

Abstract
Interstitial lung disease (ILD) is rare and encompasses a heterogeneous group of diseases, and is even rarer in children than in adults. ILDs compromise more than 100 different entities, including pulmonary alveolar proteinosis (PAP). There are many causes of PAP in children, including surfactant protein gene mutations (SFTPB, SFTPC, ABCA3, TTF-1), GMCSF receptor mutations and antigranulocyte-macrophage colony-stimulating factor autoantibodies. We report a case of a 13-year-old Chilean girl who presented with an 8-month history of progressive exercise intolerance, fatigability and diminished school performance. Physical examination revealed resting tachypnoea, a few basal bilateral inspiratory crackles, and hypoxaemia on minimal exertion. Clinical suspicion and evaluation, including international collaboration, led to the diagnosis of autoimmune PAP and specific therapy for the condition.
Background
Chronic interstitial lung diseases are a rare and heterogeneous group of chronic pulmonary diseases, and autoimmune pulmonary alveolar proteinosis (APAP) is one of these. There are few paediatric patients reported in the world.
We present the second paediatric PAP case reported in Chile and the first confirmed case of APAP treated with whole-lung lavage.
Furthermore, this case emphasises the importance of not being put off the diagnosis by a test which does not fit (first bronchoalveolar lavage (BAL)), and shows how international collaboration can result in new procedures being introduced safely into a developing world setting.
Case presentation
A 13-year-old girl presented with an 8-month history of a gradual onset of exercise intolerance, fatigability and diminished school performance. She was a term infant with no neonatal complications, and was previously healthy. She had no allergies and did not use medication. There was no family history of lung disease, and her only brother is healthy. She attended the outpatient clinic complaining of dry cough, mild fever, dyspnoea and night-time chest discomfort.
Chest radiography showed widespread interstitial abnormalities with middle and lower zone predominance. A clinical diagnosis of Mycoplasma pneumoniae infection was made and she was treated for 10 days with clarithromycin and albuterol. Her symptoms progressed and she was admitted 1 month later. Physical examination revealed tachypnoea at rest, a few basal, bilateral inspiratory crackles and intermittent hypoxaemia on minimal effort; oxygen therapy was started. The chest X-ray was unchanged (figure 1A, B) and interstitial lung disease (ILD) was suspected.
Figure 1.

Imaging studies. (A) Posteroanterior and (B) lateral chest radiographs showing widespread interstitial abnormalities, with a mid-zone and lower zone predominance and relative sparing of the apices and costophrenic angles. (C–E) High-resolution CT (HRCT) showing patchy geographic ground-glass opacities superimposed on interlobular septal thickening in multiple lobes (crazy-paving appearance). (F) Pre-WLL and (G) post-WLL HRCT showing a decrease in the density of the alveolar filling.
Investigations
High-resolution CT (HRCT) showed symmetrical, bilateral, widespread ground-glass attenuation with interlobular septal thickening (crazy-paving pattern) suggesting PAP (figure 1C–E).
Tests performed to rule out secondary PAP were normal. The first BAL was surprisingly normal. Spirometry showed mild reduction forced expiratory volume in 1 s (76%) and forced expiratory flow between 25 and 75 centile (FEF25–75) (50%) with no response to inhaled albuterol given through a large volume spacer.
Lung biopsy was performed by video-assisted thoracoscopic surgery (VATS) and light-microscopic examination showed preserved lung parenchymal architecture with the alveoli filled with granular, eosinophilic, periodic acid-schiff (PAS)-positive material, consistent with PAP. Electron microscopy showed abundant lamellar bodies (figure 2).
Figure 2.
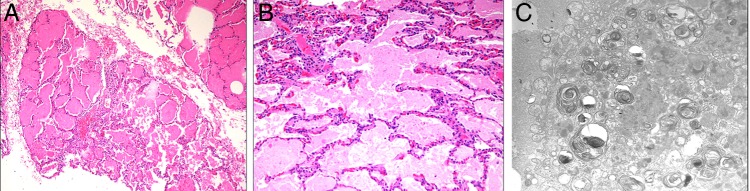
H&E staining at magnification ×100 (A) and ×200 (B) of the lung biopsy specimen revealing filling of the alveolar spaces with amorphous material and the preservation of the interstitial architecture without inflammation. Electron microscopy (C) reveals numerous alveolar cells showed abundant concentrically multilamelated phospholipid structures (lamellar bodies) and tubular myelin, widely distributed in the cell cytoplasm.
The patient travelled to Cincinnati for further evaluation, where granulocyte-macrophage colony-stimulating factor (GM-CSF) autoantibody concentration was found to be 142.7 μg/mL, reference range <3 μg/mL, confirming the diagnosis of autoimmune PAP.
Differential diagnosis
Differential diagnosis in children aged 2–18 years include ILD of known aetiology such as infections, environmental inhalants, drug or radiation-induced disorders, neoplastic or lymphoproliferative disorders and genetic surfactant or degenerative disorders; associated diseases of autoimmune mechanisms such as rheumatological conditions and vasculitis; and primary pulmonary disorders.1–4 PAP includes surfactant protein gene mutations (SFTPB, SFTPC, ABCA3, TTF-1), GMCSF receptor mutations and anti-GM-CSF autoantibodies (box 1).
Box 1. Congenital and acquired causes of pulmonary alveolar proteinosis18.
Surfactant protein system gene mutations: SFTPB, SFTPC, TTF-1 and ABCA3.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor: mutations in α-chain or β-chain of the GM-CSF receptor.
GM-CSF autoantibody disease.
Metabolic disease: lysinuric protein intolerance or rarely, of Niemann-Pick disease.
Associated with immune deficiency.
Macrophage blockade: exclusively in adults (haematological malignancy, inorganic dust inhalation).
Treatment
Whole lung lavage (WLL) was performed in the USA in July 2010. A lavage with 10 L saline solution in each lung was performed until the liquid was clear. HCRT postbilateral lung lavage showed a great improvement in tomographic findings. After several months the patient again became symptomatic and hypoxemic. Next, WLL was performed in Chile by a Chilean specialist supported by the USA specialist and successive annual WLLs were performed by Chilean specialists in Chile.
Outcome and follow-up
The outcome has been satisfactory, the patient still has normal lung function although she had required WLL annually, she attends college normally and her follow-up and treatment can be carried out in Chile.
Discussion
The clinical manifestations of paediatric ILD are often subtle and non-specific.5–16 In our patient, ILD was clinically suspected 1 month after the first consultation although she had insidious symptoms 8 months before. Differential diagnosis was discarded with specific tests.
The HRCT scan was virtually pathognomonic for PAP, but surprisingly there was no milky fluid on BAL, a result which we were unable to explain. Given the HRCT findings, despite BAL not confirming the diagnosis, we proceeded to a VATS lung biopsy which confirmed PAP. PAP comprises a heterogeneous group of diseases characterised by abnormal surfactant accumulation resulting in respiratory insufficiency and defects in alveolar macrophage and neutrophil-mediated host defense.17–36 One of them is APAP. APAP requires serum anti-GM-CSF determination, suitable only in a few international centres. In Chile, as in other Latin American countries, this assay is not yet available; hence the referral to Cincinnati. The first case diagnosed in Chile, does not access to it, for this reason the specific cause of PAP was not possible to demonstrate. Although APAP represents approximately 90% of cases of PAP, there are few paediatric cases reported. The report of a large cohort of patients with APAP in Japan showed that patients younger than 10 years of age were rare. Suzuky et al,29 reported a 6-year-old girl presented with insidious, progressive tachypnoea of several years duration and had radiological and histopathological manifestations identical to those of APAP but had a negative GM-CSF autoantibody test.
WLL has long been considered the definitive therapy for PAP. It should only be performed by a skilled anaesthesiologist and bronchoscopist.17 18 25 26
The first WLL was conducted in an international medical centre staffed with skilled and experienced specialists. WLL had previously never been performed on a paediatric patient in Chile; however, future WLLs will be. Prelavage and postlavage CT scans showed a decrease in density, which would be expected secondary to clearing of the PAS-positive material (figure 1F, G). In a few cases, GM-CSF therapy has been used,37 38 and the overall response rate appears nearly 50%; this is in contrast to the ability of WLL to produce rapid improvements in symptoms, radiological images and lung function in all patients.17 19 26 GM-CFS therapy was not considered in the reference centre where anti-GM-CSF testing was performed. Furthermore this therapy is not available in Chile due to the cost. The patient will certainly need the support of a multidisciplinary team, and successive WLL. Rituximab is another option to consider in the future,39 which has proven medically effective and useful in sporadically reported adult cases.
Learning points.
Dyspnoea and exercise intolerance in a previously healthy teenager can be autoimmune pulmonary alveolar proteinosis.
If clinical features suggest a specific diagnosis, it is important not being put off the diagnosis by a test which does not fit (first bronchoalveolar lavage).
International collaboration can result in expensive, specific diagnostic tests being performed and new procedures being introduced safely into a developing world setting.
Acknowledgments
The authors would like to thank Robin Detterding for clinical counselling, Megan Dishop for pathologist's opinion collaboration, Robert Wood for providing prelavage and postlavage CT scan images, Andrew Bush for revising and correcting the manuscript and Marcela Amtmann for the bibliographic search and collaborating with the writing.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Deutsch GH, Young LR, Deterding RR, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med 2007;176:1120–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deterding R. How to evaluate and treat children's interstitial lung disease? Paediatr Respir Rev 2006;7S:S245–7 [DOI] [PubMed] [Google Scholar]
- 3.Dinwiddie R, Sharief N, Crawford O. Idiopathic interstitial pneumonitis in children: a national survey in the United Kingdom and Ireland. Ped Pulm 2002;34:23–9 [DOI] [PubMed] [Google Scholar]
- 4.Price A, Manson D, Cutz E, et al. Pulmonary alveolar proteinosis associated with anti-GM-CSF antibodies in a child: successful treatment with inhaled GM-CSF. Pediatr Pulmonol 2006;41:367–70 [DOI] [PubMed] [Google Scholar]
- 5.Barbato A, Panizzolo C, Cracco A, et al. Interstitial lung disease in children: a multicentre survey on diagnostic approach. Eur Respir J 2000;16:509–513 [DOI] [PubMed] [Google Scholar]
- 6.Fan L, Langston C. Chronic interstitial lung disease in children. Pediatr Pulmonol 1993;16:184–96 [DOI] [PubMed] [Google Scholar]
- 7.Clement A, Nathan N, Epaud R, et al. Interstitial lung diseases in children. Orph J of Rare Dis 2010;5:22–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fauroux B, Epaud R, Clement A. Clinical presentation of interstitial lung disease in children. Paediatr Respir Rev 2004;5:98–100 [DOI] [PubMed] [Google Scholar]
- 9.Hilman B, Amaro-Galvez R. Diagnosis of interstitial lung disease in children. Paediatr Respir Rev 2004;5:101–7 [DOI] [PubMed] [Google Scholar]
- 10.Dinwiddie R. Treatment of interstitial lung disease in children. Paediatr Respir Rev 2004;5:108–15 [DOI] [PubMed] [Google Scholar]
- 11.Clement A, Eber E. Interstitial lung diseases in infants and children. Eur Respir J 2008;31:658–66 [DOI] [PubMed] [Google Scholar]
- 12.Copley SJ, Coren M, Nicholson AG, et al. Diagnostic accuracy of thin section CT and chest radiography of pediatric interstitial lung disease. AJRAm J Roentgenol 2000;174:549–54 [DOI] [PubMed] [Google Scholar]
- 13.Copley SJ, Padley SPG. High-resolution CT of paediatric lung disease. Eur Radiol 2001;11:2564–75 [DOI] [PubMed] [Google Scholar]
- 14.Boros P, Franczuk M, Wesolowski S. Value of spirometry in detecting volume restriction in interstitial lung disease patients. Respiration 2004;71:374–9 [DOI] [PubMed] [Google Scholar]
- 15.Meyer K. Bronchoalveolar lavage in the diagnosis and management of interstitial lung diseases. Clin Pulm Med 2007;14:148–56 [Google Scholar]
- 16.Munson J, Kreider M. The role of surgical biopsy in the evaluation of interstitial lung disease. Clin Pulm Med 2008;15:201–9 [Google Scholar]
- 17.Yamamoto H, Yamaguchi E, Agata H, et al. A combination therapy of whole lung lavage and GM-CSF inhalation in pulmonary alveolar proteinosis. Pediatr Pulmonol 2008;43:828–30 [DOI] [PubMed] [Google Scholar]
- 18.Bush A, Nicholson A. Paediatric Interstitial lung disease. Eur Respir Mon 2009;46:319–54 [Google Scholar]
- 19.Laplane R, Fontaine JL, Fessard C, et al. Pulmonary alveolar proteinosis in children. Presse Med 1968;76:1857–60 [PubMed] [Google Scholar]
- 20.McCook T, Kirks D, Merten D, et al. Pulmonary alveolar proteinosis in children. AJR Am J Roentgenol 1981;137:1023–7 [DOI] [PubMed] [Google Scholar]
- 21.Inoue Y, Trapnell B, Tazawa R, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med 2008;177:752–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell B, Whitsett J, Nakata K. Mechanisms of disease: pulmonary alveolar proteinosis. N Eng J Med 2003;349:2527–39 [DOI] [PubMed] [Google Scholar]
- 23.Juvet S, Hwang D, Waddell T, et al. Rare lung disease II: pulmonary alveolar proteinosis. Can Respir J 2008;15:203–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frazier A, Franks T, Cooke E, et al. From the archives of the AFIP: pulmonary alveolar proteinosis. Radiographics 2008;28:883–99 [DOI] [PubMed] [Google Scholar]
- 25.Ishii H, Trapnell B, Tazawa R, et al. Comparative study of high-resolution CT findings between autoimmune and secondary pulmonary alveolar proteinosis. Chest 2009;136:1348–55 [DOI] [PubMed] [Google Scholar]
- 26.Ioachimescu O, Kavuru M. Pulmonary alveolar proteinosis. Chron Respir Dis 2006;3:149–59 [DOI] [PubMed] [Google Scholar]
- 27.Mazzone PJ, Thomassen M, Kavuru MS. Pulmonary alveolar proteinosis: recent advances. Semin Respir Crit Care Med 2002;23:1155–6 [DOI] [PubMed] [Google Scholar]
- 28.Susuki T, Sakagami T, Rubin BK, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med 2008;205:2703–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki T, Sakagami T, Young L, et al. Hereditary pulmonary alveolar proteinosis pathogenesis, presentation, diagnosis, and therapy. Am J Respir Crit Care Med 2010;182:1292–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida K, Beck D, Yamamoto T, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med 2007;356:567–79 [DOI] [PubMed] [Google Scholar]
- 31.Sakagami T, Uchida K, Susuki T, et al. GM-CSF autoantibodies and reproduction of pulmonary alveolar proteinosis. N Eng J Med 2009;361:2679–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carey B, Trapnell B. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol 2010;135:223–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uchida K, Nakata K, Susuki T, et al. Granulocyte/macrophage–colony stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood 2009;113:2547–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kitamura T, Tanaka N, Watanabe J, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med 1999;190:875–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trapnell B, Carey B, Uchida K, et al. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol 2009;21:514–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Latzin P, Tredano M, Wüst Y, et al. Anti-GM-CSF antibodies in paediatric pulmonary alveolar proteinosis. Thorax 2005;60:39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barna B, Kavuru M, Thomassen M, et al. Multiplexed particle-based anti-granulocyte macrophage colony stimulating factor assay used as pulmonary diagnostic test. Clin Diagn Lab Immunol 2005;12:821–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meager A, Cludts I, Thorpe R, et al. Are neutralizing anti-GM-CSF autoantibodies present in all healthy persons? Blood 2010;115:433–4 [DOI] [PubMed] [Google Scholar]
- 39.Letth S, Bendstrup E, Vestergaard H, et al. Autoimmune pulmonary alveolar proteinosis: treatment options in year 2013. Respirology 2013;18:82–91 [DOI] [PubMed] [Google Scholar]