

Abstract
The complex heterogeneity of cells, and their interconnectedness with each other, are major challenges to identifying clinically relevant measurements that reflect the state and capability of the immune system. Highly multiplexed, single-cell technologies may be critical for identifying correlates of disease or immunological interventions as well as for elucidating the underlying mechanisms of immunity. Here we review limitations of bulk measurements and explore advances in single-cell technologies that overcome these problems by expanding the depth and breadth of functional and phenotypic analysis in space and time. The geometric increases in complexity of data make formidable hurdles for exploring, analyzing and presenting results. We summarize recent approaches to making such computations tractable and discuss challenges for integrating heterogeneous data obtained using these single-cell technologies.
A wide variety of analytical measurements can be used to characterize the state and capability of the immune system. The resulting data help reveal the fundamental biology of immunity, provide insight into the evolution of disease, aid the design of clinical diagnostics or interventions and establish distinct signatures for effective immune responses. Improving the resolution of our measurements to capture the full complexity encompassing time-varying states and interconnectedness of cell subsets presents a substantial challenge.
Leukocytes from both blood and tissue harbor a wealth of information, in their homeostatic state or after activation ex vivo. Cellular traits of interest include the absolute numbers and relative proportions of specific cell subsets, transcriptional states, secretory functions, proliferative capacity or cytolytic potential (Fig. 1a). Some analytical tools can provide multiparametric, albeit static, measurements of these traits, whereas others can resolve temporal variations intrinsic to an individual cell or after intercellular communication between neighboring cells. The particular design principles of a technology affect the extent to which these cellular traits can be resolved, ranging from average measurements of a population to precise and repeated measurements of single cells. Recently, there has been an increased emphasis on expanding the breadth and quality of single-cell analyses. In this Review, we examine the need for single-cell measurements, advances in the types of measurements and approaches for understanding how to integrate dense single-cell measurements computationally.
Figure 1.

Evolving landscape of cellular traits. (a) Schematic of cellular traits that can be measured in a static fashion (at a fixed point in time) and processes that imply dynamic movement of the system. (b) Combined view of activated T cells resolved by static measurements of state (left) by polychromatic flow cytometry and dynamic measurements of cytokine release (right) by serial microengraving with arrays of subnanoliter wells. T cells exhibiting different polyfunctional states (e.g., IFN-γ+, IL-2+, IFN-γ+IL-2+TNF+; color wheel) can have a range of surface-expressed markers indicating various states of maturity and differentiation based on flow cytometry (pie charts, in which each slice represents cells with a unique combination of the RA isoform of CD45 (CD45RA), CCR7, CD27 and CD57). TNF, tumor necrosis factor. Kinetic measurements of these functional states that temporally resolve dominant patterns of cytokine release show that a subset of trajectories represent the major kinetic profiles associated with T cells of varying maturity (e.g., naive, effector memory and central memory; top right). The confusion matrix shows the percentage of T cells in each differentiated state that are assigned to the correct state based only on the measured trajectories. Random assignment of state in this example is 25% (temporal trajectories and confusion matrix adapted from ref. 8).
Why do bulk measurements fail?
Many analytical technologies measure only the average response from a highly heterogeneous population of cells. These include enzyme-linked immunosorbent assays, reverse-transcription quantitative PCR (RT-qPCR) for transcriptional profiling and common assays of proliferation or cytolytic activity. Variations of these assays facilitate highly multiplexed or high-throughput analysis: bead arrays (for cytokine detection, such as Luminex technology) and gene microarrays use combinations of spectral and spatial delineation of analytes to measure dozens to thousands of parameters for a given sample. These tools for monitoring the immune system have provided exceptional insights into disease pathogenesis and immunity, including the detection (and discrimination) of respiratory viral pathogens1, high-throughput analysis of antibody responses in large studies of malaria and hepatitis B2, and multidimensional profiles of vaccine-induced T cells3. Nonetheless, these technologies make the analysis of rare subsets of cells (like antigen-specific T cells or B cells) challenging, if not impossible. Such cells contribute only a minor component of the total measurement. The implicit averaging of parameters in these measurements also masks the specific phenotypic state of these cells as well as the co-regulation of genes or proteins, the causality of events and the basic nature of interactions among cells.
To highlight the limitations of average measurements for monitoring the immune system, consider, by analogy, a game of soccer: a complex, dynamic network that evolves based on the responses and interactions of individual players, much like an immune response. An average measurement of the position of the ball (i.e., near midfield) would not reveal the importance of the goals (i.e., positioned at the extremes); a simple enumeration of the numbers of players would fail to inform the rules governing how they interact in time and situation. Similarly, a snapshot of the field at the time of a goal provides little insight into the events that led to the result. Comprehensive understanding requires fine resolution of the key players, their interactions over time and their influence on the outcome of a game, in a variety of situations.
When monitoring immune systems, the interplay between different cell types and the impact of functional responses reflect the roles of players and goals, respectively. The process can be documented at coarse or fine resolution, using either low-parameter, bulk cell analyses or high-parameter, single-cell measurements. The ability to resolve many different classes of traits as well as numerous parameters within a class is critical for understanding the state and evolution of an individual cell. For example, examination of T cells using highly multiplexed polychromatic flow cytometry (PFC4) reveals that the classification of T cells into discrete states according to function, maturity or other properties is not straightforward5, as has been shown using data from enzyme-linked immunosorbent assays, enzyme-linked immunospot or 2–3-parameter flow cytometry6. Dozens of molecules are differentially expressed across naive, memory and effector T cells, yet central memory, effector and terminal cells can all exhibit polyfunctionality (producing multiple cytokines7) when examined intracellularly (Fig. 1b). Adding temporal information to such single-cell measurements, however, can help resolve this ambiguity. Kinetic measurements of the ordering and timing of cytokines secreted by activated T cells can additionally help discriminate subsets of differentiated T cells by function (Fig. 1b)8. This example highlights how even technologies with fine resolution have limitations when used for static, snapshot measurements and implies that any single measurement may be intrinsically noisy. To enable identification of the unique and robust combination of traits that defines a critical subset of cells in an immunological response, technologies for immune monitoring should emphasize both depth and breadth of measurements with single-cell resolution.
Expanding multiplexed depth for flow cytometry
State-of-the-art systems for PFC yield up to 20 distinct channels of data for a given cell and can routinely process millions of cells per sample in a few minutes5. In the last decade, the number of parameters that can be scored simultaneously has increased dramatically, through the development of new fluorescent dyes and ‘tandems’ (conjugates of a new fluorescent dye with existing dyes), providing a variety of excitation and emission spectra5,9. The greater selection of dyes has also enabled combinatorial strategies for labeling similar variants of reagents, such as peptide-loaded major histocompatibility complex (MHC) multimers, to enhance the resolution of subsets of cells (e.g., antigen-specific T cells or B cells)10; the ability to ‘barcode’ samples (with fluorescent tags of varying brightness) has increased sample throughput11. Advances in chemistries for fluorophores and strategies for labeling will lead to increasing numbers of parameters for PFC in the coming years.
The recent commercialization of spectral flow cytometry (first reported 20 years ago by groups in Los Alamos, New Mexico, USA) provides a path toward more sophisticated analysis of fluorescence-based signals12,13. Unlike conventional flow cytometry, spectral analyzers collect the complete fluorescence emission spectrum from all fluorochromes at once. The spectrum is then deconvoluted or unmixed (functionally equivalent to compensation) to quantify individual fluorochromes. A theoretical advantage of this approach is that fluorochromes with highly overlapping emission spectra can be distinguished more accurately than by conventional flow cytometry. In addition, biological processes that affect emission spectra can be resolved, providing additional data. This technology, however, has not yet been widely adopted or tested, so it remains unclear whether spectral analyzers can expand the number of parameters available for measurement beyond those available with conventional flow cytometry.
The integration of mass spectrometry with single-cell fluidics has recently extended the number of parameters that can be measured per cell as well. Such ‘mass cytometry’ largely overcomes the challenge of spectral overlap intrinsic to fluorescence dyes by using rare-earth-metal isotopes detected distinctly (with little signal overlap)14. Currently, 37 such isotopes are available, with a projected theoretical limit of 50–100 (ref. 15). Developing staining panels is straightforward, and in principle, many independent cell-associated parameters, such as phosphorylated molecules, intracellular cytokines and surface proteins, can be interrogated simultaneously16. Reduced biological background, because of the lack of rare-earth-metal isotopes in biological systems, is another potential benefit of the technology. In practice, however, nonspecific binding of antibodies remains an intrinsic limitation of all antibody-based assays and yields a detectable background signal that tempers this theoretical advantage (Fig. 2).
Figure 2.

Antibody staining in mass cytometry. (a) Results of mass cytometry analyses, demonstrating variable staining quality among reagents. Some isotope-tagged antibodies (about 60% of those we have tested) yield very high staining indices, a measure of detection sensitivity (e.g., anti-human IFN-γ Ho165; left). This antibody does show some binding to unstimulated cells (’unstimulated’). Other isotope-tagged antibodies (about 20%) have much lower staining indices (e.g., anti-human CD45RA-Eu153; center), and nonspecific signal is seen even at low concentrations of antibody. The remaining antibodies poorly resolve from background at any concentration (e.g., anti-human CD4-Yb174; right). (b) CD19 and CD20 staining patterns by mass (left) and flow cytometry (middle and right) demonstrate the importance of light-scatter gating (LSG) to eliminate myeloid cells, which often display considerable background when analyzing lymphocytes. This morphologically related signal is not available yet in mass cytometry. (c) Discrimination of singlet signals from multiple cells by mass cytometry typically relies on DNA-intercalator staining (far left); the stringency of this gate, however, greatly influences the percentage of cells classified as positive for a given marker (middle left) but also eliminates progressively greater fractions of collected data. Staining for some markers (e.g., anti-human CD27-Er167; middle right) poorly separates from background, resulting in impure populations. These lead to an inability to discern if the presence of CD127+CD28+ cells in both CD27+ and CD27− cells (far right) reflects true biology, impure gating or poor doublet discrimination.
To date, data from mass cytometry have been reported for phenotypic and functional analyses of subsets of lymphocytes14. Studies of antigen-specific T cells, for example, have recapitulated known biology and revealed the remarkable interconnectedness of memory T cell subsets, highlighting an inherent benefit of highly multiplexed, deep profiling of cells. Results of such analyses can be examined to uncover new insights16.
Nonetheless, mass cytometry remains in its infancy. Phenotyping of cells may be limited by the poor sensitivity of many reagents. The quality of reagents can vary widely, with many providing no better resolution of positive and negative cells than fluorescence-based measurements with poor signal-to-noise ratio traits (Fig. 2). These limitations have practical consequences: the signal ranges for certain cell markers often overlap with background signals (Fig. 2), hindering classical immunophenotyping where clear definition of positive and negative signals has been essential. Likewise, first mass cytometers have a lower throughput than flow cytometers, with a 30–100-fold lower maximum rate of acquisition. As a result, enumerating rare events (for example, antigen-specific T cells) may be burdensome15, though recent approaches to barcode multiple samples by distinct mass labels for processing in parallel can substantially reduce the time required for each17.
Although these technical considerations are important to recognize, the technology has considerable power for certain lines of research. Even with overlapping positive and negative signals for some markers, it may be feasible to resolve populations of cells by clustering in other dimensions (depending on the particular biological context)14. Computational tools, such as ‘spanning-tree progression analysis of density-normalized events’ (SPADE)18, can provide unique visualization options for major cell populations but may be problematic for rare subsets of cells, such as antigen-specific cells or those associated with minimal residual disease (MRD). Notably, for MRD, a new computational tool called visual interactive stochastic neighbor embedding (viSNE) can offer more robust phenotyping of rare subsets than SPADE19. An alternative, nonclustering approach may be particularly relevant for phosphorylation studies, in which biological effects may be inferred by shifts in the intensity of the marker. Such shifts are sensitive indicators of upregulation or downregulation of cell-signaling molecules, providing a wealth of data on the effects and potency of pharmaceutical compounds17. Thus, mass cytometry has been particularly successful in the analysis of cell signaling and integrated measurements of function.
The combination of fluorescence flow cytometry with microscopy imaging is another recent development in cytometry. Two-dimensional images provide several new dimensions of data per cell beyond the magnitude of a given signal, including the morphology and spatial localization of proteins. Advances in such technologies (e.g., Imagestream) that allow both rapid acquisition and data processing enable measurements of thousands of cells per second, offer statistical and throughput advantages over conventional microscopy-based imaging platforms. Such systems have been particularly useful for monitoring cellular morphology20, nuclear localization of transcription factors during cell-signaling events21,22, protein localization to immune synapses23,24 or cellular uptake of fluorescent particles25,26.
Adding breadth to single-cell analysis
PFC, mass cytometery and imaging flow cytometry offer an extensive depth of information for single cells but provide only a single snapshot in time. A number of technologies are maturing to provide new classes of dynamic functional and transcriptional measurements. These complement and augment the types of data available from flow cytometry–based systems. The technologies rely on advanced capabilities for fabricating microliter-scale to picoliter-scale systems that permit precise handling and measurements of cells or single-cell lysates. These ‘microtools’ alone, or in conjunction with traditional single-cell technologies, are opening many new avenues of research into the functions of single cells.
To date, two dominant classes of microtools have emerged for use in monitoring the status of the immune system: microfluidic systems and simple spatial arrays of nanoliter-scale wells (Fig. 3a,b)27–29. Commercial actively valved microfluidic chips from Fluidigm now enable thousands of parallel qPCR reactions for up to 96 samples (e.g., individual cells) at a time. Similar microfluidic devices also multiplex the preparation of up to 96 cells for single-cell sequencing (whole-genome and RNA sequencing)30 and facilitate multiplexed measurements of single-cell secretomes (Fig. 3c)31. The ability to position cells and modulate fluidic environments precisely in valved microfluidics systems, however, can require extensive control elements, limiting both the total numbers of cells processed per device and their scalability to processing the large numbers of cells often needed for monitoring immune status.
Figure 3.

Classes of microtools for single-cell analysis. (a) Schematic of a microfluidic system (design is based on ref. 30) with integrated pneumatically actuated valves (orange lines). Cells, reagents and medium flow from left to right in the blue channels. Pressurization of the pneumatic lines seals regions of the blue channels to trap cells. The expanded view shows one region of trapped cells where the nominal volumes are ~1 nl. A representative region of the system is shown in the micrograph (adapted from ref. 30); numbers of cells per trap are indicated, and the overlaid fluorescence indicates regions for antibody-based immunoassays to capture cytokines (red) and reference marks (green). (b) Schematic of an array of subnanoliter wells, with the magnified region showing one block of wells with nominal volumes of ~100 pl each. The composite fluorescence micrograph shows a set of wells containing human lymphocytes with barcoded labels to identify samples. (c) Heatmaps (adapted from ref. 30) showing amounts of 12 secreted factors captured from single activated T cells (each line shows data associated with one cell) in a microfluidic system. Data are for CD8+ T cells from three healthy subjects (‘normal’; left) and MART-1+ T cell antigen receptor (TCR) transgenic T cells from a melanoma patient (right). (d) Independent cytolytic behavior of NK cells engaging human leukocyte antigen (HLA)-deficient tumor cells. Scatter plots show nuclear staining of target tumor cells measured by SYTOX staining and the intracellular staining of the target cells after 4 h in coculture with 0, 1 or 2 NK cells. MFI, mean fluorescence intensity. Insets, representative micrographs of the configurations of cells in wells scored for each condition. Graphs show measured rates of cytolytic activity (solid points and lines) compared to the predicted values based on an independence probability model (dashed lines) as a function of the number of NK cells per well. Each color represents a unique donor; the NK cells from each subject were used with no stimulation (medium) ex vivo or upon activation as indicated (reprinted from ref. 36).
In contrast, simple dense arrays that contain tens of thousands of addressable, subnanoliter compartments make it straightforward to isolate and monitor large numbers of single cells in parallel. These devices enable many new methods of studying unique aspects of single-cell phenotypes at scales relevant for detecting rare cells such as antigen-specific T cells or B cells32–34, for revealing structure-function relationships between molecular synapses and functional behaviors35, and for describing variance among highly heterogeneous populations36,37. One disadvantage of this system, however, is the limited control on the fluidic environment on a cell-by-cell basis.
An important feature of both technologies is the ability to accommodate very small clinical samples, such as cells from cytobrushes and biopsies, opening up opportunities to compare the functions and phenotypes of cells from mucosal tissues in the context of disease and interventions. Microtools also offer unique capabilities to resolve the spatiotemporal dynamics of immune cells that are not possible by flow cytometry or bulk measurements. With these tools, the exact address of each cell is determined by imaging throughout an experiment, enabling multiple measurements for the same class of data over time, or combinations of measurements of multiple classes, in the same experiment. Examples of traits measured for individual cells include protein secretion31,38–41, cell-surface markers8, motility42,43, cytotoxicity36,44,45, morphology46, proliferation47 and an endpoint analysis of intracellular protein or transcript expression48–50. The spatial confinement of cells in a microsystem allows, therefore, customized bioanalytical processes to be constructed, using modular operations to examine complex functional and phenotypic behaviors51. For example, serial measurements of activated T cells can be used to monitor the evolution of secretory states or surface-expressed proteins with time8,47. Such measurements suggest that the initiation of cytokine secretion varies among T cells, even with similar differentiation states (memory and naive) and that the kinetic trajectories of cytokines secreted by polyfunctional T cells, rather than the timing of release or cumulative set of cytokines produced, associate closely with the differentiated state of the T cells8. This new temporal resolution of complex polyfunctional states implies that observations made by intracellular staining can hide dynamic transitions of functional states and that these types of transitions themselves could provide new markers for classifying cells.
Microtools also provide a unique ability to examine intercellular communication with precision not possible in bulk co-cultures. Spatial confinement of discrete populations of individual cells in arrayed compartments makes it feasible to assess direct functional outcomes of intercellular signaling and the dynamics of interactions. For example, cocultures of effector and target cells can be combined such that individual compartments contain a distinct number of each. In this way, antigen-specific interactions between individual T cells and antigen-presenting cells can be monitored for effector events such as cytolysis and cytokine release. Such measurements have suggested that HIV-specific CD8+ T cells contacting target cells with cognate antigen can respond initially either by cytolysis or by secretion of IFN-γ36. Similar approaches to examine the motility of NK cells during engagement with target tumor cells has also revealed distinct dynamic traits sufficient for classifying subsets of cells based on these measurements37,52.
Aside from providing single-cell resolution of the spatiotemporal and functional dynamics of intercellular interactions, discrete co-cultures in microtools offer an entirely new approach to examining intercellular signaling networks. When cells are deposited into compartments, their distribution across those compartments follows Poisson statistics. As a result, there are quantized configurations of many groupings present on the same device (for example, NK cell: target ratios of 1:1, 2:1, 1:2 and so on). This feature makes it possible to measure cellular behaviors as a function of cell number. That is, unique traits such as cooperativity among cells can be assessed directly. For example, using this approach and simple probability models, it has been shown that NK cells do not cooperate in a paracrine manner to eliminate local target cells (Fig. 3d)37. A similar approach using valved microfluidics has been used to examine the cooperative effects of intercellular distances on intracellular signaling and secretion for tumor cell lines48. This ‘quantum’ approach to monitoring the immune system addresses one inherent limitation of approaches to single-cell analysis: that the behaviors of isolated cells can inform the potential range of states accessible to cells but may not fully reflect in vivo processes (in which cell-cell interactions are essential). Such capabilities provide entirely new means for assessing the cooperative behaviors of cells during such interactions, for modeling intercellular signaling networks that form the immune system and, potentially, for defining new signatures of immune status.
For monitoring the state of the immune system, applications for valved microfluidic systems and arrays of nanoliter-scale wells, as well as related systems such as ‘droplet’ microfluidics53,54, are still nascent, but these systems are poised to complement existing single-cell technologies such as flow cytometry. One example of how microtools can complement traditional flow cytometry is the combination of cell-associated immunophenotype with single-cell transcriptional profiles. Linking flow cytometry with microtools for single-cell analysis can offer two important benefits. First, it allows enrichment of specific populations of cells in a precise and scalable manner, before transcriptional analysis. This enrichment makes the analysis of rare events feasible and establishes a clear structure for comparisons among different groups55. Such classification is critical for meaningful analysis of the highly variable and multiplexed data from transcriptional studies. Second, combining orthogonal measurements, such as the expression of protein and mRNA, for the same cell may reveal discordances (for example, cells expressing a protein but not the cognate mRNA) relevant to its biological state or provide a unique correlate of response to disease or an intervention. Such cells may be in a transitional state that could not be identified if separate studies of protein and mRNA expression were performed. The presence or absence of such transitional cells underscores an important characteristic of dynamic biological systems, such as the immune system.
An important extension of transcriptomic technologies, single-cell RNA sequencing (scRNA-seq), is emerging. In principle, scRNA-seq enables genome-wide, unbiased profiling of cellular mRNA expression, increasing information content recovered per cell and improving discovery-oriented processes, relative to RT-qPCR–based approaches. The technology also enables analysis of other transcriptional features in single cells, such as splice variants and allele-specific expression, and the discovery of new genes. Though still in its infancy, it has already revealed subsets of cells not previously observed using other single-cell measurements56 and shown that cell-specific splicing56 and allele expression patterns57 can differ significantly from the pattern averaged over the population. All these parameters may have considerable effects on the function of individual cells and their influence on a population as a whole, yet they were impossible to observe in an unbiased fashion with previous single-cell methodologies. Methodologies for scRNA-seq are still immature, however. Optimal methods can only be used to acquire reliable expression estimates for transcripts expressed at medium to high levels56 and like microvalved RT-qPCR platforms, scRNA-seq is limited to the characterization of small numbers of cells (<100) at a time. Despite these current limitations, scRNA-seq paired with cell enrichment using PFC promises to greatly expand our understanding of the landscape of static single- cell phenotypes.
Data integration and analysis for understanding
Rapidly advancing technologies for single-cell analysis make it possible to generate substantial sets of data comprising many parameters and classes of data per cell. Each technology, however, presents distinctive data types, breadth and structure that encompass only portions of the landscape of cells, parameters and time (Fig. 4). Integrating measurements obtained using multiple tools, such as flow cytometry–based proteomics, PCR-based transcriptomics and time-resolved microtool-enabled measurements of function, can expand the breadth of coverage but also amplifies the complexity of datasets. The nature of these data emphasizes the need for the concurrent development of tools for data integration and thoughtful approaches to analysis that facilitates biological understanding of single-cell measurements.
Figure 4.

Relative structure of data from single-cell analyses. Each major class of technology for single-cell analysis is plotted schematically in the three-dimensional data cube represented by the numbers of cells measured, the numbers of parameters scored and breadth of temporal resolution afforded. The specific axes of cells and measurements are not drawn to scale but emphasize relative differences in each measurement. Overlapping regions indicate opportunities to integrate complementary technologies and highlight orthogonality in associated data structures that may arise from each.
When the data structures of single-cell analysis tools are examined, the extent to which the landscape of potential states is sampled becomes evident. For example, an experiment using 15-color flow cytometry can generate over 14 million different possible phenotypes, given that a phenotype could be defined based on the presence, absence or ignorance of each marker (i.e., 315 combinations). Typically, measurements on hundreds of thousands of single cells are collected per experiment, thereby sampling a tiny fraction of the potential phenotypic space. This problem grows exponentially with measurement capacity. For example, by measuring the expression of 96 genes using Fluidigm platform results in 1045 possible states. Clearly, with such complexity, each cell could occupy a unique cluster; thus, a prime problem for data analysis is a reduction in dimensionality without loss of pertinent information. The development of new computational algorithms and techniques is an active area of research; currently, our ability to generate data far outstrips our ability to analyze it.
With highly multiplexed single-cell data, new computational approaches are necessary to identify the key features that distinguish experimental groups. Tools commonly used in the analysis of gene expression such as unsupervised hierarchical clustering have, in our experience, not typically been successful for identifying biologically meaningful clusters of cells with data from PFC, microtools or 96-parameter RT-qPCR. Pairwise comparisons for differential expression of markers across groups can be used, but the sensitivity of this method is limited by the statistical corrections needed for multiple comparisons. Moreover, the low frequency of expression for many markers may be below the limit of detection for the technology.
Members of the FlowCAP58 consortium tested a variety of new approaches to the analysis of complex data sets. Two approaches, flow-Type with RchyOptimyx59,60 and SPADE18, ranked highly for their ability to reproduce manual gating results and/or identify correlates of clinical outcomes58. These tools, however, differ in one key respect: the flowType pipeline is designed to examine all markers measured in an experiment, whereas SPADE includes a screening step to reduce the noise contributed by irrelevant markers. Results can vary, depending on the approach used; therefore, the biological question must be carefully considered when choosing data-visualization or analysis tools and conclusions must be validated or rationalized appropriately. Both tools can be used in higher-parameter, gene expression data sets, although the number of events present for any given multidimensional phenotype can be far too low to provide any statistical power.
These complexities should be considered, not only when analyzing high-dimension data from studies monitoring status of the immune system but also when designing experiments. In particular, reagent panels must be carefully constructed, to ensure that the quality and utility of data is maximized55,61–63. To ease this process, qualified reagents can be grouped into ‘cassettes’ based on the shared or complementary biology of the markers they detect. Thus, various cassettes can be constructed to include markers of regulatory T cells, cytotoxic T cells or monocytes, for example, and the cassette best-suited to the experimental setting can be chosen for ‘off-the-shelf ’ use. Over time, each cassette can be refined to include new markers of interest or exclude markers that provide no meaningful information. There are several advantages to this approach, including reduced complexity of unsupervised analyses, simplified data interpretation, less stringent multiple-comparisons adjustments and decreased noise from irrelevant markers.
Conclusions and outlook
The technologies available for analyzing immune responses with single-cell resolution are advancing rapidly, with the creation of new classes of fluorescent probes to extend parameterization in flow cytometry, the innovation of (rare element) mass-based labeling and the revolution of new classes of microfabricated technologies that offer alternative dimensions of data (for example, kinetics, transcriptomics and cell-cell communication). Across the suite of technologies (Box 1 and Fig. 5), it is now feasible to generate dense data on the state and behaviors of single cells, for a broad range of clinical samples from blood to mucosal tissues. Although advancement of specific technologies will continue, the wealth of measurements available from the combination of these tools offer new opportunities to define the underlying state of an individual cell of the immune system and its role in the context of both diseases and interventions.
Box 1. Traversing the evolving landscape of single-cell technologies.
When selecting the best single-cell technologies to address a given biological question, a number of factors should be considered:
The breadth and accessibility of technologies for single-cell analysis is rapidly expanding the scope of characterization that is feasible.
The maturity of technologies ranges from decades for flow cytometry to only a couple years for single-cell transcriptional analysis by high-throughput sequencing.
Technologies in the early stages of maturation (microtools, mass cytometry and scRNA-seq), still require commercial development of reagents and software for data analysis, as well as further validation in relevant clinical studies, before they are routinely used assays.
Emerging technologies offer unique capabilities such as longitudinal single-cell measurements (microtools) and highly multiplexed characterization (mass cytometry and RNA-seq).
Characterizing cells in solitary confinement ex vivo may not reflect in vivo processes that result from an integrated summation of cooperative actions from many cells over time.
Massively parameterized data reflecting protein or gene expression for single cells can add substantial complexities in data analysis, as data structures and classes may be novel.
Large datasets from single-cell measurements can hinder throughput and introduce statistical-analysis challenges arising from multiple comparisons.
All single-cell technologies are, in general, more costly compared to bulk measurements.
For many large clinical studies, simpler and less costly bulk measurements may more easily establish clinical correlates (for example, antibody titers for vaccine responsiveness).
Single-cell analysis is most useful when seeking insights into how unique subsets of cells may associate with outcomes, when rare cells are essential for conferring protection or inducing pathology of disease (for example, antigen-specific T cells) or when establishing systems-level models that describe mechanisms of responses.
Figure 5.
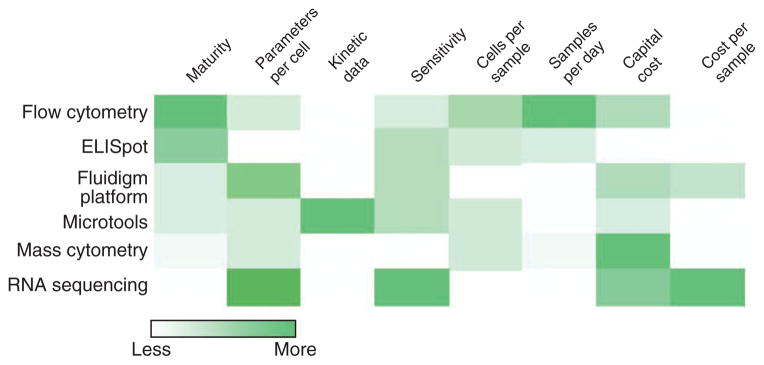
Comparative analysis of different single-cell technologies.
Static measurements of cells, such as antigen-specific T cells, reveal that they exist in a complex landscape of conditions, and dynamic measurements of these states are beginning to uncover how cells transition among states in less stochastic and more programmatic ways than previously recognized. These insights suggest the state and behaviors of individual cells are—by themselves—‘noisy’ when considered on the basis of small numbers of measurements. Embracing the integration of single-cell measurements, in combination with computational tools for data analysis and modeling, provides an experimental framework for understanding the essential features of immune responses that correlate with useful outcomes and for helping establish minimal sets of measurements critical in clinical monitoring.
Acknowledgments
This work was supported by the W.M. Keck Foundation and the US National Institute of Allergy And Infectious Diseases (1U19AI089992, 1R56AI104274 and 5R21AI106025). The content is solely the responsibility of the authors and does not necessarily represent the official views of the US National Institute of Allergy And Infectious Diseases or the US National Institutes of Health. We thank A. Shalek for helpful comments on scRNA-seq and N. Aghaeepour for discussions about data-analysis tools. J.C.L. is a Camille Dreyfus Teacher-Scholar. We acknowledge the service to the MIT community of the late Sean Collier.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Mahony J, et al. Development of a respiratory virus panel test for detection of twenty human respiratory viruses by use of multiplex PCR and a fluid microbead-based assay. J Clin Microbiol. 2007;45:2965–2970. doi: 10.1128/JCM.02436-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambrosino E, et al. A multiplex assay for the simultaneous detection of antibodies against 15 Plasmodium falciparum and Anopheles gambiae saliva antigens. Malar J. 2010;9:317. doi: 10.1186/1475-2875-9-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaucher D, et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J Exp Med. 2008;205:3119–3131. doi: 10.1084/jem.20082292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 5.Chattopadhyay PK, et al. Quantum dot semiconductor nanocrystals for immunophenotyping by polychromatic flow cytometry. Nat Med. 2006;12:972–977. doi: 10.1038/nm1371. [DOI] [PubMed] [Google Scholar]
- 6.Chattopadhyay PK, Roederer M. Good cell, bad cell: flow cytometry reveals T-cell subsets important in HIV disease. Cytometry A. 2010;77:614–622. doi: 10.1002/cyto.a.20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73:975–983. doi: 10.1002/cyto.a.20643. This review describes the traits and functions of classically defined T cell subsets, highlighting the uncertainties of profiling the immune system. The authors demonstrate that different pathogens induce different profiles of T cell responses, that there is no consensus for naming and describing T cell subsets, that functional attributes can vary upon activation and that there is little data that comprehensively correlate phenotypic or functional traits with effective immunity. [DOI] [PubMed] [Google Scholar]
- 8.Han Q, et al. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc Natl Acad Sci USA. 2012;109:1607–1612. doi: 10.1073/pnas.1117194109. Arrays of subnanoliter compartments are used to monitor the kinetic expression of four cytokines over 17 hours from thousands of activated T cells, demonstrating that the kinetic trajectory of cytokine release can predict T cell effector phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chattopadhyay PK, et al. Brilliant violet fluorophores: a new class of ultrabright fluorescent compounds for immunofluorescence experiments. Cytometry A. 2012;81:456–466. doi: 10.1002/cyto.a.22043. [DOI] [PubMed] [Google Scholar]
- 10.Newell EW, Klein LO, Yu W, Davis MM. Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat Methods. 2009;6:497–499. doi: 10.1038/nmeth.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat Methods. 2006;3:361–368. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- 12.Nolan JP, Condello D. Current Protocols in Cytometry. John Wiley & Sons, Inc; 2001. Spectral flow cytometry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanders CK, Mourant JR. Advantages of full spectrum flow cytometry. J Biomed Opt. 2013;18:037004. doi: 10.1117/1.JBO.18.3.037004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. This article introduced mass cytometry by demonstrating the power of highly multiparametric technology and analysis. From a single sample, the entire hematopoietic system could be recapitulated and each subset examined for differences in cell signalling profiles. The paper also introduced SPADE (ref. 18), a tool commonly recommended for analysis of mass cytometry datasets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends Immunol. 2012;33:323–332. doi: 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–152. doi: 10.1016/j.immuni.2012.01.002. Mass cytometry was used to demonstrate the heterogeneity of antigen-specific T cell responses and the complexity of T cell differentiation pathways. The authors propose that T cells specific for different viruses reside in different niches in a complex data space, and that the combination of mass cytometry and an adapted form of principal-component analysis can reveal these niches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30:858–867. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu P, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amir el AD, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Safeukui I, et al. Surface area loss and increased sphericity account for the splenic entrapment of subpopulations of Plasmodium falciparum ring-infected erythrocytes. PLoS ONE. 2013;8:e60150. doi: 10.1371/journal.pone.0060150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maguire O, Collins C, O’Loughlin K, Miecznikowski J, Minderman H. Quantifying nuclear p65 as a parameter for NF-κB activation: correlation between ImageStream cytometry, microscopy, and Western blot. Cytometry A. 2011;79:461–469. doi: 10.1002/cyto.a.21068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity. 2012;36:374–387. doi: 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beum PV, et al. Quantitative analysis of protein co-localization on B cells opsonized with rituximab and complement using the ImageStream multispectral imaging flow cytometer. J Immunol Methods. 2006;317:90–99. doi: 10.1016/j.jim.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 24.Wabnitz GH, et al. L-plastin phosphorylation: a novel target for the immunosuppressive drug dexamethasone in primary human T cells. Eur J Immunol. 2011;41:3157–3169. doi: 10.1002/eji.201041366. [DOI] [PubMed] [Google Scholar]
- 25.Catania A, Barrajon-Catalan E, Nicolosi S, Cicirata F, Micol V. Immunoliposome encapsulation increases cytotoxic activity and selectivity of curcumin and resveratrol against HER2 overexpressing human breast cancer cells. Breast Cancer Res Treat. 2013;141:55–65. doi: 10.1007/s10549-013-2667-y. [DOI] [PubMed] [Google Scholar]
- 26.Kamphuis MM, et al. Targeting of cancer cells using click-functionalized polymer capsules. J Am Chem Soc. 2010;132:15881–15883. doi: 10.1021/ja106405c. [DOI] [PubMed] [Google Scholar]
- 27.Lindstrom S, Andersson-Svahn H. Overview of single-cell analyses: microdevices and applications. Lab Chip. 2010;10:3363–3372. doi: 10.1039/c0lc00150c. [DOI] [PubMed] [Google Scholar]
- 28.Yin HB, Marshall D. Microfluidics for single cell analysis. Curr Opin Biotechnol. 2012;23:110–119. doi: 10.1016/j.copbio.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Yalcin A, Yamanaka YJ, Love JC. Analytical technologies for integrated single-cell analysis of human immune responses. Methods Mol Biol. 2012;853:211–235. doi: 10.1007/978-1-61779-567-1_16. [DOI] [PubMed] [Google Scholar]
- 30.Moonsamy PV, et al. High throughput HLA genotyping using 454 sequencing and the Fluidigm Access Array System for simplified amplicon library preparation. Tissue Antigens. 2013;81:141–149. doi: 10.1111/tan.12071. [DOI] [PubMed] [Google Scholar]
- 31.Ma C, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–743. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradshaw EM, et al. Concurrent detection of secreted products from human lymphocytes by microengraving: cytokines and antigen-reactive antibodies. Clin Immunol. 2008;129:10–18. doi: 10.1016/j.clim.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varadarajan N, et al. Rapid, efficient functional characterization and recovery of HIV-specific human CD8(+) T cells using microengraving. Proc Natl Acad Sci USA. 2012;109:3885–3890. doi: 10.1073/pnas.1111205109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sendra VG, Lie A, Romain G, Agarwal SK, Varadarajan N. Detection and isolation of auto-reactive human antibodies from primary B cells. Methods. 2013;64:153–159. doi: 10.1016/j.ymeth.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torres AJ, Contento RL, Gordo S, Wucherpfennig KW, Love JC. Functional single-cell analysis of T-cell activation by supported lipid bilayer-tethered ligands on arrays of nanowells. Lab Chip. 2013;13:90–99. doi: 10.1039/c2lc40869d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varadarajan N, et al. A high-throughput single-cell analysis of human CD8(+) T cell functions reveals discordance for cytokine secretion and cytolysis. J Clin Invest. 2011;121:4322–4331. doi: 10.1172/JCI58653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamanaka YJ, et al. Single-cell analysis of the dynamics and functional outcomes of interactions between human natural killer cells and target cells. Integr Biol. 2012;4:1175–1184. doi: 10.1039/c2ib20167d. [DOI] [PubMed] [Google Scholar]
- 38.Zhu H, et al. Detecting cytokine release from single T-cells. Anal Chem. 2009;81:8150–8156. doi: 10.1021/ac901390j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han Q, Bradshaw EM, Nilsson B, Hafler DA, Love JC. Multidimensional analysis of the frequencies and rates of cytokine secretion from single cells by quantitative microengraving. Lab Chip. 2010;10:1391–1400. doi: 10.1039/b926849a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin A, et al. A rapid and efficient single-cell manipulation method for screening antigen-specific antibody-secreting cells from human peripheral blood. Nat Med. 2009;15:1088–1092. doi: 10.1038/nm.1966. [DOI] [PubMed] [Google Scholar]
- 41.Lu Y, et al. High-throughput secretomic analysis of single cells to assess functional cellular heterogeneity. Anal Chem. 2013;85:2548–2556. doi: 10.1021/ac400082e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khorshidi MA, et al. Analysis of transient migration behavior of natural killer cells imaged in situ and in vitro. Integr Biol. 2011;3:770–778. doi: 10.1039/c1ib00007a. [DOI] [PubMed] [Google Scholar]
- 43.Hong S, Pan Q, Lee LP. Single-cell level co-culture platform for intercellular communication. Integr Biol. 2012;4:374–380. doi: 10.1039/c2ib00166g. [DOI] [PubMed] [Google Scholar]
- 44.Guldevall K, et al. Imaging immune surveillance of individual natural killer cells confined in microwell arrays. PLoS ONE. 2010;5:e15453. doi: 10.1371/journal.pone.0015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schiffenbauer YS, et al. A cell chip for sequential imaging of individual non-adherent live cells reveals transients and oscillations. Lab Chip. 2009;9:2965–2972. doi: 10.1039/b904778f. [DOI] [PubMed] [Google Scholar]
- 46.Frisk TW, Khorshidi MA, Guldevall K, Vanherberghen B, Onfelt B. A silicon-glass microwell platform for high-resolution imaging and high-content screening with single cell resolution. Biomed Microdevices. 2011;13:683–693. doi: 10.1007/s10544-011-9538-2. [DOI] [PubMed] [Google Scholar]
- 47.Zaretsky I, et al. Monitoring the dynamics of primary T cell activation and differentiation using long term live cell imaging in microwell arrays. Lab Chip. 2012;12:5007–5015. doi: 10.1039/c2lc40808b. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, et al. Quantitating cell-cell interaction functions with applications to glioblastoma multiforme cancer cells. Nano Lett. 2012;12:6101–6106. doi: 10.1021/nl302748q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gong Y, Ogunniyi AO, Love JC. Massively parallel detection of gene expression in single cells using subnanolitre wells. Lab Chip. 2010;10:2334–2337. doi: 10.1039/c004847j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi Q, et al. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc Natl Acad Sci USA. 2012;109:419–424. doi: 10.1073/pnas.1110865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love JC. Integrated process design for single-cell analytical technologies. AIChE J. 2010;56:2496–2502. [Google Scholar]
- 52.Vanherberghen B, et al. Classification of human natural killer cells based on migration behavior and cytotoxic response. Blood. 2013;121:1326–1334. doi: 10.1182/blood-2012-06-439851. [DOI] [PubMed] [Google Scholar]
- 53.Brouzes E, et al. Droplet microfluidic technology for single-cell high-throughput screening. Proc Natl Acad Sci USA. 2009;106:14195–14200. doi: 10.1073/pnas.0903542106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konry T, Dominguez-Villar M, Baecher-Allan C, Hafler DA, Yarmush ML. Droplet-based microfluidic platforms for single T cell secretion analysis of IL-10 cytokine. Biosens Bioelectron. 2011;26:2707–2710. doi: 10.1016/j.bios.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dominguez MH, et al. Highly multiplexed quantitation of gene expression on single cells. J Immunol Methods. 2013;391:133–145. doi: 10.1016/j.jim.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shalek AK, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. RNA-seq was used to identify new subsets of dendritic cells missed by other single-cell assays, and this work provides a quantitative analysis of the limitations of RNA-seq accuracy when measuring transcripts with low expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang F, et al. Deterministic and stochastic allele specific gene expression in single mouse blastomeres. PLoS ONE. 2011;6:e21208. doi: 10.1371/journal.pone.0021208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aghaeepour N, et al. Critical assessment of automated flow cytometry data analysis techniques. Nat Methods. 2013;10:228–238. doi: 10.1038/nmeth.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aghaeepour N, et al. Early immunologic correlates of HIV protection can be identified from computational analysis of complex multivariate T-cell flow cytometry assays. Bioinformatics. 2012;28:1009–1016. doi: 10.1093/bioinformatics/bts082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aghaeepour N, et al. RchyOptimyx: cellular hierarchy optimization for flow cytometry. Cytometry A. 2012;81:1022–1030. doi: 10.1002/cyto.a.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Finak G, et al. Mixture models for single-cell assays with applications to vaccine studies. Biostatistics. 2014;15:87–101. doi: 10.1093/biostatistics/kxt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahnke YD, Roederer M. Optimizing a multicolor immunophenotyping assay. Clin Lab Med. 2007;27:469–485. doi: 10.1016/j.cll.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McDavid A, et al. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics. 2013;29:461–467. doi: 10.1093/bioinformatics/bts714. [DOI] [PMC free article] [PubMed] [Google Scholar]