

Abstract
Mechanical strain provides an anti-adipogenic, pro-osteogenic stimulus to mesenchymal stem cells (MSC) through generating intracellular signals and via cytoskeletal restructuring. Recently, mTORC2 has been shown to be a novel mechanical target critical for the anti-adipogenic signal leading to preservation of β-catenin. As mechanical activation of mTORC2 requires focal adhesions (FAs), we asked whether proximal signaling involved Src and FAK, which are early responders to integrin-FA engagement. Application of mechanical strain to marrow-derived MSCs was unable to activate mTORC2 when Src family kinases were inhibited. Fyn, but not Src, was specifically required for mechanical activation of mTORC2 and was recruited to FAs after strain. Activation of mTORC2 was further diminished following FAK inhibition, and as FAK phosphorylation (Tyr-397) required Fyn activity, provided evidence of Fyn/FAK cooperativity. Inhibition of Fyn also prevented mechanical activation of RhoA as well as mechanically induced actin stress fiber formation. We thus asked whether RhoA activation by strain was dependent on mTORC2 downstream of Fyn. Inhibition of mTORC2 or its downstream substrate, Akt, both prevented mechanical RhoA activation, indicating that Fyn/FAK affects cytoskeletal structure via mTORC2. We then sought to ascertain whether this Fyn-initiated signal pathway modulated MSC lineage decisions. siRNA knockdown of Fyn, but not Src, led to rapid attainment of adipogenic phenotype with significant increases in adipocyte protein 2, peroxisome proliferator-activated receptor gamma, adiponectin, and perilipin. As such, Fyn expression in mdMSCs contributes to basal cytoskeletal architecture and, when associated with FAs, functions as a proximal mechanical effector for environmental signals that influence MSC lineage allocation.
Keywords: Src, Fyn, FAK, RhoA, Mesenchymal stem cells, Adipogenesis
Introduction
The mesenchymal stem cell (MSC) pool within bone marrow serves as a critical repository for lineages that support bone remodeling throughout life. Along with chemical and hormonal influences on MSC fate, mechanical signals provide significant regulatory control of MSC lineage allocation. Both static [1,2] and dynamic [3] mechanical cues modulate MSC differentiation in vitro, both through increasing cytoskeletal architecture [4,5] and by activation of β-catenin, which protects multipotentiality of the stem cell [6], preventing adipogenesis [7], and allowing entry into the osteogenic lineage [8]. Force activation of mTORC2 is a required proximal event for β-catenin activation [9]. Mechanical activation of pathways affecting MSC lineage allocation is initiated at focal adhesions (FAs) [5], where cellular tension is generated through the interface of the external microenvironment with the actin cytoskeleton, via integrin attachments [10,11].
mTORC2 is a multiprotein complex composed of subunits including mTOR, mSin1, GβL, and rictor. Association of rictor within the mTOR complex defines mTORC2, whereas mTORC1 contains raptor. The rictor versus raptor association defines mTOR substrate specificity. Mechanical activation of mTORC2 induces phosphorylation of Akt at Ser-473; in contrast, mTORC1 is not involved [9]. Akt phosphorylation is an essential event in mechanical repression of adipogenesis, acting through inhibition of GSK3β via Ser-9 phosphorylation to enhance availability of β-catenin. Furthermore, mTORC2 has been implicated in cytoskeletal organization [12] and cells deficient in the mTORC2-specific protein rictor display altered morphology with impaired migration [13]. As mechanical loading induces changes in the actin cytoskeleton through activation of RhoA, a GTPase fundamentally associated with actin cytoskeletal dynamics [4,14], we considered the possibility that mTORC2 might regulate cytoskeletal dynamics through effects on RhoA. Growth factor signaling, distinct from mechanical activation, can also induce mTORC2 activity: insulin potently induces Akt Ser-473 phosphorylation through activation of mTORC2. Our previous studies demonstrated that while mechanical inhibition of GSK3β was prevented in the absence of mTORC2 activity, GSK3β inhibition via insulin did not require Akt Ser-473 phosphorylation [9]; this suggests that the requirement for mTORC2 in transmitting mechanical information is unique. The upstream signaling events regulating mechanical activation of mTORC2/Akt remain unclear.
Mechanical strain activates multiple kinases within the FA complex at the plasma membrane including extracellular regulated kinase 1/2 (ERK1/2), focal adhesion kinase (FAK), and Src family kinases (SFKs). Activation of SFKs and FAK are early events following integrin–extracellular matrix engagement [15]. During cell migration, where cytoskeletal reorganization propels the cell forward, SFKs transiently translocate to newly formed FAs [16]. New FAs are also generated in response to mechanical strain along with the development of radial actin stress fibers [5], suggesting SFKs might be involved in mechanical cytoskeletal restructuring, paralleling their involvement in integrin-induced RhoA activation [17]. Cell adhesion and spreading, events that initiate FA formation, require both SFKs and FAK for full activation and recruitment of paxillin to the FA complex [18]. FAK functions cooperatively with SFKs during mechanical signaling, and cyclical strain increases the Src-FAK association [19]. Both FAK and SFKs are involved in actin cytoskeleton dynamics in response to mechanical force [20,21]. We hypothesized that mTORC2 regulates cytoskeletal rearrangement in response to mechanical strain through FAK and SFKs.
In this work, we used marrow-derived MSCs (mdMSCs) to identify a role for the SFK member Fyn in mechanical activation of mTORC2/Akt. We showed that the SFK isoform Fyn is necessary for mechanical mTORC2 activation. Strain activation of mTORC2 also requires FAK, which cooperates with Fyn for maximal activation. Furthermore, we demonstrate that Fyn and mTORC2 are crucial for mechanical activation of RhoA, and that mTORC2’s effect is conferred through Akt. This critical signaling pathway not only decreases mdMSC entrance into adipocyte lineage through enhancing β-catenin signaling as we have shown previously [9] but importantly reinforces cytoskeletal architecture by activating RhoA.
Materials and Methods
Reagents
Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Atlanta, GA, www.atlantabio.com). Culture media, trypsin-EDTA, antibiotics, and phalloidin-Alexa488 were from Invitrogen (Carlsbad, CA, www.invitrogen.com). Insulin, dexamethasone, indomethacin, rapamycin, Akti1/2, KU63794, PF573228, and PP2 were purchased from Sigma Aldrich (St. Louis, MO, www.sigmaaldrich.com). SU6656 and U0126 inhibitors were obtained from Calbiochem (La Jolla, CA, www.emdmillipore.com).
RNA-Mediated Interference
mdMSCs were transfected with gene-specific small interfering RNA (siRNA) or control siRNA (20 nM) using PepMute Plus transfection reagent (SignaGen Labs, Rockville, MD, www.signagen.com). Medium was replaced at 18 hours with Iscove’s Modified Dulbecco’s Medium (IMDM) containing FBS (10%, v/v) and penicillin/streptomycin (100 μg/ml). Mechanical strain was applied 72 hours after initial transfection; adipogenic media was added 18 hours after transfection. The following Stealth Select siRNAs (Invitrogen) were used in this study: negative control for rictor 5′-GCCCUCGUUGACUGAAA GAAUCUGA-3′; rictor 5′-UCAUCUUUCUGACUAAGCGAAG GGC-3′; negative control for Src 5′-GCCUCGUACAGAAGAA ACGCCGAAU-3′; Fyn 5′-UAAAGCGCCACAAACAGUGUCACUC-3′; Src 5′-AUUCGUUGUCUUCUAUGAGCCGGGC-3′; Yes 5′-CAGAUCGCUGAUGGCAUGGCGUAUA-3′.
Antibodies
The following antibodies were purchased from Cell Signaling (Danvers, MA, www.cellsignal.com): Akt (#4685), pAkt Ser-473 (#4058L), rictor (#D16H9), Src (#2110), pSrc Tyr-418 (#2101), Yes (#3201), and perilipin (#9349). Antibodies against FAK (sc-558), pFAK Tyr-397 (sc-11765-R), and RhoA (sc-418) were purchased from Santa Cruz Biotechnology (Dallas, TX, www.scbt.com). Anti-Fyn antibody (610163) was from BD Biosciences (San Jose, CA, www.bdbiosciences.com). The vinculin antibody (ab91459) was obtained from Abcam (Cambridge, MA, www.Abcam.com). Antibody recognizing adipocyte protein 2 (aP2) (XG6174) was purchased from ProSci, Inc. (Poway, CA, www.prosci-inc.com). The anti-adiponectin antibody (PA1-054) was from Affinity BioReagents (Rockford, IL, www.pierce-antibodies.com).
Cells and Culture Conditions
mdMSCs [7] were maintained in IMDM with FBS (10%, v/v) and penicillin/streptomycin (100 μg/ml). For experiments, cells were plated at a density of 6,000–10,000 cells per square centimeter on collagen-I coated silicone membrane plates (Flexcell International, Hillsborough, NC, www.flexcellint.com) and cultured for 2–4 days before beginning experiments. Cells were serum starved overnight in α-Minimal Essential Medium lacking serum before all assays. Adipogenic medium included dexamethasone (0.1 μM), insulin (5 μg/ml), and indomethacin (50 μM). Pharmacological inhibitors were added 1 hour before strain experiments at the following concentrations: rapamycin (30 nM), SU6656 (2.5 μM), PP2 (10 μM), U0126 (10 μM), PF573228 (3 μM), Akti1/2 (40 μM), and KU0063794 (2 μM).
Murine embryonic fibroblasts (MEFs) lacking the Src kinases members Src, Yes, and Fyn (SYF) and wild-type MEFs were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with FBS (10%, v/v) and penicillin/streptomycin (100 μg/ml). SYF cells harboring stably transfected constructs, expressing Src or Fyn, were grown under the same conditions and were generated as described previously [17].
Mechanical Strain
Uniform biaxial strain was applied to mdMSCs plated on six-well Bioflex Collagen-I coated plates using the Flexcell FX-4000 system (Flexcell International). A regimen of 2% strain was delivered at 10 cycles per minute for a total of 100 cycles (10 minutes) for all experiments. Cells used for phosphorylation induction and RhoA activity assays were lysed directly following the 100 cycles of strain, whereas cells stained for actin cytoskeleton were allowed to rest for 3 hours following 100 cycles of strain to enable actin stress fiber formation.
Western Blotting
Whole cell lysates were prepared using radio immunoprecipitation assay lysis buffer (150 mM NaCl, 50 mM Tris HCl, 1 mM EGTA, 0.24% sodium deoxycholate, 1% Igepal, pH 7.5) containing NaF (25 mM) and Na3VO4 (2 mM). Aprotinin, leupeptin, pepstatin, and phenylmethylsulfonylfluoride (PMSF) were added before each lysis. Whole cell lysates (20 μg) were separated on polyacrylamide gels (7, 9 or 12%) and transferred to polyvinylidene difluoride membranes. Membranes were blocked with milk (5%, w/v) diluted in Tris-buffered saline containing Tween20 (TBS-T, 0.05%). Blots then were incubated overnight at 4°C with the appropriate primary antibodies, washed and incubated with horseradish peroxidase-conjugated secondary antibody (1:5,000 dilution; Cell Signaling) at room temperature (RT) for 1 hour. Chemiluminescence was detected with ECL plus (Amersham Biosciences, Piscataway, NJ, www.gelifesciences.com) and developed. Images were acquired with a Hewlett Packard Scanjet scanner, and densitometry was determined using NIH ImageJ software version 1.45s.
RhoA Assay
Purification of recombinant proteins and construction of the pGEX4T-1 prokaryotic expression constructs containing the Rho-binding domain (RBD) of Rhotekin has been described [22]. Briefly, expression of the fusion proteins in Escherichia coli was induced using isopropyl β-D-1-thiogalactopyranoside (100 μM) for 12–16 hours at RT. Bacterial cells were lysed in lysis buffer containing Tris HCl (50 mM, pH 7.6), NaCl (150 mM), MgCl2 (5 mM), dithiothreitol (1 mM), aprotinin (10 μg/ml), leupeptin (10 μg/ml), and PMSF (1 mM). Recombinant proteins were purified by incubation with glutathione-sepharose 4B beads (GE Healthcare, Piscataway, NJ, www.GEHealthcare.com) at 4°C. Pull down of active RhoA, using glutathione-S-transferase-RBD (GST-RBD) beads, was performed as described [23]. mdMSC cells were lysed in buffer containing Tris HCl (50 mM, pH 7.6), NaCl (500 mM), Triton X-100 (1%, v/v), SDS (0.1%, v/v), sodium deoxycholate (0.5%, w/v), MgCl2 (10 mM), orthovanadate (200 μM), and protease inhibitors. Lysates were clarified by centrifugation, equalized for total volume and protein concentration and rotated at 4°C for 30 minutes with 50 μg of purified GST-RBD bound to glutathione-sepharose beads. The bead pellets were washed in lysis buffer three times, followed by pelleting of the beads by centrifugation between each wash, and subsequently processed by SDS-polyacrylamide gel electrophoresis.
Immunofluorescence
Following strain and/or treatment with pharmacological inhibitors, cells were fixed with paraformaldehyde (4%, v/v) for 20 minutes, permeabilized with Triton X-100 (0.1%, v/v) for 5 minutes at RT, and donkey serum (5%, v/v) blocking buffer diluted in TBS was added for 30 minutes to block nonspecific epitopes. Cells were washed three times for 10 minutes each with TBS. The silicone culture membranes were detached from the BioFlex plates using a scalpel and transferred to wells of six-well plates. Cells were incubated with phalloidin-conjugated Alexa Fluor-488 (Invitrogen) diluted in TBS (1:100) 30 minutes at RT. Cells were washed three times for 10 minutes each and the silicone membranes were set on glass slides, covered, and sealed with mounting medium containing dapi (Invitrogen). Images were taken using a Zeiss LSM 710 confocal laser scanning microscope running ZEN 2011 software (Carl Zeiss Microscopy, Inc., Thornwood, NY, www.microscopy.zeiss.com), excitation 488 nm line of the argon ion laser, emission 493–630 nm, objective lens 20× 0.95 Plan Apo.
Statistical Analysis
Statistical variance was expressed as the means ± SE. Statistical significance was evaluated using a natural log transformation of the data to make the distribution more symmetric, followed by analysis using either a t-test or a one-way ANOVA, allowing for unequal variance (Prism GraphPad, LA Jolla, CA, www.graphpad.com). All experiments were replicated at least twice to assure reproducibility. Densitometry data, where given, were compiled from at least three separate biological replicates.
Results
Src-like Kinases Are Critical for Mechanical, but Not Insulin, Activation of Akt
We demonstrated previously that Akt was critical to mechanical inhibition of GSK3β and that mechanical activation of Akt was mTORC2 dependent [9]. Here, we confirmed that Akt Ser-473 phosphorylation was increased following 100 cycles of biaxial strain (Fig. 1A, 1B) and that strain-induced phosphorylation of Akt Ser-473 was disrupted by pharmacological inhibition of mTOR activity (KU0063794) (Fig. 1A). Our previous study showed that inhibition of mTORC1 activity, using rapamycin, failed to block mechanical Akt Ser-473 phosphorylation [9]. To confirm that mechanical activation of Akt Ser-473 was mTORC2-specific, and independent of mTORC1, the mTORC2-associated subunit rictor was knocked down using siRNA, and compared with cultures treated with a control siRNA. Rictor-deficient mdMSCs demonstrated dramatically reduced Akt Ser-473 phosphorylation after strain compared with cells treated with control siRNA (Fig. 1B). To determine whether Src-like kinases were necessary for strain activation of mTORC2, mdMSCs were treated with the SFK inhibitor PP2 (10 μM). Mechanical phosphorylation of Akt was suppressed in the presence of PP2 compared with vehicle treated controls (Fig. 1C). We confirmed the role of SFKs in mechanical phosphorylation of Akt by treating mdMSCs with the SFK inhibitor SU6656 (2.5 μM) before strain application. While strain significantly (p < .05) increased pAkt Ser-473, inhibiting SFK activity prevented Akt Ser-473 phosphorylation by strain. Densitometry demonstrated the significant increase in pAkt Ser-473 in strained compared with nonstrained cells and the inhibition of Akt phosphorylation following treatment with SU6656 (Fig. 1D). In contrast to treatment with SFK inhibitors, the ERK1/2 inhibitor U0126 (10 μM) did not prevent strain-induced phosphorylation of Akt Ser-473; this was verified by densitometry (Fig. 1E).
Figure 1.

Mechanical activation of mTORC2 requires Src kinases. (A): Mechanical treatment of marrow-derived mesenchymal stem cells (mdMSCs; 2% strain, 100 cycles) induces Akt Ser-473 phosphorylation. Pharmacological inhibition of mTOR (KU0063794, 2 μM) prevented strain-induced Akt phosphorylation. (B): Strain-induced phosphorylation of Akt Ser-473 was not observed after knockdown of the mTORC2 component rictor with siRNA. (C): Inhibition of SFKs (PP2, 10 μM) prevented strain activation of Akt Ser-473. (D): The SFK inhibitor SU6656 (2.5 μM) blocked mechanical Akt Ser-473 phosphorylation. Densitometry shows that pAkt Ser-473 was significantly reduced (n = 3, p < .05). (E): Blocking ERK1/2 activity (U0126, 10 μM) did not suppress the mechanical activation of Akt. Densitometry showed a significant increase in Akt phosphorylation following strain in the absence or presence of the ERK1/2 inhibitor (n = 3, p < .05). (F): Akt Ser-473 phosphorylation after insulin stimulation was significantly increased (p < .0001) and was not prevented by SFK inhibition (SU6656, 2.5 μM). Significance was confirmed by densitometry analysis. Abbreviation: DMSO, dimethyl sulfoxide.
Furthermore, we asked whether SFKs were necessary for insulin activation of mTORC2. mTORC2 plays a unique role in Akt Ser-473 phosphorylation in response to insulin stimulation [24], an effect that appears to be PI3K-dependent, enabling Akt relocation to the plasma membrane [25]. Insulin treatment of mdMSCs produced a robust increase in pAkt Ser-473, as expected; however, pretreatment with the SFK inhibitor SU6656 failed to repress Akt Ser-473 phosphorylation (Fig. 1F). As such, the requirement for SFKs in phosphorylation of Akt Tyr-473 specifically requires FA contacts that mediate “outside-in” mechanical signaling, as is distinct from Akt activation by soluble growth factors.
Fyn Is Required for Mechanical Activation of mTORC2 in mdMSCs
Several SFK isoforms can be activated by mechanical force including Src and Fyn [17]. To determine the SFK member required for mechanical activation of mTORC2, siRNA was used to selectively knockdown individual SFK members. Treatment with siRNA against Fyn reduced Fyn protein to undetectable levels. In the absence of Fyn, strain was not able to induce phosphorylation of Akt Ser-473 (Fig. 2A). Densitometry analysis demonstrated a significant increase in pAkt Ser-473 in strained cells compared with nonstrained cells transfected with a control siRNA, while there was no significant difference in pAkt Ser-473 between strained and non-strained cells following treatment with siRNA against Fyn (Fig. 2A, p < .05). In contrast, knockdown of the Src isoform with siRNA failed to repress mechanical activation of pAkt Ser-473 (Fig. 2B). As Akt is mTORC2’s effector, in response to mechanical strain, this data suggests that Fyn’s role is proximal to mTORC2 during mechanical activation of Akt.
Figure 2.

Mechanical activation of mTORC2 requires Fyn. (A): Mechanical strain (2%, 100 cycles) of marrow-derived mesenchymal stem cells (mdMSCs) significantly increased Akt Ser-473 phosphorylation. Knockdown of Fyn with siRNA blocked mechanical phosphorylation of Akt. Densitometry confirms significance levels (n = 3, p < .05). (B): Mechanical activation of Akt Ser-473 was equivalently enhanced in mdMSCs following with either control siRNA or siRNA knockdown of Src. Densitometry demonstrates significance (n = 3, p < .05). (C): Transfection of siRNA targeting the Yes isoform did not reduce mechanical phosphorylation of Akt Ser-473 in mdMSCs. Compared with MCF7 human breast adenocarcinoma cell lysates, mdMSCs expressed little to none of the Yes isoform. (D): Mechanical strain (2%, 100 cycles) of MEFs increased Akt Ser-473 phosphorylation. pAkt Ser-473 was not enhanced following strain in MEFs lacking the SFKs Src, Yes, and Fyn (SYF). Re-expression of Src (+Src) rescued the mechanical activation of Akt Ser-473, whereas Akt phosphorylation remained suppressed with re-expression of Fyn (+Fyn). Abbreviations: mdMSC, marrow-derived mesenchymal stem cell; MEF, murine embryonic fibroblast; SYF, Src, Yes, and Fyn.
The contribution of the SFK member Yes to mechanical activation of mTORC2 was also evaluated. Knockdown of Yes did not alter mechanical phosphorylation of Akt Ser-473 in mdMSCs (Fig. 2C). Further analysis demonstrated that mdMSCs expressed little or none of the Yes isoforms; MCF7 human breast adenocarcinoma cell lysates were used as a positive control for Yes expression (Fig. 2C).
MEFs lacking the SFK members SYF were used to further test the requirement for SFKs in the mechanical activation of Akt. Control MEF cells demonstrated increased Akt phosphorylation at Ser-473 following 100 cycles of mechanical strain. MEFs lacking SYF displayed no increase in pAkt Ser-473 following mechanical strain, confirming that SFKs participate in mechanical activation of Akt (Fig. 2D). Stable transfection of the Src isoform into SYF cells rescued mechanical phosphorylation of Akt Ser-473, while overexpression of Fyn failed to enhance Akt Ser-473 phosphorylation in response to strain (Fig. 2D). This suggests that participation of SFK isoforms in mechanical Akt signaling is cell specific and that mechanical activation of Fyn is distinct from Src in MSCs.
Fyn and FAK Cooperate in Mechanical mTORC2 Activation
Following mechanical strain or cell attachment, FAK is recruited to integrin/FA sites where FAK is autophosphorylated on Tyr-397 [26]. FAK activation creates a high affinity binding site for Src kinases [26,27] enabling SFKs to phosphorylate FAK, resulting in full activation of both kinases. The cooperative activity between Src kinases and FAK led us to ask whether FAK might contribute to mechanical activation of mTORC2. We found that pharmacological inhibition of FAK (PF573228, 3 μM) prevented mechanical activation of mTORC2, as determined by Akt Ser-473 phosphorylation (Fig. 3A). Mechanical strain induced a significant (p < .05) increase in FAK Tyr-397 phosphorylation, which did not occur in mdMSCs pretreated with the SFK inhibitor SU6656 (2.5 μM) (Fig. 3B).
Figure 3.

Force activation of mTORC2 requires cooperative activation of Fyn and focal adhesion kinase (FAK). (A): Strain significantly increased Akt Ser-473 phosphorylation following strain. Treatment with the FAK inhibitor PF573228 (3 μM) blocked force-induced Akt phosphorylation. Densitometry confirms significance (n = 3, p < .05). (B): FAK Tyr-397 phosphorylation was significantly increased after 100 cycles of strain (2%). Inhibition of Src kinases (SU6656, 2.5 μM) significantly reduced FAK phosphorylation as shown in densitometry measurements (n-3, p < .05). (C): Compared with nonstrained controls, Src/Fyn phosphorylation at Tyr-418 was not significantly increased, densitometry n = 3. (D): The FA protein vinculin was immunoprecipitated from marrow-derived mesenchymal stem cell lysates and Western blotting was used to detect Fyn pulled down in the precipitate. There was significantly increased association of Fyn with the FA protein vinculin following mechanical strain, as shown by densitometry (n = 3, p < .05). Abbreviation: DMSO, dimethyl sulfoxide.
SFK phosphorylation at Tyr-418 is known to occur during integrin ligation [28] and following force activation [17,29]; surprisingly, in mdMSCs mechanical strain did not induce phosphorylation at this site following strain durations of 2–30 minutes (10 cycles per minute) (Fig. 3C, showing 100 cycles of strain). Fyn is located within FA complexes [30] and association of Fyn with FAs, at sites of integrin attachment, is known to cooperate in SFK activation [26]. To determine whether mechanical strain directed Fyn localization, we evaluated its association with FAs by immunoprecipitating the FA-associated protein vinculin. Mechanical strain induced a significant (p < .05) association between Fyn and vinculin when compared with nonstrained control samples (Fig. 2E), suggesting that mechanical strain promotes Fyn recruitment to sites of focal contacts. The absence of Fyn phosphorylation at Tyr-418 suggests an alternate mechanism of Fyn activation, perhaps dependent on the relocalization of Fyn to adhesion sites.
Mechanical RhoA Activity in mdMSCs Is Fyn Dependent
Src kinases play an important role in cytoskeletal rearrangement by regulating RhoA activity [31]. Fyn contributes to the mechanical activation of RhoA by regulating the exchange of GDP/GTP via the guanine exchange factor LARG [17]. We demonstrated previously that mechanical strain activates RhoA in mdMSCs [5]. Here, we examined whether SFKs were necessary for strain-dependent RhoA activation. When compared with nonstrained control cells (Fig. 4A), strained cells developed numerous actin stress fibers spanning the cell (Fig. 4B). Inhibition of SFK activity prevented actin stress fiber formation (Fig. 4D) compared with unstrained (static) control cells (Fig. 4C).
Figure 4.

Mechanical formation of actin stress fibers requires Fyn activation of RhoA. (A–D): Marrow-derived mesenchymal stem cells (mdMSCs) were stained with Alexa488-conjugated Phalloidin and dapi following strain (2%, 100 cycles) (B, D) or under nonstrained conditions (A, C). Images were captured using a 20× 0.95 plan apo lens. Cells were treated with either DMSO as a vehicle control (A, B) or with the Src inhibitor SU6656 (2.5 μM) (C, D). (E): mdMSCs were treated with Src kinase inhibitor (SU6656, 2.5 μM) or vehicle control and ± strain. Active RhoA (RhoA-GTP) was pulled down from cell lysates and compared with total RhoA by Western blot. (F): Fyn expression was knocked down in mdMSCs using siRNA or treatment with a control siRNA sequence. Following strain application active RhoA was pulled down and analyzed by Western blot. Abbreviation: DMSO, dimethyl sulfoxide.
Having shown that actin stress fiber reorganization was dependent on SFKs, we next investigated the influence of SFKs on strain-dependent activation of RhoA using pharmacological inhibition of SFKs and siRNA knockdown. Cyclical strain enhanced RhoA activity as expected [5]. Importantly, the strain-dependent RhoA activation was abrogated when cells were treated with the SFK pharmacological inhibitor SU6656 (Fig. 4E). As Fyn was identified as the SFK isoform necessary for mechanical activation of mTORC2 (Fig. 2A), we tested its requirement for mechanical activation of RhoA in mdMSCs. Indeed, mechanical activation of RhoA was prevented following knockdown of Fyn with siRNA (Fig. 4F). These data indicate that actin stress fiber formation in response to mechanical strain is dependent on Fyn activation of RhoA.
mTORC2/Akt Is Required for Mechanical Activation of RhoA
The absence of the mTORC2 binding partner rictor results in abnormal cell morphology [13] and mTORC2 has been implicated in cytoskeletal reorganization [12]. Mechanical strain generates actin stress fiber struts, which span the cell and enhance rigidity and cellular structure, an adaptation that influences MSC lineage commitment [5]. We hypothesized that mTORC2 was a critical step in mechanical regulation of RhoA. To test this, active RhoA was pulled down from total cell lysates following mechanical strain. RhoA activity was significantly (p < .05) increased comparing strained to unstrained mdMSCs, while addition of the mTOR inhibitor KU0063794 prevented mechanical activation of RhoA (Fig. 5A). A previous study reported that treatment with rapamycin, which acutely inhibits mTORC1, suppressed RhoA activity [32], implicating mTOR complex 1 in the regulation of RhoA activation. Treatment of MSCs with rapamycin for 1 hour failed to suppress mechanical activation of RhoA (Fig. 5C). In contrast, siRNA-mediated knockdown of the mTORC2-specific subunit rictor prevented RhoA activation following mechanical strain (Fig. 5D). These data indicate that mTOR complex 2, but not complex 1, is involved in mechanical activation of RhoA in mdMSCs. As Akt is a target of mTORC2 activation (Fig. 1B) [9], we investigated if Akt served as an intermediary for mTORC2 activation of RhoA. Treatment with the pharmacological Akt inhibitor Akti1/2 prevented mechanical activation of RhoA (Fig. 5B). These data suggest that Akt is the mTORC2 substrate necessary for strain-induced RhoA activation.
Figure 5.

Fyn regulates RhoA activity through mTORC2/Akt. (A): RhoA activity was analyzed by pulling down active RhoA following inhibition of mTOR (KU0063794, 2 μM). Densitometry shows a significant increase in RhoA activation following strain and when marrow-derived mesenchymal stem cells (mdMSCs) were treated with the mTOR inhibitor under nonstrained conditions (n = 3, p < .05). (B): Active RhoA was pulled down following inhibition of Akt (AKTi1/2, 40 μM) and ± strain. Densitometry demonstrated a significant increase in active RhoA following strain (n = 3, p < .05), which was blocked with addition of the Akt inhibitor. (C): RhoA activity was determined following inhibition of mTORC1 with rapamycin (30 nM) and ± strain. (D): The mTORC2 binding partner rictor was knocked down with siRNA and cells were strained or left unstrained. Active RhoA was pulled down and samples were analyzed by Western blot. Abbreviation: DMSO, dimethyl sulfoxide.
Fyn Restrains Adipogenic Lineage Commitment of mdMSCs
The requirement for Fyn participation in mechanical mTORC2/Akt activation in MSCs is not shared by MEFs (Fig. 2D), suggesting that Fyn might have a unique role in determination of MSC lineage. Indeed, several studies have implicated SFKs in adipogenic lineage commitment. A previous study demonstrated that inhibition of SFK activity decreased adipogenesis in MEF cells, but it was the Src, rather than the Fyn isoform that was involved [33]. Furthermore, Fyn knockout mice displayed decreased whole-body adipose tissue [34]. In contrast to this suggested proadipogenic stimulus of Fyn, here we demonstrate that Fyn activation restrains adipogenic commitment of mdMSCs. Following knockdown of Fyn using siRNA, we found a significant increase in the adipogenic markers aP2 (p < .05), peroxisome proliferator-activated receptor gamma (PPARγ) (p < .05), adiponectin (APN) (p < .05), and perilipin (p < .01) when cells were cultured in adipogenic conditions for four days (Fig. 6A). Protein quantities for adipogenic markers were normalized to β-actin expression and fold increases (over treatment with a control siRNA) were 2.1 ± 0.3, 6.3 ± 0.9, 35.4 ± 3.4, and 89.9 ± 11.3 for aP2, PPARγ, APN, and perilipin, respectively (Fig. 6B). In contrast to the enhanced adipogenic commitment of mdMSCs following knockdown of Fyn, siRNA treatment of Src resulted in no observable differences in aP2, PPARγ, or perilipin by Western blot, while expression of APN was increased in the absence of Src (Fig. 6C). These data demonstrate that Fyn is a critical regulator of adipogenic lineage commitment decisions in MSCs from the marrow of murine long bones, where Fyn serves to restrain adipocyte formation.
Figure 6.
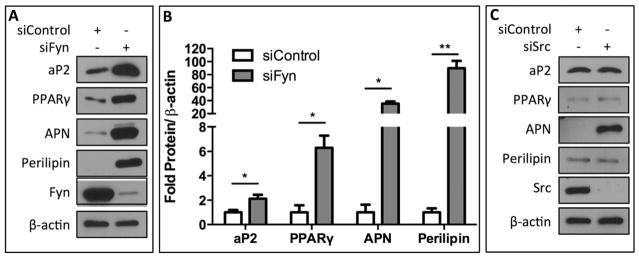
Fyn restrains adipogenic commitment in mdMSCs. (A): Fyn expression was knocked down using siRNA and cells were cultured in adipogenic media for 4 days. Protein expression of adipocyte protein 2 (aP2), peroxisome proliferator-activated receptor gamma (PPARγ), adiponectin (APN), and perilipin by Western blot. (B): Densitometry measurements of three separate experiments showed a significant increase in expression of each adipogenic marker when treatment with Fyn siRNA was compared with control siRNA (*p < .05, **p < .01). Protein expression was normalized to β-actin. (C): Src was knocked down using siRNA and cells were cultured in adipogenic media for 4 days. Expression of adipogenic markers was determined by Western blotting. Abbreviations: aP2, adipocyte protein 2; APN, adiponectin; PPARγ, peroxisome proliferator-activated receptor gamma.
Discussion
The lineage fate of mdMSCs has a critical influence on the health and maintenance of bone, where bone density is inversely proportionate to adipogenic commitment of these progenitor cells [8,35]. Lineage decisions are dependent on cell architecture, where the structure of the cytoskeleton influences how cells respond to the local physical environment [2]. We have shown here that mechanical activation of a specific SFK isoform, Fyn, cooperating with FAK, is required for a signaling pathway that, through mTORC2/Akt, controls MSC cytoskeletal architecture by activating RhoA. This indicates that Fyn/FAK signaling participates in MSC sensing of its physical environment, and reinforces signaling events that are important for lineage selection. Fyn/FAK activation is a crucial proximate step in mechanical mTORC2 activation, and is distinct from mTORC2 activation by growth factors. As such, Fyn is a critical early activator of mechanical signals emanating from FAs that define cytoskeletal structure and lineage decisions.
Src kinases are recruited to FAs following strain or cell adhesion where they cooperate with FAK, inducing reciprocal activations. Here, we show that FAK is also necessary for mechanical activation of Akt. The cooperative activation of FAK and SFKs is initiated upon autophosphorylation of FAK Tyr-397, creating a high affinity binding site for SFKs [26]. Cyclical strain increases the Src-FAK association in osteoblasts [19] and SFK involvement in strain-induced FAK phosphorylation is consistent with previous work showing that inhibition of SFKs prevented FAK Tyr-397 phosphorylation [36].
Strain and fluid shear stress have been shown to induce Src Tyr-418 phosphorylation in intestinal epithelial cells [36] and MC3T3-E1 preosteoblastic cells [29]. Here, we did not detect phosphorylation of Fyn or Src at Tyr-418 in mdMSCs following mechanical strain, but we did show that Fyn was newly recruited to FAs where FAK is localized. This suggests that Fyn-dependent activation of mTORC2, rather than requiring phosphorylation of Tyr-418, involves a strain-dependent recruitment of Fyn to the FA complex where it comes into association and interacts with FAK.
Both FAK and SFKs participate in the regulation of actin cytoskeleton reorganization in response to mechanical force [20,21]. In mdMSCs, we now show that strain-induced actin stress fiber formation is blocked when SFKs are inhibited with SU6656. Inhibition of SFKs and knockdown of Fyn prevented mechanical activation of RhoA, an essential regulator of actin stress fiber formation. While Fyn has been shown to be partially responsible for integrin-induced LARG activation [17], we believe that Fyn’s major target is the mTORC2 kinase, which precedes Akt activation of RhoA. We showed that the mTORC2 complex requires Fyn to sense mechanical strain and to transmit that signal to RhoA. Indeed, mTORC2 has been implicated in basal cytoskeletal organization [37] as rictor-deficient cells display altered morphology with impaired migration [13]. It is known that rictor, independently mTORC2, influences the organization of the actin cytoskeleton by suppressing RhoGDI2 expression, a negative regulator of Rho GTPases [13]. As well, in mdMSCs, mechanical activation of RhoA was dependent on mTOR complex two but not complex 1, consistent with previous work from our lab that rapamycin treatment (mTORC1-specific) did not suppress strain-induced Akt Tyr-473 phosphorylation [9]. These results suggest that while mechanical activation of RhoA via mTORC2 is a generalizable phenomenon, that the proximal signal, Fyn/FAK in the case of mdMSCs, may provide cell specificity.
While mechanical strain of mdMSCs consistently produced a rise in RhoA activity, we also found an unexpected rise in basal RhoA activation following treatment with mTOR or Akt inhibitors. A similar basal enhancement of RhoA activity was seen when SFKs were inhibited or Fyn expression was knocked down. This effect was not observed following treatment with rapamycin (specific for mTORC1). A previous study also demonstrated high constitutive activation of RhoA following knockdown of the mTORC2-specific binding partner rictor [38], suggesting that the Fyn/mTORC2/Akt pathway might supply a tonic inhibitory control over basal RhoA activity. RhoA activity is modulated by a variety of regulatory molecules such as guanine nucleotide dissociation inhibitors (GDIs) [39], guanine exchange factors (GEFs), and GTPase activating proteins (GAPs) [40]. Rho GDIs function to sequester RhoA in the cytosol, limiting its ability to be fully activated. It is possible that the absence of mTORC2 signaling reduces GDI sequestration of RhoA in the cytosol, making RhoA readily available for GEF-mediated activation. Alternatively, suppression of mTORC2/Akt activity might directly activate Rho GAPs, leading to decreased RhoA activity.
Mechanical regulation of MSC differentiation has a direct effect on cell lineage, as both static [1,2] and dynamic [3] mechanical cues modulate lineage commitment in vitro, in part through stiffening of the cell cytoskeleton [4]. These direct effects on the cytoskeleton of uncommitted marrow MSCs likely contribute to the increased osteogenesis that occurs during exercise-induced skeletal loading [41], while reduced loading results in increases in marrow adipocytes [42,43]. RhoA, which directly modulates the cytoskeleton, also affects lineage selection, as inhibition of RhoA suppresses osteogenesis and enhances adipogenesis [44]. Our findings add detail to the cytoskeletal control of MSC lineage selection. We found that Fyn knockdown led to a dramatic enhancement of mdMSC adipogenic lineage commitment. Additionally, knockdown of Src had little effect on adipogenesis: expression of aP2, PPARγ, and Perilipin were not altered following Src knockdown. Interestingly, APN expression was specifically increased with Src deficiency, suggesting a direct effect not related to promotion of adipogenic phenotype. The involvement of Fyn, but not Src, in adipogenesis of mdMSCs was indeed consistent with our data showing that Fyn, but not Src, was required for mechanical activation of mTORC2. As Fyn altered cytoskeletal assembly in response to mechanical strain, and was required for RhoA activation, it is likely that Fyn modulates mdMSC lineage through its effects on cytoskeletal structure.
Src family kinases may have tissue and activator-specific roles in controlling cell differentiation. In mdMSCs, knock down of Src had little influence on adipogenesis in comparison with the robust increase in fat formation when Fyn was deficient. In contrast to our findings, Fyn depleted 3T3-L1 preadipocytes and Fyn null MEFs displayed suppressed adipocyte formation [34]. Adding further complexity, mice with global Fyn knock-out had reduced adipose mass and adipocyte size [34] but showed increased subcutaneous adipose formation [45], highlighting the variable effects of Fyn in different fat depots. Finally, deleting all SFK isoforms from MEFs decreased their adipogenic potential, yet re-expression of the Src isoform in these cells rescued adipogenesis [33]. This may indicate that the contribution of a specific SFK iso-form to lineage selection is not only dependent on the incoming stimulus (e.g., physical vs. soluble), but also on responses specific to the cell type (e.g., mdMSC vs. MEF).
This study provides in vitro evidence of the role of Fyn in mechanical activation of an anti-adipogenic pathway and demonstrates the ability of this SFK isoform to inhibit adipogenesis in mesenchymal progenitors from the bone marrow compartment. These findings should provide the basis for further in vivo studies, with the goal of identifying physiologically relevant loading regimens or pharmacological targets that can restrain adipose formation within the bone marrow cavity. Activating these anti-adipogenic pathways should lead to enhanced skeletal integrity by increasing the available pool of osteogenic progenitors.
Summary
In summary, the cytoskeletal structure of mdMSCs is influenced by, and responsive to, its mechanical environment, and contributes to the promulgation of signaling events that influence lineage commitment. FA assembly and stress fiber formation provide a more rigid cellular framework that restrains adipogenesis and promotes osteogenic differentiation. We have identified the Src family kinase Fyn, cooperating with FAK, as a novel proximate participant in mechanical activation of mTORC2. mTORC2, via its downstream substrate Akt, induced mechanical activation of RhoA. RhoA ensures that the cytoskeleton adapts to mechanical signals. In conclusion, mechanical strain utilizes Fyn as a critical regulator of mTORC2/Akt/RhoA signaling to influence lineage commitment decisions of mdMSCs.
Acknowledgments
We thank Dr. Mark Weaver of NC TraCS for his assistance with the statistical analysis of our data and Ms. Lisa Sharek for her assistance with preparation of the RBD beads. This study was supported by AR042360 and AR056655 (to J.R.), AR064133 (W.R.T.). and GM029860 (K.E.B.).
Footnotes
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
Author contributions: W.T.: concept/design, collection/assembly of data, data analysis/interpretation, manuscript writing, final approval of manuscript; C.G., S.Y., B.S., G.U., M.S., N.C., and K.B.: data analysis/interpretation, final approval of manuscript; Z.X. and K.B.: collection/assembly of data; J.R.: concept/design, financial support, data analysis/interpretation, manuscript writing, final approval of manuscript.
References
- 1.Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science. 2009;324:1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engler AJ, Sen S, Sweeney HL, et al. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 3.Sen B, Styner M, Xie Z, et al. Mechanical loading regulates NFATc1 and beta-catenin signaling through a GSK3beta control node. J Biol Chem. 2009;284:34607–34617. doi: 10.1074/jbc.M109.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McBeath R, Pirone DM, Nelson CM, et al. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 5.Sen B, Guilluy C, Xie Z, et al. Mechanically induced focal adhesion assembly amplifies anti-adipogenic pathways in mesenchymal stem cells. Stem Cells. 2011;29:1829–1836. doi: 10.1002/stem.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Case N, Thomas J, Xie Z, et al. Mechanical input restrains PPAR-gamma2 expression and action to preserve mesenchymal stem cell multipotentiality. Bone. 2012 doi: 10.1016/j.bone.2012.08.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Case N, Xie Z, Sen B, et al. Mechanical activation of beta-catenin regulates phenotype in adult murine marrow-derived mesenchymal stem cells. J Orthop Res. 2010;28:1531–1538. doi: 10.1002/jor.21156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cawthorn WP, Bree AJ, Yao Y, et al. Wnt6, Wnt10a And Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a beta-catenin-dependent mechanism. Bone. 2012;50:477–489. doi: 10.1016/j.bone.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Case N, Thomas J, Sen B, et al. Mechanical regulation of glycogen synthase kinase 3beta (GSK3beta) in mesenchymal stem cells is dependent on Akt protein serine 473 phosphorylation via mTORC2 protein. J Biol Chem. 2011;286:39450–39456. doi: 10.1074/jbc.M111.265330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shemesh T, Geiger B, Bershadsky AD, et al. Focal adhesions as mechanosensors: a physical mechanism. Proc Natl Acad Sci U S A. 2005;102:12383–12388. doi: 10.1073/pnas.0500254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roca-Cusachs P, Del Rio A, Puklin-Faucher E, et al. Integrin-dependent force transmission to the extracellular matrix by alpha-actinin triggers adhesion maturation. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1220723110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal NK, Chen CH, Cho H, et al. Rictor regulates cell migration by suppressing RhoGDI2. Oncogene. 2012 doi: 10.1038/onc.2012.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyamoto S, Teramoto H, Coso OA, et al. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan KB, Swedlow JR, Morgan DO, et al. c-Src enhances the spreading of src−/− fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- 17.Guilluy C, Swaminathan V, Garcia-Mata R, et al. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol. 2011;13:722–727. doi: 10.1038/ncb2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown MC, Cary LA, Jamieson JS, et al. Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol Biol Cell. 2005;16:4316–4328. doi: 10.1091/mbc.E05-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boutahar N, Guignandon A, Vico L, et al. Mechanical strain on osteoblasts activates autophosphorylation of focal adhesion kinase and proline-rich tyrosine kinase 2 tyrosine sites involved in ERK activation. J Biol Chem. 2004;279:30588–30599. doi: 10.1074/jbc.M313244200. [DOI] [PubMed] [Google Scholar]
- 20.Leve F, Marcondes TG, Bastos LG, et al. Lysophosphatidic acid induces a migratory phenotype through a crosstalk between RhoA-Rock and Src-FAK signalling in colon cancer cells. Eur J Pharmacol. 2011;671:7–17. doi: 10.1016/j.ejphar.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Destaing O, Sanjay A, Itzstein C, et al. The tyrosine kinase activity of c-Src regulates actin dynamics and organization of podosomes in osteoclasts. Mol Biol Cell. 2008;19:394–404. doi: 10.1091/mbc.E07-03-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu BP, Burridge K. Vav2 activates Rac1, Cdc42, And Rhoa downstream from growth factor receptors but not beta1 integrins. Mol Cell Biol. 2000;20:7160–7169. doi: 10.1128/mcb.20.19.7160-7169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell. 2001;12:2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gan X, Wang J, Su B, et al. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 2011;286:10998–11002. doi: 10.1074/jbc.M110.195016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaller MD, Hildebrand JD, Shannon JD, et al. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlaepfer DD, Hanks SK, Hunter T, et al. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 28.Izawa T, Zou W, Chappel JC, et al. c-Src links a RANK/alphavbeta3 integrin complex to the osteoclast cytoskeleton. Mol Cell Biol. 2012;32:2943–2953. doi: 10.1128/MCB.00077-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan JM, Wong A, Genetos DC, et al. Src is sufficient, but not necessary, for osteopontin induction in osteoblasts. Biorheology. 2011;48:65–74. doi: 10.3233/BIR-2011-0582. [DOI] [PubMed] [Google Scholar]
- 30.Yeo MG, Oh HJ, Cho HS, et al. Phosphorylation of Ser 21 in Fyn regulates its kinase activity, focal adhesion targeting, and is required for cell migration. J Cell Physiol. 2011;226:236–247. doi: 10.1002/jcp.22335. [DOI] [PubMed] [Google Scholar]
- 31.Peng F, Zhang B, Ingram AJ, et al. Mechanical stretch-induced RhoA activation is mediated by the RhoGEF Vav2 in mesangial cells. Cell Signal. 2010;22:34–40. doi: 10.1016/j.cellsig.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Liu L, Luo Y, Chen L, et al. Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J Biol Chem. 2010;285:38362–38373. doi: 10.1074/jbc.M110.141168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y, Ma YC, Huang J, et al. Requirement of SRC-family tyrosine kinases in fat accumulation. Biochemistry. 2005;44:14455–14462. doi: 10.1021/bi0509090. [DOI] [PubMed] [Google Scholar]
- 34.Bastie CC, Zong H, Xu J, et al. Integrative metabolic regulation of peripheral tissue fatty acid oxidation by the SRC kinase family member Fyn. Cell Metab. 2007;5:371–381. doi: 10.1016/j.cmet.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Thompson WR, Rubin CT, Rubin J. Mechanical regulation of signaling pathways in bone. Gene. 2012;503:179–193. doi: 10.1016/j.gene.2012.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaturvedi LS, Gayer CP, Marsh HM, et al. Repetitive deformation activates Src-independent FAK-dependent ERK motogenic signals in human Caco-2 intestinal epithelial cells. Am J Physiol Cell Physiol. 2008;294:C1350–1361. doi: 10.1152/ajpcell.00027.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 38.Liu L, Das S, Losert W, et al. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev Cell. 2010;19:845–857. doi: 10.1016/j.devcel.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Mata R, Burridge K. Catching a GEF by its tail. Trends Cell Biol. 2007;17:36–43. doi: 10.1016/j.tcb.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 41.Ozcivici E, Luu YK, Adler B, et al. Mechanical signals as anabolic agents in bone. Nat Rev Rheumatol. 2010;6:50–59. doi: 10.1038/nrrheum.2009.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trudel G, Payne M, Madler B, et al. Bone marrow fat accumulation after 60 days of bed rest persisted 1 year after activities were resumed along with hemopoietic stimulation: the Women International Space Simulation for Exploration study. J Appl Physiol. 2009;107:540–548. doi: 10.1152/japplphysiol.91530.2008. [DOI] [PubMed] [Google Scholar]
- 43.Menuki K, Mori T, Sakai A, et al. Climbing exercise enhances osteoblast differentiation and inhibits adipogenic differentiation with high expression of PTH/PTHrP receptor in bone marrow cells. Bone. 2008;43:613–620. doi: 10.1016/j.bone.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 44.Meyers VE, Zayzafoon M, Douglas JT, et al. RhoA and cytoskeletal disruption mediate reduced osteoblastogenesis and enhanced adipogenesis of human mesenchymal stem cells in modeled microgravity. J Bone Miner Res. 2005;20:1858–1866. doi: 10.1359/JBMR.050611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee TW, Kwon H, Zong H, et al. Fyn deficiency promotes a preferential increase in subcutaneous adipose tissue mass and decreased visceral adipose tissue inflammation. Diabetes. 2013;62(5):1537–1546. doi: 10.2337/db12-0920. [DOI] [PMC free article] [PubMed] [Google Scholar]