

Abstract
Pure erythroid leukaemia is a rare subtype of acute myeloid leukaemia (AML) and its occurrence at acute lymphoblastic leukaemia (ALL) relapse has not been reported earlier. A 39-year-old man received chemotherapy for Philadelphia-negative B cell ALL. Subsequently, he developed pure erythroid leukaemia with >80% immature erythroid precursors in bone marrow showing block positivity on periodic acid-Schiff stain, expressing CD71, CD34 but lacking CD235a. The interval between exposure to multidrug chemotherapy including cyclophosphamide and AML diagnosis was 2 years and 9 months. No cytogenetic abnormality was detected at the time of relapse. The patient died 2 weeks after starting AML chemotherapy. The relatively narrow time interval (usually 5–10 years) between chemotherapy and AML development and normal karyotype at relapse raises a possibility of lineage switch besides therapy-related AML as the likely pathogenesis. Further exploration of such cases may unravel the pathways responsible for lineage assignment in pluripotent stem cells.
Background
Pure erythroid leukaemia is an extremely rare subtype of acute myeloid leukaemia (AML), acute erythroid leukaemia type.1 The occurrence of pure erythroid leukaemia at the time of acute lymphoblastic leukaemia (ALL) relapse has not been reported earlier.2–7 Diagnosis may pose difficulties due to its resemblance with cytotoxic chemotherapy-related megaloblastic erythroid proliferation in the bone marrow. The usual latency period between exposure to alkylating agents such as cyclophosphamide and the development of secondary leukaemias is 5–10 years with chromosomal abnormalities in the majority of patients.7 8 Our case raises a possibility of lineage switch as the likely pathogenesis due to a shorter latency period and absence of cytogenetic abnormalities at relapse.
Case presentation
A 39-year-old male patient presented to our hospital 3 years ago with low-grade fever of 2-month duration along with symptoms of weight loss, intermittent vomiting and abdominal distension. He had haemoglobin (Hb) of 118 g/L, total leucocyte count (TLC) 9.7×109/L and reduced platelets at 56×109/L. There was cervical and axillary lymph node enlargement. Liver and spleen were palpable 12 and 6 cm below right and left costal margins, respectively. Upper gastrointestinal endoscopic examination revealed gastric and duodenal polyps showing infiltration by atypical lymphoid cells microscopically. Bone marrow examination showed 70% blasts (figure 1) exhibiting bright expression of CD10 and CD19 on immunophenotyping. The blasts were negative for CD13, CD33 and myeloperoxidase (MPO). The BCR-ABL fusion gene was not detected on reverse transcription PCR (RT-PCR). Diagnosis of B cell ALL, Philadelphia negative was made.
Figure 1.
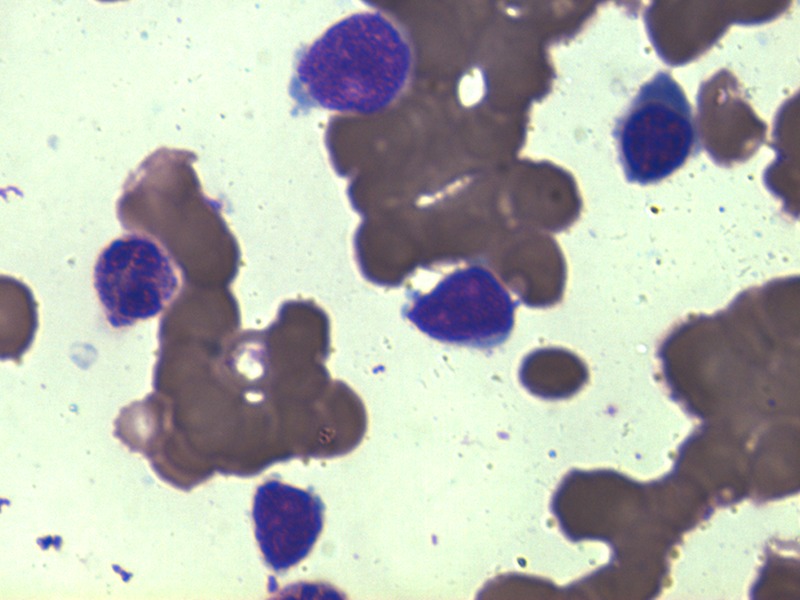
Bone marrow aspirate smear showing blasts along with a neutrophil and an intermediate erythroid precursor (Jenner-Giemsa ×1000).
Treatment
ALL chemotherapy was started as per the institutional protocol. The patient went into complete remission after induction chemotherapy with vincristine, daunorubicin and prednisolone. He then received consolidation chemotherapy consisting of cyclophosphamide at a dose of 600 mg/m2 on days 1, 15 and 29 besides cytarabine, 6-mercaptopurine (6-MP), intrathecal methotrexate (MTX) and prophylactic cranial irradiation. The patient was put on oral maintenance chemotherapy with 6-MP and MTX at the end of consolidation and reintensification phases. However, maintenance chemotherapy had to be stopped 3 months before scheduled completion due to low-blood counts (Hb 80 g/L, TLC 3.6×109/L and platelets 76×109/L). Bone marrow aspiration and imprint smears showed near total replacement by immature erythroid precursors with many of them having prominent nucleoli. There were dysplastic changes in neutrophils with few Pelger-Huet cells (5%) in the peripheral smear. The patient was kept on haematological follow-up.
Outcome and follow-up
Over the next 2 months, the patient had a further drop in the blood counts (Hb 60 g/L, TLC 1.8×109/L and platelets 44×109/L) and developed low-grade fever, weakness and joint pains. A repeat bone marrow examination was performed to rule out relapsed acute leukaemia. Bone marrow aspirate and imprint smears showed near total replacement by immature erythroid precursors with many of them having prominent nucleoli. Erythroid maturation was absent. There was cytoplasmic vacuolation in most of these erythroid precursors and some of the cells had coalescing cytoplasmic vacuoles (figure 2). MPO was negative in these cells. Periodic acid-Schiff (PAS) stain showed cytoplasmic block positivity in the erythroid precursors (figure 2, inset). Only occasional myeloid precursors and megakaryocytes were noted. On immunophenotypic analysis by multiparametric flow cytometry, the gated CD45 negative/low side scatter cells expressed CD36 (86%), CD34 (50%) and CD71 (heterogeneous, 40%) and were CD235a negative. The cells were also negative for CD13, CD33, CD117, CD14, CD64, CD10, CD19, CD20, CD22, CD38 and CD138. Conventional cytogenetics of bone marrow cells revealed normal karyotype (46,XY). The overall clinical, morphological, cytochemical and immunophenotypic findings suggested a diagnosis of pure erythroid leukaemia (subtype of AML, acute erythroid leukaemia type) as per the WHO (2008) criterion which requires the presence of >80% immature erythroblasts in the bone marrow.1
Figure 2.
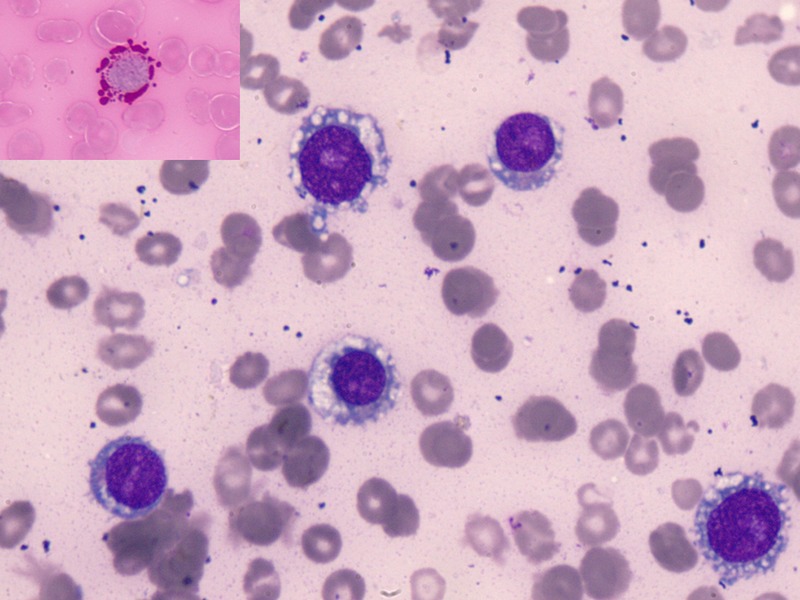
Bone marrow aspirate smear showing immature erythroid precursors with coalescing cytoplasmic vacuoles and prominent nucleoli (Jenner Giemsa ×1000). Periodic acid-Schiff stain showing pink cytoplasmic globules (inset; ×1000).
Chemotherapy was started as the standard 3+7 induction chemotherapy for AML, but the patient started having fever from the first day. On investigation, blood and urine cultures were negative but chest X-ray showed bilateral nodular opacities. With persistent fever and onset of sepsis, his blood pressure also dropped and he required ionotropic support. Renal and liver functions deteriorated rapidly with development of disseminated intravascular coagulation. Coagulation profile revealed deranged prothrombin time (37 s with control of 13 s), deranged activated partial thromboplastin time (57 s with control of 30 s), normal thrombin time (18 s with control of 16 s), low platelets 11×109/L, normal fibrinogen (4.2 g/L) and raised D-dimer level. Blood component support with fresh frozen plasma, platelet concentrates and packed RBC was given. However, the patient died due to sepsis and multiorgan dysfunction after 2 weeks of starting chemotherapy.
Discussion
To the best of our knowledge, the occurrence of pure erythroid leukaemia at ALL relapse has not been reported earlier.2–7 Pure erythroid leukaemia is clinically aggressive with rapid progression and death, as in our case. Thus, its early and accurate detection is important. At the same time, the possibility of chemotherapy-related megaloblastic proliferation should be ruled out. Our patient was also receiving cytotoxic chemotherapy known to result in inhibition of DNA synthesis and megaloblastic erythroid maturation like folate antagonist (methotrexate), purine antagonist (6-MP), pyrimidine antagonist (cytosine arabinoside) and alkylating agent (cyclophosphamide) creating a diagnostic dilemma. So, in these situations, careful attention must be paid to the various morphological, cytochemical and immunophenotypic clues for accurate diagnosis and differentiation. The abnormal erythroid precursors in erythroid leukaemia may display coalescing cytoplasmic vacuoles. Moreover, the nuclear chromatin is not as fine as in true megaloblasts and a greater proportion of the erythroid precursors are more immature in erythroid leukaemia than in megaloblastic anaemia.9 PAS stain also helps in confirming the abnormal erythroid precursors since cytoplasmic block positivity is not seen in normal precursors. Flow cytometry can help in identification of abnormal erythroid precursors based on aberrant expression patterns of CD71 (transferrin receptor) and CD235a (glycophorin A). CD235a is usually negative in immature erythroid precursors unlike more differentiated forms, as was seen in our case.1
With regard to the pathogenesis, two possibilities arise as in our case. First is the alkylating agent-related secondary pure erythroid leukaemia. Therapy-related AMLs (t-AML) are mostly seen in solid cancers (breast, ovary, uterus, lung, etc), lymphomas and multiple myeloma with a median latency period of 5–10 years after exposure to alkylating agents with/without radiation.7 8 Our case had a relatively narrow time interval of 2 years and 9 months between first exposure to an alkylating agent and AML diagnosis. Moreover, chromosomal abnormalities are seen in the majority of t-AML, particularly chromosome 5, 7 and 11q defects, but our case had a normal karyotype at the time of relapse. Second, it may be a case of lineage switch in which the leukaemic cell lineage at onset converts to another lineage at a later time. The frequency of lineage switch among patients with acute leukaemias is estimated to be about 6–9%.3 The precise mechanism of lineage switch of acute leukaemia remains obscure. One of the various hypotheses is that it is a type of mixed lineage leukaemia where chemotherapy suppresses the leukaemic clone apparent at the diagnosis, but subsequently allows the expansion of a subclone with a different phenotype. Another possible mechanism of lineage switch is the presence of common myeloid/lymphoid progenitor cells as seen in chronic myeloid leukaemia with the potential for expressing lymphoid and myeloid phenotypes in blast crisis. In our case, however, the BCR-ABL fusion gene was not found by RT-PCR analysis. Although prognosis is dismal for t-AML (median survival 4–7 months)7 10 and cases with lineage switch, such cases are of interest especially because they afford the possibility of exploring the pathways responsible for lineage assignment in pluripotent stem cells.
Learning points.
Pure erythroid leukaemia may morphologically mimic cytotoxic chemotherapy-related megaloblastic proliferation.
Block/globular periodic acid-Schiff positivity is an indicator of abnormal erythroid precursors.
The flow cytometry immunophenotypic features of erythroid cells include negative/low CD45, low side scatter and expression of glycophorin A (CD235a), CD71 (transferrin receptor) and CD36.
The median latency period of therapy-related acute myeloid leukaemia (t-AML) varies with the type of chemotherapy and is 5–10 years after exposure to alkylating agents with/without radiation.
Prognosis is poor in t-AML cases with a median survival of 4–7 months.
Acknowledgments
All the authors thank Dr Jogamaya Pattnaik, Senior Resident, Department of Medical Oncology, AIIMS.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Arber DA, Brunning RD, Orazi A, et al. Acute myeloid leukaemia, not otherwise specified. In: Swerdlow SH, Campo E, Harris NL, et al., eds WHO classification of tumours of haematopoietic and lymphoid tissues. 4th edn Lyon: IARC, 2008:130–9 [Google Scholar]
- 2.Dorantes-Acosta E, Pelayo R. Lineage switching in acute leukemias: a consequence of stem cell plasticity? Bone Marrow Res 2012;2012:406796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stass S, Mirro J, Melvin S. Lineage switch in acute leukemia. Blood 1984;64:701–6 [PubMed] [Google Scholar]
- 4.Park M, Koh KN, Kim BE, et al. Lineage switch at relapse of childhood acute leukemia: a report of four cases. J Korean Med Sci 2011;26:829–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung HJ, Park CJ, Jang S, et al. A case of lineage switch from acute lymphoblastic leukemia to acute myeloid leukemia. Korean J Lab Med 2007;27:102–5 [DOI] [PubMed] [Google Scholar]
- 6.Imataki O, Ohnishi H, Yamaoka G, et al. Lineage switch from precursor B cell acute lymphoblastic leukemia to acute monocytic leukemia at relapse. Int J Clin Oncol 2010;15:112–15 [DOI] [PubMed] [Google Scholar]
- 7.Mauritzson N, Albin M, Rylander L, et al. Pooled analysis of clinical and cytogenetic features in treatment-related and de novo adult acute myeloid leukemia and myelodysplastic syndromes based on a consecutive series of 761 patients analyzed 1976–1993 and on 5098 unselected cases reported in the literature 1974–2001. Leukemia 2002;16:2366–78 [DOI] [PubMed] [Google Scholar]
- 8.Jagasia MH. Complications of hematopoietic neoplasms. In: Greer JP, Foerster J, Rodgers GM, et al. eds Wintrobe's clinical hematology. 12th edn Philadelphia: Lippincott Williams & Wilkins, 2009:1669–93 [Google Scholar]
- 9.Vardiman JW, Arber DA, Brunning RD, et al. Therapy-related myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al. eds. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th edn Lyon: IARC, 2008:127–9 [Google Scholar]
- 10.Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood 2003;102:43–52 [DOI] [PubMed] [Google Scholar]