L.; Nannipieri P. Biol. Fertil. Soils 2012, 48, 123–149. [Google Scholar]
- Barbour M. G.; Burk J. H.; Pitts W. D.; Gilliam F. S.; Schwartz M. W.. Terrestrial Plant Ecology; Addison Wesley Longman: Menlo Park, CA, 1999; Chapter 7, pp 149–177. [Google Scholar]
- Arneson L. P. S.; Fagerlund A.; Granum P. E.. FEMS Microbiol. Rev. 2008, 32, 579–606. [DOI] [PubMed] [Google Scholar]
- Executive Summary of the Centers for Disease Control and Prevention. Antibiotic Resistance in the United States, 2013. http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf.
- Boucher H. W.; Corey G. R. Clin. Infect. Dis. 2008, 46, S344–S349. [DOI] [PubMed] [Google Scholar]
- Perencevich E. N.; Diekema D. J. JAMA 2010, 304, 687–689. [DOI] [PubMed] [Google Scholar]
- Brown D. F. J. J. Antimicrob. Chemother. 2001, Suppl. S165–70. [DOI] [PubMed] [Google Scholar]
- Gibbons S. Nat. Prod. Rep. 2004, 21, 263–277. [DOI] [PubMed] [Google Scholar]
- Selwitz R. H.; Ismail A. I.; Pitts N. B. Lancet 2007, 369, 51–59. [DOI] [PubMed] [Google Scholar]
- Loesche W. J. Microbiol. Rev. 1986, 50, 353–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushnie T. P. T.; Lamb A. J. Int. J. Antimicrob. Agents 2005, 26, 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch R. E.; Guo B.; Timper P.; Wilson J. Pest Manage. Sci. 2003, 59, 718–727. [DOI] [PubMed] [Google Scholar]
- Cruz I.; Turpin F. T. J. Econ. Entomol. 1983, 76, 1052–1054. [Google Scholar]
- Metcalf R. A.; Metcalf R. L.. Destructive and Useful Insects: Their Habits and Control, 5th ed.; McGraw Hill, Co.: New York, 1995. [Google Scholar]
- Dowd P. F.; Johnson E. T.; Pinkerton T. S. J. Agric. Food Chem. 2007, 55, 3421–3428. [DOI] [PubMed] [Google Scholar]
- Williamson S. M.; Wright G. A. J. Exp. Biol. 2013, 216, 1799–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer M. J.; Moffat C.; Saranzewa N.; Harvey J.; Wright G. A.; Connolly C. N. Nat. Commun. 2013, 4, 1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmquist K.; Salatas J.; Fairbrother A. In Insecticides–Advances in Integrated Pest Management; Perveen F., Ed.; InTech: Rijeka, Croatia, 2011; Chapter 11; pp 1–29; DOI: 10.5772/29495. [DOI] [Google Scholar]
- Relyea R. A. Ecol. Appl. 2005, 15, 618–627. [Google Scholar]
- Bouchard M. F.; Bellinger D. C.; Wright R. O.; Weisskopf M. G. Pediatrics 2010, 125, e1270–e1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S. J. Pestic. Biochem. Physiol. 1991, 39, 84–91. [Google Scholar]
- De Araujo R. M.; Lima M. A. S.; Silveira E. R. J. Braz. Chem. Soc. 2009, 20, 935–938. [Google Scholar]
- Gaffield W. Tetrahedron 1970, 26, 4093–4108. [Google Scholar]
- Slade D.; Ferreira D.; Marais J. P. J. Phytochemistry 2005, 66, 2177–2215. [DOI] [PubMed] [Google Scholar]
- Iinuma M.; Ohyama M.; Tanaka T. Phytochemistry 1995, 38, 539–543. [Google Scholar]
- Snatzke G. Tetrahedron 1965, 21, 439–448. [DOI] [PubMed] [Google Scholar]
- Gao S.; Xu Y.-M.; Valeriote F. A.; Gunatilaka A. A. L. J. Nat. Prod. 2011, 74, 852–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyengi L.; Lee I.-S.; Mar W.; Fong H. H. S.; Pezzuto J. M.; Kinghorn A. D. Phytochemistry 1994, 36, 1523–1526. [Google Scholar]
- Yenesew A.; Midiwo J. O.; Waterman P. G. Phytochemistry 1998, 47, 295–300. [Google Scholar]
- Ngamga D.; Yankep E.; Tane P.; Bezabih M.; Ngadjui B. T.; Fomum T.; Abegaz B. M. Bull. Chem. Soc. Ethiop. 2005, 19, 75–80. [Google Scholar]
- Dagne E.; Bekele A. Phytochemistry 1990, 29, 2679–2682. [Google Scholar]
- Yenesew A.; Midiwo J. O.; Waterman P. G. Phytochemistry 1996, 41, 951–955. [Google Scholar]
- Ollis W. D.; Rhodes C. A.; Sutherland I. O. Tetrahedron 1967, 23, 4741–4760. [Google Scholar]
- Cushnie T. T. P.; Lamb A. J. Int. J. Antimicrob. Agents 2011, 38, 99–107. [DOI] [PubMed] [Google Scholar]
- Eumkeb G.; Richards R. M. E. Acta Hortic. 2005, 678, 171–178. [Google Scholar]
- Morimoto M.; Kumeda S.; Komai K. J. Agric. Food Chem. 2000, 48, 1888–1891. [DOI] [PubMed] [Google Scholar]
- Dowd P. F.; Berhow M. A.; Johnson E. T. J. Chem. Ecol. 2011, 37, 443–449. [DOI] [PubMed] [Google Scholar]
- Cespedes C. L.; Calderon J. S.; Lina L.; Aranda E. J. Agric. Food Chem. 2000, 48, 1903–1908. [DOI] [PubMed] [Google Scholar]
- Rattan R. S. Crop. Prot. 2010, 29, 913–920. [Google Scholar]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard - Seventh Edition; Clinical and Laboratory Standards Institute document M7-A7; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2006.
- Shock C. C. Oregon State University, Malheur Experiment Station, Ontario, OR, Sept. 2013; personal communication. Fresh plant material, in triplicate, oven-dried (60 °C, 24 h).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.