

Abstract
Purpose
Idiotypes (Ids), the unique portions of tumor immunoglobulins, can serve as targets for passive and active immunotherapies for lymphoma. We performed a multicenter, randomized trial comparing a specific vaccine (MyVax), comprising Id chemically coupled to keyhole limpet hemocyanin (KLH) plus granulocyte macrophage colony-stimulating factor (GM-CSF) to a control immunotherapy with KLH plus GM-CSF.
Patients and Methods
Patients with previously untreated advanced-stage follicular lymphoma (FL) received eight cycles of chemotherapy with cyclophosphamide, vincristine, and prednisone. Those achieving sustained partial or complete remission (n = 287 [44%]) were randomly assigned at a ratio of 2:1 to receive one injection per month for 7 months of MyVax or control immunotherapy. Anti-Id antibody responses (humoral immune responses [IRs]) were measured before each immunization. The primary end point was progression-free survival (PFS). Secondary end points included IR and time to subsequent antilymphoma therapy.
Results
At a median follow-up of 58 months, no significant difference was observed in either PFS or time to next therapy between the two arms. In the MyVax group (n = 195), anti-Id IRs were observed in 41% of patients, with a median PFS of 40 months, significantly exceeding the median PFS observed in patients without such Id-induced IRs and in those receiving control immunotherapy.
Conclusion
This trial failed to demonstrate clinical benefit of specific immunotherapy. The subset of vaccinated patients mounting specific anti-Id responses had superior outcomes. Whether this reflects a therapeutic benefit or is a marker for more favorable underlying prognosis requires further study.
INTRODUCTION
The survival of patients with follicular lymphoma (FL) has improved with the introduction of rituximab, a passive immunotherapy targeting CD20-expressing B cells.1–4 However, most patients with FL remain incurable with currently available therapies.5 B-cell tumors can be targeted more specifically with antibodies against their unique surface immunoglobulins (ie, idiotype [Id]).6 Active vaccination of patients with the Id protein from their tumor has potential advantages over passive immunotherapy.7 An evoked immune response (IR) would be polyclonal and therefore broader than a single monoclonal antibody, could include T cells as well as antibodies,8 would be more durable than passive therapies, and would spare normal B cells.7,9–12
Id proteins can be obtained through hybridoma or recombinant methods and can be rendered immunogenic by chemical coupling to a foreign protein, such as keyhole limpet hemocyanin (KLH). In phase II trials, vaccinated patients mounting anti-Id antibody responses had longer freedom from progression and improved survival compared with patients not mounting such responses.10,13 These phase II results motivated randomized studies of Id vaccination, including our phase III study as well as two previously completed trials.14,15 Here, we present results of a randomized trial comparing a patient-specific recombinant Id coupled to KLH (MyVax), coadministered with granulocyte macrophage colony-stimulating factor (GM-CSF)16 to a control immunotherapy comprising KLH and GM-CSF.
PATIENTS AND METHODS
Patients
This was a randomized, double-blinded phase III trial conducted at 32 centers in Canada and the United States (Data Supplement). Eligible patients were age ≥ 18 years with untreated FL requiring therapy and had Ann Arbor stage III or IV disease, WHO histologic grade 1, 2, or 3,17 Eastern Cooperative Oncology Group performance score < 3, and FL histology confirmed by central pathologic review. Exclusion criteria included serologic evidence of HIV, hepatitis B or C, history of autoimmune disease or treatment with immunosuppressive agents including corticosteroids within 12 months, pregnancy or breastfeeding, and prior malignancy other than cutaneous basal cell carcinoma or cervical carcinoma in situ. Patients underwent computed tomography (CT) scans of the chest, abdomen, and pelvis and submitted a fresh tumor tissue sample for MyVax manufacturing before initiation of chemotherapy.
Study Design
All patients received chemotherapy with CVP (cyclophosphamide 1,000 mg/m2, intravenous vincristine 1.4 mg/m2, and oral prednisone 100 mg per day for 5 days) every 21 days for eight cycles. Patients were restaged 4 to 8 weeks after end of chemotherapy with physical examination and CT scans, with all scans evaluated by central radiologic review. A bone marrow biopsy was repeated in patients whose marrow was initially positive. Patients achieving partial (PR) or complete remission (CR) that persisted at an evaluation 22 weeks after the completion of CVP were randomly assigned at a ratio of 2:1 to MyVax or control immunotherapy stratified by study site and disease response status (CR unconfirmed/PR v CR). To maintain blinding, a specific vaccine was manufactured for all randomly assigned patients, including those assigned to control immunotherapy. An unblinded statistician at the contract research organization (ICON Clinical Research, Redwood City, CA) determined the treatment assignments and instructed designated personnel in quality assurance at Genitope (Fremont, CA). All other personnel at Genitope and ICON Clinical Research, central radiologic reviewers, physicians, and patients were blinded for the duration of the study.
Manufacture of MyVax and Control Immunotherapies
Id proteins were derived from each individual patient's FL by recombinant methodology.16 Each Id was conjugated to KLH (Biosyn, Carlsbad, CA) with 0.1% glutaraldehyde (Sigma, St Louis, MO) at room temperature for 60 minutes, then dialyzed into normal saline and stored at −80°C. The conjugations were performed at 0.5 mg/mL each of Id and KLH. For the control immunotherapy, KLH was self-conjugated (KLH-KLH) at 0.5 mg/mL using the same conditions, such that patients in both study arms received the same total dose of KLH and were subjected to identical conjugation and handling.
Immunization
Randomly assigned patients received seven subcutaneous immunizations with 1.0 mg Id-KLH (MyVax) or 0.5 mg KLH-KLH (control) at 4-week intervals over a period of 24 weeks. Each dose was split and injected bilaterally into the anterior thighs. Additionally, each immunization site received GM-CSF 125 μg on days 1 to 4.
Humoral IR
Anti-Id and anti-KLH enzyme-linked immunosorbent assays were performed on sera from all study patients across both arms of the study in a blinded fashion, as detailed in the Data Supplement. A positive anti-Id humoral IR was predefined as postimmunization serum having a value greater than that of preimmunization serum plus two standard deviations from the mean of replicate wells at four-fold greater dilution.
End points and statistical methods.
Clinical response was assessed using International Working Group Criteria.18 Patients were evaluated by physical examination throughout the immunization period at 2 to 4 weeks after the seventh immunization and at months 3, 6, 9, 12, 18, and 24. Patients were also evaluated by CT scans at 2 to 4 weeks after the seventh immunization as well as at months 6, 12, 18, and 24 or any other time as clinically indicated. The primary end point was progression-free survival (PFS), defined as time from random assignment to the earliest time point identifying progression or death resulting from any cause. Progressions during the first 30 months after random assignment were determined by central radiographic review and at later time points by local investigators. Treating physicians remained blinded to treatment arm and central radiographic reviews. Patients who received subsequent antilymphoma therapy (SALT) before disease progression were censored on date of first SALT. Secondary efficacy end points included SALT-free survival (SALT-FS; defined as time from random assignment to SALT or death resulting from lymphoma) and IRs.
Analyses of PFS and SALT-FS were conducted using the log-rank test with results expressed as Kaplan-Meier plots. A Cox proportional hazards analysis of PFS included demographic and baseline characteristics. As prespecified, the trial was unblinded when all patients completed evaluation at 2 years postimmunization. The statistical plan assumed median PFS of 22 months from random assignment for the control arm and 43 months for the experimental group, with accrual duration of 15 months. The actual accrual duration was 42 months for the entire cohort. Using a two-sided α of 0.01, the trial had a power of 96% to detect a 21-month difference and at least 80% power to detect a ≥ 14-month difference between groups.
The relationship of humoral IRs to MyVax was assessed as a continuous variable using A univariable Cox proportional hazards model, with PFS as the dependent variable.19 Using the coxph function in the R statistical package (http://www.r-project.org), the Wald test was used to assess the significance of covariates. We used Schoenfeld residuals to assess violation of proportional hazards assumption (cox.zph function) and confirmed proportionality of hazards for associations between PFS and IR and between SALT and IR. Additional details are as outlined in the Data Supplement.
RESULTS
A total of 675 patients were registered and assessed for eligibility, with 75 patients (11%) lacking a clonal Id and thus not eligible for vaccine production (Fig 1); 513 patients met initial screening criteria and were enrolled. Among this group, 226 patients (44%) started chemotherapy but did not achieve a PR or progressed before random assignment (Fig 1). After eight cycles of chemotherapy and a 6-month recovery period, the remaining 287 patients were randomly assigned to receive either MyVax or control immunotherapy at a 2:1 ratio, and this group was evaluable for clinical outcome on an intent-to-treat (ITT) basis (Data Supplement). The first patient was screened in November 2000 and started immunization in November 2001, and the last patient started immunization in June 2005. The database was locked, and the trial was unblinded in December 2007 after all patients had completed the prespecified evaluation at 2 years postimmunization.
Fig 1.

CONSORT diagram. Of patients with tumor progression before random assignment (n = 189), 52 received < eight cycles of CVP (cyclophosphamide 1 g/m2 on day 1, vincristine 1.4 mg/m2 on day 1, and prednisone 100 mg per day on days 1 to 5) chemotherapy, 20 had progressive disease (PD) immediately after chemotherapy, and 117 achieved response but had PD between postchemotherapy assessment and random assignment. Of those randomly assigned, nine patients in MyVax arm were not immunized because of PD before planned first immunization; one patient in control immunotherapy arm erroneously received single injection of MyVax but was nonetheless considered among those in control immunotherapy arm in intent-to-treat analyses. GM-CSF, granulocyte macrophage colony-stimulating factor; Id, idiotype; Id-KLH, idiotype conjugated to keyhole limpet hemocyanin.
Although patients were not stratified based on prognostic risk, there was good balance between arms regarding FL International Prognostic Index (FLIPI) score20 (Table 1). Of 287 patients in the ITT population, nine were not immunized, because of disease progression during the 4 weeks preceding first planned vaccination; all nine of these nonimmunized patients were in the MyVax group. Of the remaining 278 immunized patients, eight received fewer than the prespecified four immunizations, leaving 270 patients evaluable for IR. One patient randomly assigned to the control immunotherapy group mistakenly received MyVax but remained in the control immunotherapy group for analysis, per the ITT principle.
Table 1.
Patient Demographic and Clinical Characteristics
| Characteristic | MyVax (n = 192) |
Control Immunotherapy (n = 95) |
||
|---|---|---|---|---|
| No. | % | No. | % | |
| Age, years | ||||
| < 40 | 26 | 13.5 | 15 | 15.8 |
| 40 to 50 | 60 | 31.3 | 33 | 34.7 |
| 50 to 60 | 56 | 29.2 | 25 | 26.3 |
| ≥ 60 | 50 | 26.0 | 23 | 24.2 |
| Median | 50 | 50 | ||
| Range | 22-80 | 25-80 | ||
| Sex | ||||
| Female | 106 | 55.2 | 51 | 53.7 |
| Male | 86 | 44.8 | 44 | 46.3 |
| ECOG performance status | ||||
| 0 | 135 | 70.3 | 66 | 69.5 |
| 1 | 55 | 28.6 | 27 | 28.4 |
| Not evaluable/missing | 2 | 1.1 | 2 | 2.1 |
| WHO histologic grade | ||||
| Local review | ||||
| 1 | 111 | 57.8 | 59 | 62.1 |
| 2 | 68 | 35.4 | 31 | 32.6 |
| 3 | 12 | 6.3 | 5 | 5.3 |
| Not evaluable/missing | 1 | 0.5 | ||
| Central review | ||||
| 1 | 97 | 50.5 | 49 | 51.6 |
| 2 | 75 | 39.1 | 37 | 38.9 |
| 3 | 13 | 6.8 | 7 | 7.4 |
| Not evaluable/missing | 7 | 3.6 | 2 | 2.1 |
| Ann Arbor disease stage | ||||
| III | 80 | 41.7 | 36 | 37.9 |
| IV | 112 | 58.3 | 59 | 62.1 |
| FLIPI risk group | ||||
| Low | 23 | 12.0 | 6 | 6.3 |
| Intermediate | 100 | 52.1 | 53 | 55.8 |
| High | 67 | 34.9 | 36 | 37.9 |
| Not evaluable/missing | 2 | 1.0 | 0 | 0.0 |
| Bone marrow involvement | 128 | 66.7 | 66 | 69.5 |
| B symptoms | 31 | 16.1 | 17 | 17.9 |
| Bulky disease | 57 | 29.7 | 16 | 16.8 |
| Elevated LDH | 29 | 15.1 | 17 | 17.9 |
| ≥ One extranodal site | 53 | 27.6 | 25 | 26.3 |
| Median duration of watchful waiting, days | 100 | 98 | ||
Abbreviations: ECOG, Eastern Cooperative Oncology Group; FLIPI, Follicular Lymphoma International Prognostic Index; LDH, lactate dehydrogenase.
Safety
Immunizations were generally well tolerated (Table 2), with only one grade 4 toxicity of back pain and muscle spasm, occurring in the control arm. The most common toxicities were related to injection site reactions, including ecchymosis, erythema, pruritus, and pain. The most common systemic adverse events were fatigue, fevers, chills, nausea, muscle pain, and diarrhea, all grade ≤ 2. Adverse events occurred at similar rates in both study arms.
Table 2.
Adverse Events by Maximum Grade Occurring Within 4 Days of Immunization
| Toxicity Level (grade) | MyVax (n = 184) |
Control Immunotherapy (n = 94) |
||
|---|---|---|---|---|
| No. | % | No. | % | |
| 1 | 60 | 32.6 | 31 | 33.0 |
| 2 | 103 | 56.0 | 51 | 54.3 |
| 3 | 21 | 10.9 | 9 | 9.6 |
| 4 | 0 | 0.0 | 1 | 1.1 |
NOTE. Per Common Terminology Criteria for Adverse Events (version 3.0).
PFS and Subsequent Lymphoma Therapy
Among all randomly assigned patients, the median PFS was 19.9 months, and the median time to SALT was 42.8 months (Fig 1). There was no difference in PFS between patients receiving MyVax versus control immunotherapy (P = .89; Fig 2A). Even among patients achieving CR or CR unconfirmed before vaccination (MyVax, n = 62; control group, n = 28), PFS was not different between arms (hazard ratio, 1.18; 95% CI, 0.66 to 2.12; P = .56). After a median follow-up of 48 months postchemotherapy, 49.5% of patients had not received SALT. There was no significant difference in the time to SALT (prespecified secondary end point) between the two trial arms (P = .36; Fig 2B).
Fig 2.
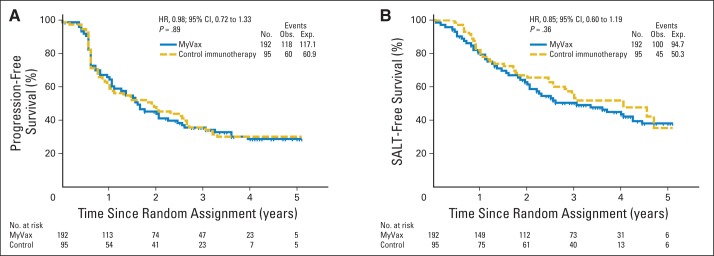
Comparison of treatment arms for (A) primary end point (progression-free survival [PFS]) and (B) prespecified secondary end point (time to subsequent antilymphoma therapy [SALT]). Kaplan-Meier curves show patients randomly assigned to MyVax versus control immunotherapy, with corresponding end points measured from time of random assignment for intent-to-treat population (n = 287). (A) Steep drop in PFS at 6 months reflects timing of first radiographic response assessment. SALT was administered in blinded fashion. Neither end point was significantly different between study arms. Exp, expected; HR, hazard ratio; Obs, observed.
IRs
The magnitude of anti-KLH IR (surrogate for immunologic health) was not significantly different between patients randomly assigned to specific versus control immunotherapy (Data Supplement). Among patients receiving MyVax, 178 (93%) were evaluable for the prespecified IR end point, having received ≥ four vaccinations, and among this group, 73 patients (41%) were classified as mounting significant anti-Id antibody IRs (IR positive) by predefined criteria. The magnitude of humoral anti-Id IRs (Data Supplement) did not show a relationship to complete versus partial clinical response after chemotherapy (Data Supplement).
IR-positive patients who received MyVax had superior PFS and time to subsequent therapy compared with IR-negative patients (Figs 3A and 3B). However, the observed relationship between antibody responses and clinical outcome might have been confounded by a guarantee time bias, because patients who remained free of progression would have had more opportunity to respond to continued vaccination, whereas patients with an early progression would have been taken off study and not received as many vaccinations.21,22 However, we could not identify such a confounding effect, neither in separately conducting landmark analyses (Figs 3A and 3B) nor in applying adjusted survival models accounting for the timing of these IRs in relation to follow-up and outcome (Data Supplement). Patients mounting anti-Id IRs by a prespecified landmark (fourth immunization) had both superior PFS (median, 2.9 v 1.2 years; P = .02; Fig 3A) and superior SALT-FS (median, not reached v 2.7 years; P = .03; Fig 3B) when compared with all other MyVax-immunized patients. Finally, although FLIPI retained univariable prognostic significance in this randomly assigned cohort for both PFS and SALT-FS (P = .024 and P = .021, respectively), MyVax-induced IRs remained independently prognostic of FLIPI risk for both end points in multivariable models (P = .002 and P = .016, respectively).
Fig 3.

Superior outcomes among patients mounting humoral immune response (IR) to idiotypic vaccination. Patients randomly assigned to receive MyVax were assessed for anti-idiotypic antibody responses and stratified into IR-positive (IR+) and IR-negative (IR−) designations based on prespecified criteria on serial serologic assessments, solely considering seroconversion before prespecified landmark for such evaluation. Kaplan-Meier curves show (A) progression-free survival and (B) subsequent antilymphoma therapy (SALT) –free survival of IR-positive versus IR-negative patients treated with MyVax. Exp, expected; HR, hazard ratio; Obs, observed.
DISCUSSION
B-cell lymphomas are attractive targets for immunotherapy because they have a unique surface immunoglobulin distinguishing tumor cells from normal cells. This prospective, randomized phase III trial was designed to evaluate the efficacy of personalized immunotherapy after chemotherapy. Similar to two related randomized studies of Id vaccination for FL,23 this study showed no significant difference between specific and control immunotherapy groups regarding PFS, the primary end point.
Our study examined the relationship between humoral IRs and clinical outcomes, a secondary end point. Notably, in the MyVax arm, patients mounting anti-Id IRs had significantly superior PFS when compared with patients without such responses and with patients in the control arm. This result replicates observations from previous phase II trials,10,13 confirming that patients with stronger IRs have better outcomes. Indeed, our trial is unique among the three randomized studies of Id vaccination for FL24,25 in relating any IR to clinical outcome. Nonetheless, even if humoral IRs were predictive of therapeutic benefit from specific immunotherapy, they currently occur in only 41% of patients, and ideally, future studies would need to improve immunogenicity of the vaccine product. Several phase II studies have demonstrated utility of cellular IRs as biomarkers of possible therapeutic benefit from Id vaccination.9,26,27 Because these assays require live cells from responding patients, they were impractical to perform in our multicenter setting.
An alternative explanation for the better outcomes of patients mounting IRs to MyVax is that such IRs are a complex biomarker for unmeasured intrinsic patient factors, in the absence of any therapeutic effect. For instance, IR could simply be a proxy for other host factors influencing the natural history of disease,28,29 such as each patient's general immunologic health, and these factors might have similarly stratified the control cohort if they could be prospectively measured. Thus, humoral response to any antigen might similarly stratify patient outcomes with no relation to therapeutic benefit.
Patients in the two study arms had nearly identical outcomes as measured by PFS, despite 41% of patients receiving MyVax mounting such IRs. In the MyVax arm, although not statistically significant, the subset of patients who did not mount such responses (ie, IR negative) seemed to have a trend toward inferior outcomes relative to patients in the control immunotherapy group. These differences may reflect a detrimental effect of MyVax in a subgroup of patients, as also suggested in another study.24 One potential mechanism for a negative effect might be the induction of suppressive T cells that prevent anti-Id antibodies and foster disease progression.
Including our study, there have been three randomized studies testing Id-KLH vaccines in patients with FL.24,25 All three studies used GM-CSF as an immunologic adjuvant to enhance IR. A study sponsored by Favrille (San Diego, CA) reported a median time to progression of 9 months for the active immunotherapy group, compared with 12.6 months in the control group (P = .02), suggesting a detrimental effect from active vaccination.24 However, this difference was attributed to the higher number of high-risk patients (by FLIPI) randomly assigned to the Id-KLH plus GM-CSF arm, because no difference was observed once the results were corrected for FLIPI risk. Furthermore, this trial used rituximab for remission induction, which is known to interfere with the capacity to generate anti-Id antibody responses.26
Another recently completed randomized study reported slightly improved outcomes for eligible patients who had a sustained CR with induction chemotherapy and received Id-KLH vaccines (Biovaxid; Biovest International, Tampa, FL) produced by rescue fusion technology.25 There were a number of differences between our study and the Biovaxid study. For instance, the Biovaxid study employed higher-intensity chemotherapy and also restricted eligibility to patients in CR, which for some patients needed to be sustained for longer than a year before their vaccine was available. In an unplanned analysis of our study, we noted that patients achieving sustained CR had similar PFS in the two arms. In the Biovaxid study, patients who had immunoglobulin M (IgM) –expressing tumors and received the IgM product had superior outcomes compared with patients who had IgG tumors and received the IgG product.25
In our trial, all patients received a product using the same IgG3 isotype, allowing us to test outcomes in relation to the isotype of the tumor. A reanalysis of our data according to the isotype of the tumors failed to confirm a relationship to clinical outcome, neither in our study nor in prior phase II studies. Nevertheless, the selection of the IgG3 constant region for manufacture of recombinant vaccines in our study may have obscured therapeutic effect. Host factors may contribute to differences in vaccine-induced immunity among patients, because genetic determinants and disease-related influences are known to affect vaccination responses.
Our study employed a chemotherapy induction regimen no longer considered standard of care for patients with advanced FL, resulting in a sustained overall response of only 56%. Future trials of vaccination after remission induction would likely employ more effective regimens, making more patients eligible. Superior induction chemotherapy regimens have now emerged that significantly improve responses when compared with the CVP regimen we employed or with the anthracycline-based regimen used in the Biovaxid study. In addition, all three randomized studies used GM-CSF as an immunologic adjuvant in both arms, an agent which may have affected clinical outcomes, suggesting the need for additional control arms in future trial designs.
Our study commenced before rituximab had become standard of care in therapy for FL.4,30 The other studies demonstrated significant improvements in outcomes when rituximab was used during or after chemotherapy of FL, with the magnitude of therapeutic effect from rituximab being at least as great as any possible benefit seen in our trial. Future trials of vaccination for FL will thus need to interdigitate with rituximab, which can blunt memory humoral IRs but has less effect on primary IRs.31,32 One approach to prime such responses could be to vaccinate patients after cytotoxic therapy but before B-cell depletion with rituximab. Here, it is notable that humoral IRs can indeed be augmented using immunomodulatory doses of cytotoxic agents,33 which have been proposed to exert this effect by inhibiting suppressive T regulatory cells and increasing humoral responses to KLH-conjugated vaccines.34–37 Alternatively, using newer technologies for rapid vaccine generation,38 it might even be possible to vaccinate as a primary maneuver, before the initiation of other therapies.
In conclusion, this trial failed to demonstrate a clinical benefit for patients receiving MyVax versus control immunotherapy, the primary end point of the study. However, unlike two prior randomized studies of Id vaccination for FL,24,25 our study confirmed a relationship between evoked humoral IRs and clinical outcome. Whether these superior outcomes are attributable to therapeutic benefit from immunization or are proxies for underlying host immunologic factors remains uncertain. The discovery and validation of biomarkers of immunologic and clinical responses are critical for identifying patients more likely to benefit from such therapy. Candidate biomarkers could include gene expression signatures of tumors, host genotypes, and specific features of the Ids themselves. In future Id vaccine studies, it will be important to develop more immunologically potent vaccine formulations and identify prospectively those patients most likely to mount anti-Id IRs and who thereby have a better chance of benefitting from such vaccination.
Supplementary Material
Acknowledgment
We thank the patients and families who participated in this study for their generosity and support as well as all the dedicated employees of Genitope, the nurses, patient coordinators, and staff at each of the participating Cancer Centers who made this work possible.
Footnotes
See accompanying editorial on page 1757
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00017290.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Dan W. Denney Jr, Genitope (C) Consultant or Advisory Role: None Stock Ownership: Dan W. Denney Jr, Genitope Honoraria: None Research Funding: Ronald Levy, Genitope; Ian W. Flinn, Genitope; Joseph M. Connors, F. Hoffmann-La Roche; John M. Timmerman, Genitope Expert Testimony: None Patents, Royalties, and Licenses: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Ronald Levy, Julie M. Vose, Joseph M. Connors, Lori A. Kunkel, Diane E. Ingolia, Ash A. Alizadeh, Dan W. Denney Jr
Financial support: Dan W. Denney Jr
Administrative support: Ronald Levy, John P. Leonard, Julie M. Vose, Ian W. Flinn, Joseph M. Connors, Neil L. Berinstein, Andrew R. Belch, Nancy L. Bartlett, Craig Nichols, John M. Timmerman, Stephanie A. Gregory, Brian K. Link, David J. Inwards, Arnold S. Freedman, Jeffrey V. Matous, Michael J. Robertson, Lori A. Kunkel, Ash A. Alizadeh, Dan W. Denney Jr
Provision of study materials or patients: Ronald Levy, Kristen N. Ganjoo, John P. Leonard, Julie M. Vose, Ian W. Flinn, Richard F. Ambinder, Joseph M. Connors, Neil L. Berinstein, Andrew R. Belch, Nancy L. Bartlett, Craig Nichols, Christos E. Emmanouilides, John M. Timmerman, Stephanie A. Gregory, Brian K. Link, David J. Inwards, Arnold S. Freedman, Jeffrey V. Matous, Michael J. Robertson, Lori A. Kunkel, Ash A. Alizadeh, Dan W. Denney Jr
Collection and assembly of data: Ronald Levy, Kristen N. Ganjoo, John P. Leonard, Julie M. Vose, Ian W. Flinn, Richard F. Ambinder, Joseph M. Connors, Neil L. Berinstein, Andrew R. Belch, Nancy L. Bartlett, Craig Nichols, Christos E. Emmanouilides, John M. Timmerman, Stephanie A. Gregory, Brian K. Link, David J. Inwards, Arnold S. Freedman, Jeffrey V. Matous, Michael J. Robertson, Lori A. Kunkel, Diane E. Ingolia, Chih Long Liu, Ash A. Alizadeh, Dan W. Denney Jr
Data analysis and interpretation: Ronald Levy, Kristen N. Ganjoo, John P. Leonard, Julie M. Vose, Ian W. Flinn, Richard F. Ambinder, Joseph M. Connors, Neil L. Berinstein, Andrew R. Belch, Nancy L. Bartlett, Craig Nichols, Christos E. Emmanouilides, John M. Timmerman, Stephanie A. Gregory, Brian K. Link, David J. Inwards, Arnold S. Freedman, Jeffrey V. Matous, Michael J. Robertson, Lori A. Kunkel, Diane E. Ingolia, Andrew J. Gentles, Robert Tibshirani, Ash A. Alizadeh, Dan W. Denney Jr
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Hiddemann W, Kneba M, Dreyling M, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: Results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood. 2005;106:3725–3732. doi: 10.1182/blood-2005-01-0016. [DOI] [PubMed] [Google Scholar]
- 2.Hochster H, Weller E, Gascoyne RD, et al. Maintenance rituximab after cyclophosphamide, vincristine, and prednisone prolongs progression-free survival in advanced indolent lymphoma: Results of the randomized phase III ECOG1496 study. J Clin Oncol. 2009;27:1607–1614. doi: 10.1200/JCO.2008.17.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcus R, Imrie K, Belch A, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood. 2005;105:1417–1423. doi: 10.1182/blood-2004-08-3175. [DOI] [PubMed] [Google Scholar]
- 4.Marcus R, Imrie K, Solal-Celigny P, et al. Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol. 2008;26:4579–4586. doi: 10.1200/JCO.2007.13.5376. [DOI] [PubMed] [Google Scholar]
- 5.Friedberg JW. Treatment of follicular non-Hodgkin's lymphoma: The old and the new. Semin Hematol. 2008;45(suppl):S2–S6. doi: 10.1053/j.seminhematol.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller RA, Maloney DG, Warnke R, et al. Treatment of B-cell lymphoma with monoclonal anti-idiotype antibody. N Engl J Med. 1982;306:517–522. doi: 10.1056/NEJM198203043060906. [DOI] [PubMed] [Google Scholar]
- 7.Kwak LW, Campbell MJ, Czerwinski DK, et al. Induction of immune responses in patients with B-cell lymphoma against the surface-immunoglobulin idiotype expressed by their tumors. N Engl J Med. 1992;327:1209–1215. doi: 10.1056/NEJM199210223271705. [DOI] [PubMed] [Google Scholar]
- 8.Eichmann K, Rajewsky K. Induction of T and B cell immunity by anti idiotypic antibody. Eur J Immunol. 1975;5:661–666. doi: 10.1002/eji.1830051002. [DOI] [PubMed] [Google Scholar]
- 9.Bendandi M, Gocke CD, Kobrin CB, et al. Complete molecular remission induced by patient-specific vaccination plus granulocyte-monocyte colony-stimulating factor against lymphoma. Nat Med. 1999;5:1171–1177. doi: 10.1038/13928. [DOI] [PubMed] [Google Scholar]
- 10.Hsu FJ, Caspar CB, Czerwinski D, et al. Tumor-specific idiotype vaccines in the treatment of patients with B-cell lymphoma: Long-term results of a clinical trial. Blood. 1997;89:3129–3135. [PubMed] [Google Scholar]
- 11.Inogès S, Rodrìguez-Calvillo M, Zabalegui N, et al. Clinical benefit associated with idiotypic vaccination in patients with follicular lymphoma. J Natl Cancer Inst. 2006;98:1292–1301. doi: 10.1093/jnci/djj358. [DOI] [PubMed] [Google Scholar]
- 12.Timmerman JM, Czerwinski DK, Davis TA, et al. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: Clinical and immune responses in 35 patients. Blood. 2002;99:1517–1526. doi: 10.1182/blood.v99.5.1517. [DOI] [PubMed] [Google Scholar]
- 13.Ai WZ, Tibshirani R, Taidi B, et al. Anti-idiotype antibody response after vaccination correlates with better overall survival in follicular lymphoma. Blood. 2009;113:5743–5746. doi: 10.1182/blood-2009-01-201988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuster SJ, Neelapu SS, Gause BL, et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J Clin Oncol. 2011;29:2787–2794. doi: 10.1200/JCO.2010.33.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freedman A, Neelapu SS, Nichols C, et al. Placebo-controlled phase III trial of patient-specific immunotherapy with mitumprotimut-T and granulocyte-macrophage colony-stimulating factor after rituximab in patients with follicular lymphoma. J Clin Oncol. 2009;27:3036–3043. doi: 10.1200/JCO.2008.19.8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timmerman JM, Vose JM, Czerwinski DK, et al. Tumor-specific recombinant idiotype immunisation after chemotherapy as initial treatment for follicular non-Hodgkin lymphoma. Leuk Lymphoma. 2009;50:37–46. doi: 10.1080/10428190802563355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaffe ES, Harris NL, Stein H, et al., editors. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2001. [Google Scholar]
- 18.Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas: NCI-sponsored international working group. J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 19.Cox D. Regression models and life-tables. J R Stat Soc B. 1972;34:187–220. [Google Scholar]
- 20.Solal-Céligny P, Roy P, Colombat P, et al. Follicular lymphoma international prognostic index. Blood. 2004;104:1258–1265. doi: 10.1182/blood-2003-12-4434. [DOI] [PubMed] [Google Scholar]
- 21.Anderson JR, Cain KC, Gelber RD. Analysis of survival by tumor response. J Clin Oncol. 1983;1:710–719. doi: 10.1200/JCO.1983.1.11.710. [DOI] [PubMed] [Google Scholar]
- 22.Bouwhuis MG, Suciu S, Collette S, et al. Autoimmune antibodies and recurrence-free interval in melanoma patients treated with adjuvant interferon. J Natl Cancer Inst. 2009;101:869–877. doi: 10.1093/jnci/djp132. [DOI] [PubMed] [Google Scholar]
- 23.Bendandi M. Idiotype vaccines for lymphoma: Proof-of-principles and clinical trial failures. Nat Rev Cancer. 2009;9:675–681. doi: 10.1038/nrc2717. [DOI] [PubMed] [Google Scholar]
- 24.Freedman A, Neelapu SS, Nichols C, et al. Placebo-controlled phase III trial of patient-specific immunotherapy with mitumprotimut-T and granulocyte-macrophage colony-stimulating factor after rituximab in patients with follicular lymphoma. J Clin Oncol. 2009;27:3036–3043. doi: 10.1200/JCO.2008.19.8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuster SJ, Neelapu SS, Gause BL, et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J Clin Oncol. 2011;29:2787–2794. doi: 10.1200/JCO.2010.33.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neelapu SS, Kwak LW, Kobrin CB, et al. Vaccine-induced tumor-specific immunity despite severe B-cell depletion in mantle cell lymphoma. Nat Med. 2005;11:986–991. doi: 10.1038/nm1290. [DOI] [PubMed] [Google Scholar]
- 27.Houot R, Levy R. Vaccines for lymphomas: Idiotype vaccines and beyond. Blood Rev. 2009;23:137–142. doi: 10.1016/j.blre.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 28.Cerhan JR, Wang S, Maurer MJ, et al. Prognostic significance of host immune gene polymorphisms in follicular lymphoma survival. Blood. 2007;109:5439–5446. doi: 10.1182/blood-2006-11-058040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dave SS, Wright G, Tan B, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351:2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 30.Salles G, Seymour JF, Offner F, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): A phase 3, randomised controlled trial. Lancet. 2011;377:42–51. doi: 10.1016/S0140-6736(10)62175-7. [DOI] [PubMed] [Google Scholar]
- 31.Takata T, Suzumiya J, Ishikawa T, et al. Attenuated antibody reaction for the primary antigen but not for the recall antigen of influenza vaccination in patients with non-Hodgkin B-cell lymphoma after the administration of rituximab-CHOP. J Clin Exp Hematop. 2009;49:9–13. doi: 10.3960/jslrt.49.9. [DOI] [PubMed] [Google Scholar]
- 32.Bedognetti D, Zoppoli G, Massucco C, et al. Impaired response to influenza vaccine associated with persistent memory B cell depletion in non-Hodgkin's lymphoma patients treated with rituximab-containing regimens. J Immunol. 2011;186:6044–6055. doi: 10.4049/jimmunol.1004095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berd D, Mastrangelo MJ, Engstrom PF, et al. Augmentation of the human immune response by cyclophosphamide. Cancer Res. 1982;42:4862–4866. [PubMed] [Google Scholar]
- 34.Bass KK, Mastrangelo MJ. Immunopotentiation with low-dose cyclophosphamide in the active specific immunotherapy of cancer. Cancer Immunol Immunother. 1998;47:1–12. doi: 10.1007/s002620050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacLean GD, Miles DW, Rubens RD, et al. Enhancing the effect of THERATOPE STn-KLH cancer vaccine in patients with metastatic breast cancer by pretreatment with low-dose intravenous cyclophosphamide. J Immunother Emphasis Tumor Immunol. 1996;19:309–316. doi: 10.1097/00002371-199607000-00006. [DOI] [PubMed] [Google Scholar]
- 36.Miles D, Roché H, Martin M, et al. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist. 2011;16:1092–1100. doi: 10.1634/theoncologist.2010-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miles D, Towlson K, Graham R, et al. A randomised phase II study of sialyl-Tn and DETOX-B adjuvant with or without cyclophosphamide pretreatment for the active specific immunotherapy of breast cancer. Br J Cancer. 1996;74:1292–1296. doi: 10.1038/bjc.1996.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanter G, Yang J, Voloshin A, et al. Cell-free production of scFv fusion proteins: An efficient approach for personalized lymphoma vaccines. Blood. 2007;109:3393–3399. doi: 10.1182/blood-2006-07-030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.