

Abstract
CLC-type exchangers mediate transmembrane Cl– transport. Mutations altering their gating properties cause numerous genetic disorders. However, their transport mechanism remains poorly understood. In conventional models two gates alternatively expose substrate(s) to the intra- or extra-cellular solutions. In the CLCs, a glutamate was identified as the only gate; suggesting that they function according to a non-conventional mechanism. Here we show that transport in CLC-ec1, a prokaryotic homologue, is inhibited by crosslinks constraining movement of helix O far from the transport pathway. Crosslinked CLC-ec1 adopts a wild type-like structure, indicating stabilization of a native conformation. Movements of helix O are transduced to the ion pathway via a direct contact between its C-terminus and a tyrosine, a constitutive element of the second gate of CLC transporters. Therefore, the CLC exchangers have two gates that are coupled through conformational rearrangements outside the ion pathway.
Introduction
The CLC channels and transporters form a widespread family of membrane proteins whose primary task is to mediate anion transport1,2. The CLCs form dimers, with each monomer delimiting separate Cl– translocation pathways. These proteins function in two fundamentally different ways: the channels allow rapid and passive anion passage across membranes, while the transporters mediate the stoichiometric exchange of one or two anions for a proton3-8. The human genome encodes for nine CLC homologues that belong to both functional subtypes and mutations in at least four CLC genes lead to genetically inherited diseases1.
While opening of the CLC channels and exchangers is regulated by voltage, H+ and Cl– concentrations1,2, the conformational changes underlying gating remain poorly understood. High resolution crystal structures of four CLC transporters have been solved9-11, but have shed limited mechanistic insights into this process: the transmembrane regions of all four homologues adopt nearly identical conformations (Fig. 1a). The only mechanistically telling difference is the position of a highly conserved glutamate side chain, Gluex, which competes with the Cl– ions for occupancy of two of the three substrate binding sites which define the anion transport pathway11,12. Upon protonation, Gluex moves out of the pathway thereby opening it to the extracellular side (Fig. 1b). This, together with extensive functional studies3,12-15, led to the conclusion that Gluex is a gate in the CLCs. The lack of other, crystallographically resolved, structural rearrangements led to the hypothesis that movement of Gluex is the only relevant conformational change occurring during CLC channel and transporter gating11,12,16. In support of this minimalistic gating mechanism functional studies showed that the two subunits function independently19 and that transport does not require major conformational rearrangements17,18. The idea that a single gate regulates ion transport by the CLCs rationalizes how the family could have diverged into channels and secondary active transporters, and explains key features of CLC channel gating. However, a single-gate exchange mechanism is incompatible with the basic tenet of alternating-access transport, which postulates that bound substrates cannot be in simultaneous contact with both sides of the membrane20. Indeed, large conformational rearrangements underlie the alternate exposure of substrates to either side of the membrane in most transporters21-28. Disruption of these coordinated movements decouples the two gates and can result in channel-like behavior of the transporters, causing the unwanted dissipation of substrate gradients, with potentially catastrophic consequences29. A recent attempt to resolve this quandary postulated that in the CLCs the Gluex gate is assisted by a static kinetic barrier11,16, possibly formed by the steric constriction of the Cl– transport pathway at the side chains of conserved residues, Ser107 and Tyr445 in CLC-ec1 (Fig. 1b). This static barrier reduces slippage by preventing Cl– diffusion11,16 so that, on average, during a protonation-deprotonation event of Gluex only 2 Cl– ions permeate through the “open” CLC transporter. Kinetic simulations showed that such a mechanism could account for the 2 Cl–:1 H+ exchange stoichiometry of the CLCs11,16. However, this model fails to explain some of the key functional properties of the CLC transporters, such as the independence of the stoichiometry with pH and with the transport rate, which differ by more than 3 orders of magnitude among different CLCs3,10,18. Furthermore, mutations scattered throughout the transmembrane region strongly affect voltage-dependent gating of CLC channels and transporters30, in some cases causing genetic disorders. Thus, regions beyond the ion pathway proper might be involved in gating31,32. Finally, several lines of evidence suggest that conformational changes in regions distal to the ion pathway might occur during transport. For example, pH and Cl– dependent environmental changes were reported in helix R33 and for Tyr419 in the P-Q loop34 of CLC-ec1 (Fig. 1c), a prokaryotic homologue. Similarly, Zn2+ binding to the N-O extracellular loop inhibits the mammalian CLC-4 transporter35 (Fig. 1c). When mapped on the three dimensional structure of CLC-ec1 these regions delimit a well-defined area (Fig. 1c), offering a hint as to what portions of the protein, in addition to the ion pathway, might regulate transport.
Figure 1. Structural arrangement of CLC-ec1.

a) Overlay of the backbone of four CLC homologues: CLC-ec1 (1OTS, black), stCLC (1KPL, dark gray), spCLC (3Q17, medium gray) and cmCLC (3ORG, light gray). b) Close up view of the Cl– binding site of CLC-ec1. Highlighted in red is the side chain of E148. The three conformations are taken from cmCLC (down), WT CLC-ec1 (middle) and E148Q CLC-ec1 (up). Cl– ions are shown as green spheres. c) Ribbon representation of a single subunit of CLC-ec1. Highlighted in red are areas that were reported in the literature to be involved in conformational changes33-35. Helices J, O and Q are shown in cyan.
We hypothesized that if this region undergoes a conformational rearrangement during the exchange cycle, then constraining this motion should inhibit transport. We focused on the helical bundle formed by helices J, O and Q that connect the ion pathway to the three regions (Fig. 1c). To test our hypothesis, we introduced cysteine pairs at their contact points and crosslinked them using divalent Hg2+, a bifunctional, thiol-specific reagent23. Crosslinking the intracellular portion of helix O to helices Q or J reduces Cl– transport by 30-100 fold indicating that movement of helix O is required for transport. The crosslinked protein adopts a WT-like conformation, showing that the disulfide bond does not distort the structure of the protein. Movement of helix O is transduced to the Cl– pathway via a direct contact between its C-terminal end and Tyr445. Our results show that transport mediated by CLC-ec1 requires movement of helix O and that Tyr445 forms the inner gate of CLC-ec1.
Results
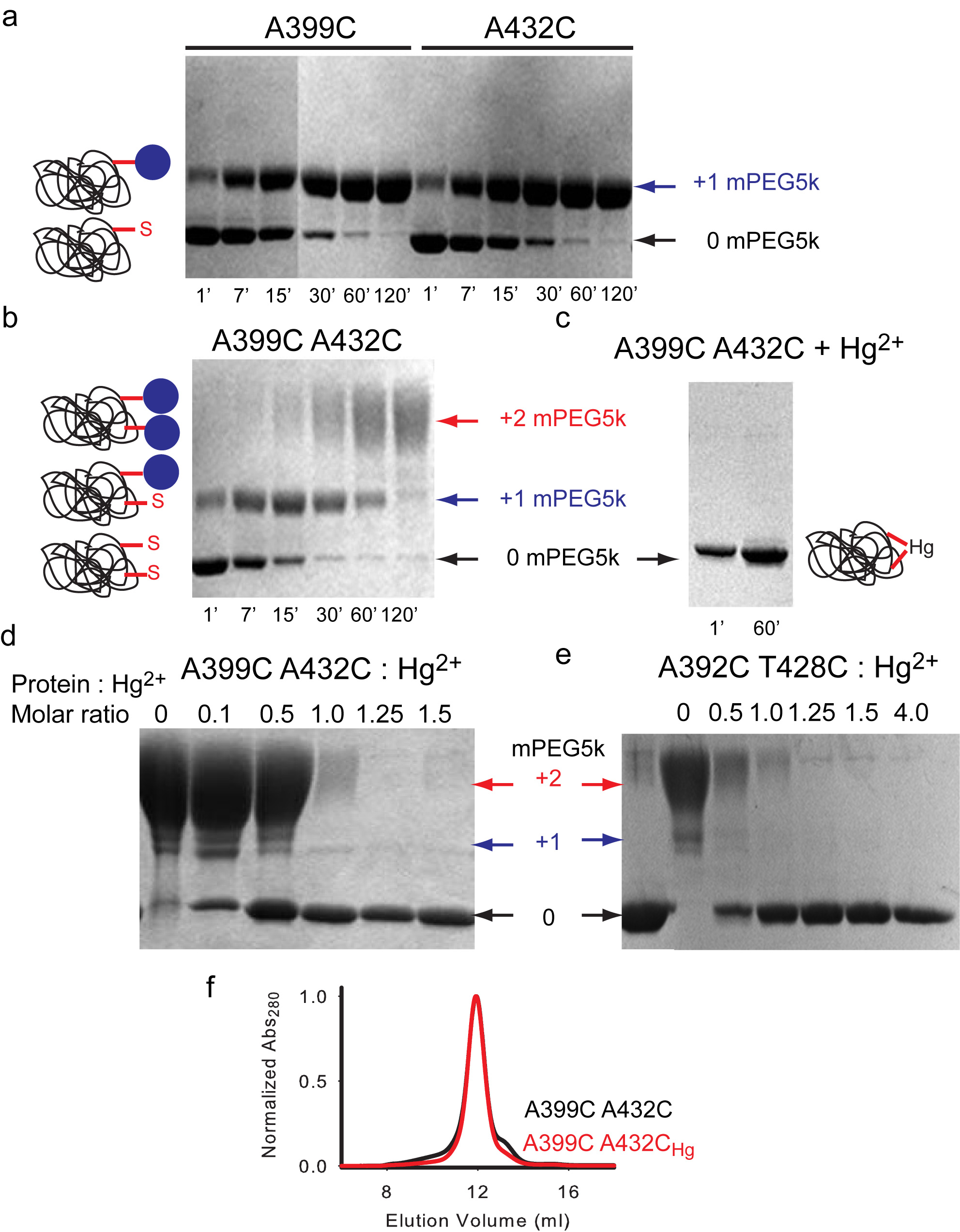
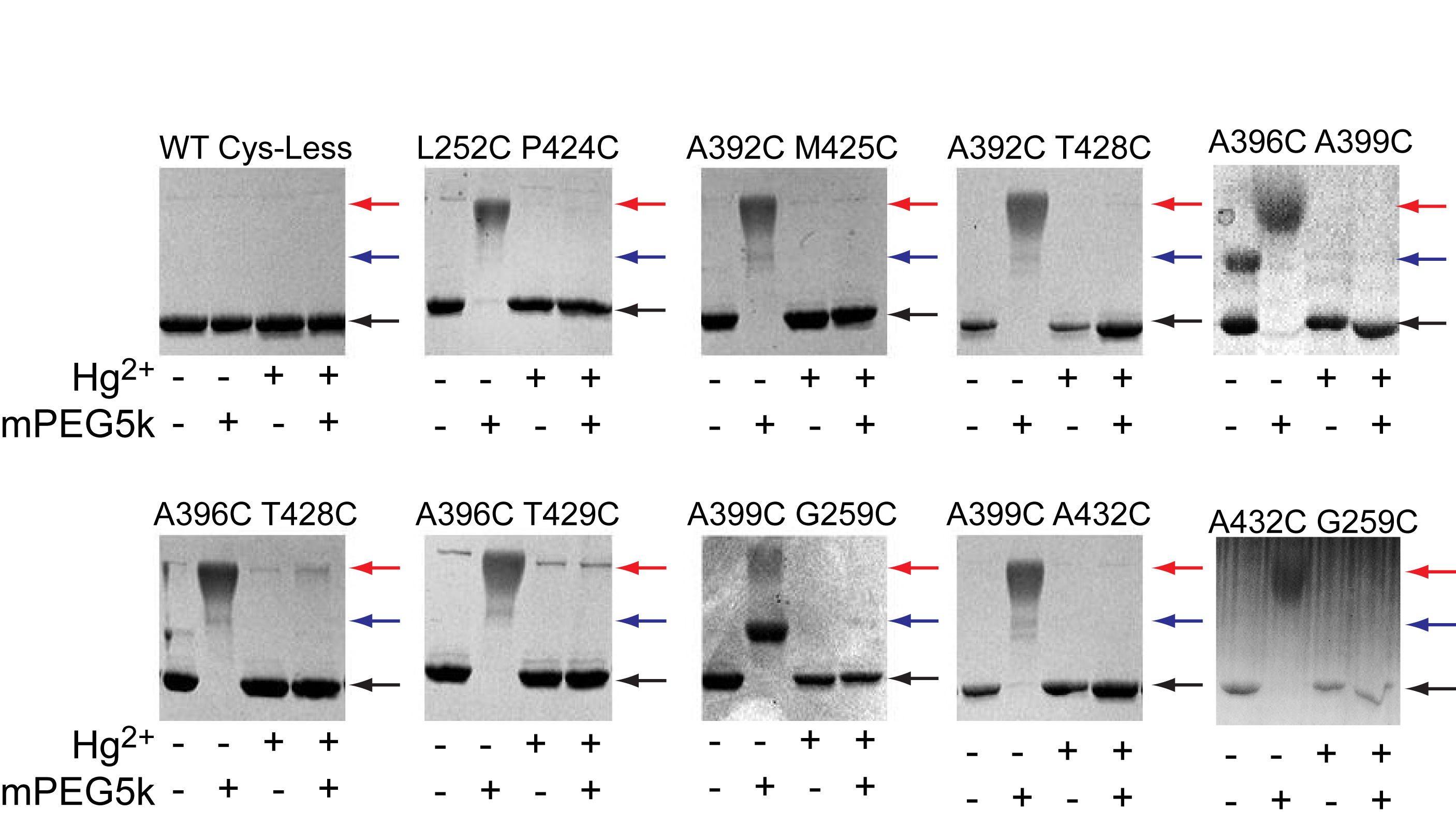
Our experiments were conducted in the background of a well-characterized Cys-less variant of CLC-ec1 that mediates H+/Cl– exchange with wild-type (WT) properties17. We identified 7 points of contact between helices J, O and Q (Fig. 2a) and introduced cysteine pairs at these positions, distributed in three regions –external (Fig. 2b), central (Fig. 2c) and internal (Fig. 2d). All double cysteine mutants, when uncrosslinked, mediate robust Cl– transport (Table 1), with the least active mutant (G259C A399C) being ~5-fold slower than the WT protein. This reduction is caused by the G259C mutation (Table 1, Supplementary Fig. 1), while the A399C substitution mediates Cl– transport at WT-like rates (Table 1, Supplementary Fig. 1). The extent of Hg2+-induced crosslink was assessed by counting the number of free cysteines by reacting the protein with maleimide 5,000 polyethylene-glycol (mPEG5K) and using a gel shift assay (Supplementary Fig. 2, 3). All cysteine pairs were nearly completely crosslinked after incubation with a 4-fold molar excess of Hg2+ for ~1 hr. PEGylation of the G259C A399C mutant is incomplete even after incubation for 4 hours due to poor accessibility of the G259C residue to mPEG5k. In the remainder of this work we will use the Hg subscript to indicate crosslinked mutants, i.e. A399C A432CHg.
Figure 2. Functional effects of crosslinking helices J, O and Q.

a-d) View of the relative position of the Cl– binding site, helices J, O, Q and R (a). The positions of the crosslinks are indicated by dashed lines and the residues are shown in blue (b, external region), green (c, central region) or red (d, internal region). The residues S107, E148 and Y445 are shown as yellow sticks. Cl– ions are shown as green spheres. e-h) Representative time courses of Cl– efflux from proteoliposomes reconstituted with WT CLC-ec1 (e), L252C P424C (f), A396C T428C (g) or A399C A432C (h) mutants CLC-ec1 before (black) and after (red) treatment with Hg2+ to induce crosslink formation. For clarity only traces for one mutant from each group are shown, traces of the other mutants are shown in Supplementary Figure 4. i) Average reduction of the transport rate after Hg2+-induced crosslink formation. Bars are color-coded as in panels a-d. The means ± s.e.m. of the unitary transport rates and number of repeats are reported in Table 1.
Table 1.
Cl– transport rates of WT and mutant CLC-ec1.
| Protein | γ (ion s–1) | γHg (ion s–1) | γHg/γ | n | nHg | Crosslinked Helices |
|---|---|---|---|---|---|---|
| WT | 2337±378 | 1341±224 | 0.57±0.19 | 6 | 8 | -- |
| L252C P424C | 694±77 | 528±48 | 0.76±0.15 | 7 | 7 | J-Q |
| A392C M425C | 1136±66 | 753±97 | 0.66±0.12 | 7 | 7 | O-Q |
| A392C T428C | 1734±229 | 1177±235 | 0.68±0.23 | 7 | 7 | O-Q |
| A396C T428C | 811±96 | 394±59 | 0.49±0.13 | 7 | 7 | O-Q |
| A396C G429C | 714±90 | 500±77 | 0.70±0.20 | 6 | 4 | O-Q |
| A399C A432C | 1357±87 | 16±36 | 0.01±0.03 | 10 | 10 | O-Q |
| G259C A399C | 552±83 | 32±19 | 0.06±0.04 | 6 | 6 | J-O |
| A399C A396C | 680±26 | 227±46 | 0.33±0.08 | 6 | 6 | O-O |
| G259C A432C | 517±78 | 320±70 | 0.62±0.23 | 15 | 15 | J-Q |
| A399C A432C E148A Y445A | 23260±1480 | 22255±2191 | 0.96±0.16 | 4 | 3 | O-Q |
| A399C A432C Y445A | 218±37 | 72±21 | 0.33±0.15 | 12 | 11 | O-Q |
| A399C A432C E148A | 564±26 | 244±21 | 0.43±0.06 | 9 | 9 | O-Q |
| A399C A432C I402G | 909±215 | 572±65 | 0.63±0.22 | 6 | 6 | O-Q |
| A399C A432C I402A | 668±57 | 248±92 | 0.37±0.17 | 6 | 6 | O-Q |
| A399C A432C I402S | 967±137 | 629±54 | 0.65±0.15 | 8 | 8 | O-Q |
| G259C | 257±57 | -- | -- | 4 | -- | -- |
| A399C | 1652±160 | -- | -- | 3 | -- | -- |
| A432C | 1268±77 | -- | -- | 4 | -- | -- |
Cl– transport rates measured using the efflux assay before (γ) and after Hg2+ treatment (γHg). Reported values are the average ± s.e.m.; n and nHg are the number of repeats for the untreated and treated mutants respectively, each from at least 2 independent preparations.
Transport requires a rearrangement outside the Cl– pathway
We assessed the function of the crosslinked proteins and found a striking pattern. Constraining the extracellular portions of helices J, O and Q (Fig. 2b) has only modest functional effects. The reduction in transport rate of the crosslinked L252C P424CHg, A392C M425CHg and A392C T428CHg mutants is ~30-40% and comparable to that seen for the WT protein after Hg2+-treatment (Fig. 2e, f; Supplementary Fig. 4a, b; Table 1). This reduction in rate likely reflects a non-specific effect of Hg2+ on the protein. Crosslinks in the middle of the transmembrane region, A396C G429CHg and A396C T428CHg, elicit a moderate reduction, ~30-50% (Fig. 2g; Supplementary Fig. 4c; Table 1). In contrast to these mild effects, constraints placed at the intracellular portions of helices J, O and Q abolish transport almost completely: crosslinking helices O and J (A399C G259CHg) or helices O and Q (A399C A432CHg) reduces transport by ~15- and ~100-fold, respectively (Fig. 2h; Supplementary Fig. 4d; Table 1). In the remainder of our work, we will use the A399C A432C mutant because, before Hg2+-treatment, it has WT- like transport properties whereas the other, A399C G259C, is~5-fold slower (Table 1). These results (Fig. 2i) suggest that during transport the relative movement of the intracellular portions of helices J, O and Q is required for Cl–/H+ exchange. Gly259, Ala399 and Ala432, are located far from the Cl– and H+ binding sites (11-22 Å) suggesting that crosslinking them does not directly affect the ion pathway but rather that these residues allosterically control ion access to and from the pathway. We now seek to determine what movement is constrained by the crosslinks and whether and how it is relayed to the Cl– pathway.
Structure of the A399C A432CHg mutant
We crystallized the A399C A432CHg mutant and solved its structure to 3.5 Å resolution (Fig. 3ac; Table 2). As expected from our experimental design, the backbone of the A399C A432CHg mutant superimposes well to that of WT CLC-ec1 (Fig. 3a) with an r.m.s.d. of 0.48 Å. The ion binding region of the A399C A432CHg mutant is similar to that of the WT transporter (Fig. 3b; Supplementary Fig. 5a), with Ser107, Glu148 and Tyr445 occupying their normal positions and with an extra electron density which we chose to model as a Cl– atom given its overlap with the central ion binding site (Supplementary Fig. 5a). Thus, the crosslink does not induce global rearrangements and the ion pathway is intact.
Figure 3. Structure of the A399C A432C crosslinked mutant.

a) Overlay of the backbones of WT CLC-ec1 (black, PDB ID: 1OTS) and A399C A432CHg mutant (red, PDB ID: 4MQX). b) Structure of the ion binding region of the A399C A432CHg mutant. Green sphere represents a Cl– ion modeled in the central binding site. The electron density is contoured at 1 σ. c) Structure of helices O and Q in the A399C A432CHg mutant. The electron density is contoured at 1.7 σ. The fo-fc electron density is shown in red and contoured at 3.3 σ. d-e) Thermograms of Cl– binding to A399C A432C (d) and A399C A432CHg (e). Upper panels show the heats released upon ion binding. Lower panels show the integrated heats (circles) and the solid line is the fit to a single site isotherm. Averaged thermodynamic parameters are reported in Supplementary Table 1.
Table 2.
Crystallographic statistics for the A399C A432CHg mutant.
| CLC-ec1 Cys-less A399C A432CHg | |
|---|---|
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 233.5, 94.5, 170.6 |
| α, β, γ (°) α, β,γ (°) | 90, 131.75, 90 |
| Resolution (Å) | 43.56 (3.52) *)* |
| Rsym or Rmerge (%) | 11.5 (52.2))* |
| I / σ/σI | 6.57 (1.90))* |
| Completeness (%) | 98.76 (97.88))* |
| Redundancy | 3.3 (3.2))* |
| Refinement | |
| Resolution (Å) | 43.56-3.52 |
| No. reflections | 34345 |
| Rwork / Rfree | 0.22/0.26 |
| No. atoms | |
| Protein | 13225 |
| Ligand/ion | 4 |
| Water | 0 |
| B factors | |
| Protein | 91.5 |
| Ligand/ion | 90.9 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.620 |
Values in parentheses are forrefer to the outermost shell. * Data was collected from a single crystal. *Values in parentheses are for highest-resolution shell.
In the crosslinked protein helices O and Q are unperturbed (Fig. 3c) with the exception of extra electron density bridging them between residues Cys399 and Cys432 (Fig. 3c; Supplementary Fig. 5b), confirming the formation of a crosslink. In an fo-fc difference map, the peak intensity of this extra density is ~5 σ (Fig. 3c), suggesting either low occupancy by Hg2+, which is in contrast to the results seen from the PEG-ylation experiments (Supplementary Fig. 2, 3), or that Hg2+ catalyzed the formation of a disulfide bond between the two cysteines. Therefore, we chose to model this electron density region as a disulfide bond between Cys399 and Cys432 (Fig. 3c).
We measured ion binding to the A399C A432C mutant before and after crosslink formation to rule out that subtle distortions of the Cl– pathway, obfuscated by the limited resolution of our structure, might impair ion binding. The unreacted A399C A432C mutant binds Cl– with WT-like characteristics, Kd~0.9 mM (Fig. 3d; Supplementary Table 1)36. After Hg2+ treatment, Cl– affinity is slightly lowered, Kd(A399C A432CHg)~3.2 mM (Fig. 3e; Supplementary Table 1). This modest effect confirms that the structural integrity of the Cl– pathway is preserved in the crosslinked protein and supports modeling the extra electron density in the central binding site with a Cl– ion (Fig. 3b). The ~100-fold reduction of transport the crosslinked mutant implies that during transport CLC-ec1 adopts a conformation different from that of the WT and that this difference involves a rearrangement of helices O and Q.
Helix O moves as a rigid body
The two “high-impact” crosslinks we identified tether helix O to helix J, G259C A399CHg (Supplementary Fig. 4d) or to helix Q, A399C A432CHg (Fig. 2d, h). To determine whether helix O moves relative to J and Q or vice versa, we crosslinked helices J and Q, G259C A432CHg (Supplementary Fig. 6a) and tested if transport is inhibited. This is not the case: the G259C A432CHg mutant is less than 2-fold slower than its uncrosslinked counterpart (Supplementary Fig. 6b; Table 1). This, together with the observation that crosslinking helix Q at a nearby position (T433) to the other subunit in the CLC-ec1 dimer does not impair transport17, suggests that helices J and Q are static while helix O moves.
We reasoned that an intra-helix crosslink between residues Ala396 and Ala399 (Supplementary Fig. 6c) might constrain a complex rearrangement of helix O, such as bending or a partial unwinding while a rigid-body motion of the helix (i.e. rotation-translation) should not be affected. The A396C A399CHg crosslinked mutant is only ~3-fold slower than the uncrosslinked A396C A399C protein (Supplementary Fig. 6d; Table 1), suggesting that helix O moves as a rigid body, at least below Ala396.
Movement of helix O is coupled to the Cl– pathway
To test whether helix O is coupled to the Cl– pathway, we introduced the A399C A432CHg crosslink in the background of the channel-like mutant E148A Y445A where the external gate and internal constriction have been removed37. The A399C A432CHg crosslink has no effect on Cl– transport mediated by this channelized mutant (Fig. 4a; Table 1; Supplementary Fig. 7) indicating that removal of the Glu148 and Tyr445 side chains decouples Cl– transport from the conformational cycle of CLC-ec1.
Figure 4. Movement of helix O is coupled to the Cl– gates.

a-c) Representative traces of Cl– efflux mediated by the E148A Y445A (a), Y445A (b) and E148A (c) mutants before (black) and after (red) the Hg2+-induced formation of the crosslink at A399C A432C. The average values are reported in Table 1. d) Schematic representation of the Cl– uptake assay. e) 36 Cl– uptake mediated by the A399C A432C mutant before (black circles) and after (red triangles) Hg2+- induced crosslink formation. Symbols represent the average of n=3-6 replicate time points. The errors are the s.e.m. Solid lines represent the fits to a rising exponential (for A399C A432C) of the form C(t)= aM*(1–exp(–t/τ) with a =0.28±0.08 and τ=0.77±0.09 min–1 for an initial rate K(A399C A432C)=0.22±0.06 min–1. The data for the crosslinked A399C A432CHg was fit to a line of the form a +K*t with a0=0.007±0.03 and K(A399C A432CHg)=0.0017±0.0002 min .The errors on the fitted values represent the uncertainty on the fit rather than the s.e.m.
If Glu148 is the only gate (i.e. moving part) regulating Cl– transport then the A399C A432CHg crosslink should inhibit transport of the A399C A432C Y445A mutant to the same extent as seen in the WT protein, since in both constructs the Gluex gate is intact. This is not the case: transport by the crosslinked Y445A mutant is ~3-fold slower than by its unconstrained counterpart, as opposed to the ~100-fold reduction seen with at tyrosine at position 445 (Fig. 4b; Table 1; Supplementary Fig. 7). This result has two implications: first, Glu148 cannot be the only gate regulating Cl– transport in the CLCs, as the crosslink does not inhibit the A399C A432C Y445AHg mutant. Second, it shows that a second gate exists and that Tyr445 is its critical component.
To determine whether helix O is coupled only to the internal gate through Tyr445 or if it is also coupled to the outer gate we tested whether removal of the latter, via the E148A mutation, relieves the inhibitory effect of the crosslink. Transport mediated by the E148A A399C A432CHg mutant is only ~2.3-fold slower than by its un-reacted counterpart (Fig. 4c; Table 1; Supplementary Fig. 7), suggesting that movement of helix O is not exclusively coupled to the inner gate, but rather that it controls Cl– transport through both gates. Alternatively, the crosslink might act through the H+ pathway. In this case, the crosslinked protein should be capable of completing a half-cycle38 and therefore mediate electro-neutral Cl–/Cl– exchange (Fig. 4d). In contrast, if the crosslink affects the Cl– pathway by “locking” one or both gates, then exchange should be inhibited to the same extent as net transport. Indeed, Cl–/Cl– exchange mediated by the A399C A432CHg protein is nearly 40-fold slower than that mediated by its uncrosslinked version (Fig. 4e). These results suggest that movement of helix O is relayed to the Cl– pathway and is essential for the coordinated alternate opening of the two gates formed by Glu148 and Tyr445.
Movement of helix O is relayed to the Y445 inner gate
In the structures of WT CLC-ec1 and of the crosslinked A399C A432CHg mutant the C-terminal residue of helix O, Ile402, sterically interacts with the aromatic ring of Tyr445, a key constituent of the inner gate (Fig. 5a). If this interaction is important for coupling helix O to the inner gate, then its disruption should have three effects on transport: it should impair turnover, remove the inhibitory effect of the A399C A432CHg crosslink and alter the transport stoichiometry. Results meet expectations on all fronts: replacing Ile402 with the smaller side chains Gly, Ala or Ser leads to a ~2-4 fold reduction in turnover rate, the A399C A432CHg crosslink exerts a < 2-fold inhibitory effect (Fig. 5b-d; Table 1; Supplementary Fig. 7) and the transport stoichiometry of the I402S mutant is degraded by ~50%, to ~3 Cl–: 1H+ (Fig. 6). These effects are comparable to those caused by mutations of the inner-gate residue Tyr445, on the transport rate, stoichiometry39,40 and on the relief of the crosslink inhibition (Fig. 4b). Thus, the contact between Ile402 and Tyr445 appears to be a key structural element to efficiently transduce movement of helix O to the inner gate and to prevent slippage, suggesting that this interaction is part of the allosteric pathway coupling the two gates of the CLC exchangers. Further work is needed to determine whether Aadditional residues in this region play a role in these processes, for example by coupling helix O to the external gate.
Figure 5. I402 couples helix O to Y445.

a) View of the relative position of the Cl– binding site, helices J, O, Q and R, the A399C A432C crosslink (red dashed line) and the Ile402 Tyr445 contact (yellow, spacefilling representation). The gating residues Ser107 and Glu148 are shown as yellow sticks. Cl– ions are shown as green spheres. b-d) Representative traces of Cl–efflux mediated by the I402G (b), I402A (c) and I402S (d) mutants in the background of the A399C A432C mutant before (black traces) and after (red traces) formation of the crosslink. The average values are reported in Table 1.
Figure 6. Effects of crosslinking helix O on the Cl–/H+ exchange stoichiometry.

a) Schematic representation of the simultaneous Cl– and H+ flux recordings. b-e) Upper panels: simultaneous recordings of Cl– efflux into (dashed lines) and H+ efflux from (solid lines) proteoliposomes reconstituted with WT CLC-ec1 (b), A399C A432C (c), A399C A432C I402S (d) or A399C A432C Y445A (e) before (black traces) and after (red traces) formation of the crosslink. Bottom panels: time course of the stoichiometry of transport (nCl/nH) determined as the ratio of the total transported Cl– and H+ ions. Dashed cyan line indicates the average value for the traces shown. e) Average stoichiometry of transport for the A399C A432C (nCl/nH(NoRx)= 2.0±0.1, n=5; nCl/nH(Hg)= 2.1±0.1, n=8), A399C A432C I402S (nCl/nH(NoRx)= 3.1±0.1, n=7; nCl/nH(Hg)= 3.2±0.2, n=8) and A399C A432C Y445A (nCl/nH(NoRx)= 6.3±0.8, n=5; nCl/nH(Hg)= 7.8±1.4, n=4) mutants before (black bars) and after (red bars) Hg treatment. The dashed blue line indicates the WT value (nCl/nH= 2.2±0.1, n=5). Data shown is the average±s.e.m. of n experiments from 2+ independent preparations.
Helix O is coupled to the Cl– but not the H+ pathway
Our results cannot rule out the possibility that helix O controls the H+ pathway in addition to or instead of the Cl– pathway. We therefore tested whether crosslink formation alters the stoichiometry of transport and thus H+-coupling. We determined the stoichiometry of transport by simultaneously measuring Cl– efflux from and H+ influx into vesicles reconstituted at high protein density (Fig. 6a). As expected, the WT protein3 and the A399C A432C mutant mediate the stoichiometric exchange of 2 Cl– for 1 H+ (Fig. 6b, c, black traces). The A399C A432C I402S and A399C A432C Y445A39 constructs have degraded transport stoichiometries of ~3 and ~7 Cl–: 1 H+ (Fig. 6d-e, black). All crosslinked mutants mediate exchange with stoichiometries that are nearly identical to those of the untreated proteins (Fig. 6c-f, red). These results are consistent with the hypothesis that helix O is coupled to the Cl– pathway, and that effects on H+ transport are indirect, if at all present.
Discussion
We set out to test the hypothesis that H+/Cl– exchange mediated by the CLC transporters involves conformational rearrangements outside of the Cl– pathway. We found that we could inhibit transport by constraining movement of the intracellular portion of helix O via covalent crosslinks to either helix J or helix Q (Fig. 2). A direct interaction between Ile402 and Tyr445 appears to be important in coupling the rearrangement of helix O to the Cl– pathway as its disruption impairs turnover, degrades the transport stoichiometry and relieves the inhibitory effect of the A399C A432CHg crosslink (Fig. 4-6).
Our results have several important implications for the exchange mechanism of the CLC transporters. First, the conformational changes occurring during H+/Cl– exchange in a CLC transporter involve regions located far from the Cl– transport pathway. While several reports previously suggested that this might be the case33-35, the identification of these conformational rearrangements and the demonstration that they are required for transport was missing. We found that movement of helix O is necessary for transport, as preventing this motion is sufficient to shut-down exchange, and that helix O is directly coupled to Tyr445. The conformational rearrangements undergone by helix O during transport are likely to be of limited scope; point mutations disrupting the contact between Ile402 and Tyr445 are sufficient to remove the inhibitory effect of the A399C A432CHg crosslink (Fig. 4, 5). This conclusion is also consistent with our observation that constraints placed on helix O have graded effects: small on the outside, medium effects in the center and strong in the internal region (Fig. 2). This pattern raises the possibility that only the intracellular half of helix O, below Ala396, moves relative to helices J and Q. A highly conserved glycine residue kinks helix O at position 393 (Supplementary Fig. 8a, b), just above Ala396, creates a potentially flexible hinge point separating the mobile intracellular half from the fixed extracellular portion, a scenario that is also compatible with the lack of effect induced by the intra-helix crosslinks between Ala396 and Ala399 (Supplementary Fig. 7). Alternatively, the crosslinks might inhibit a conformational change that is not part of the transport cycle. For example, it might inhibit an active to inactive transition, similar to that seen in mammalian CLC channels and exchangers, which involves a concerted rearrangement of both subunits as well as of their C-termini41-45 . While our results cannot rule out this possibility, we think it is unlikely. Several published lines of evidence suggest that that transport by CLC-ec1 does not entail such a concerted motion, but rather that individual monomers are fully functional17,19. Furthermore, in CLC channels mutations at the tyrosine corresponding to Tyr445 dramatically affect this concerted transition46 while similar mutations in CLC-ec1 have minor effects on turnover40, consistent with the idea that transport by this homologue does not entail such a transition.
The second major implication of our results is that the CLC transporters have two gates: Glu148 is the external gate and Tyr445 is a key element of the internal one. If Glu148 were the only gate regulating CLC transport then movements of helix O should affect the Cl– pathway through this residue alone. In contrast, we found that the movement of helix O is transduced to the Cl– pathway via a direct interaction between Ile402 and Tyr445 and that the Y445A mutant is also sufficient to remove the inhibitory effects of the crosslink. How movement of helix O and by consequence of Tyr445 is coupled to the external gate remains to be elucidated. Two, non-mutually exclusive possibilities come to mind. The most parsimonious explanation is that the permeant ions themselves couple the two gates by a mixture of steric and electrostatic interactions within the narrow confines of the Cl– transport pathway. This would explain how maneuvers lowering the ion pathway's affinity for ions degrade transport coupling39,40,47. Alternatively, helix O might be coupled to the external gate through the complex network of helices forming the CLC topology, for example via a contact with helix N. This would explain how mutations of residues far from the ion pathway can cause diseases by severely impacting CLC channel and transporter gating and voltage dependence31,45,48-50 or how Zn2+ binding to the N-O loop of CLC-4 inhibits transport35. These possibilities are not mutually exclusive; the inner and outer gates could be coupled through both the ion pathway and the protein. This dual mechanism could allow evolution to fine tune the gating of the CLC channels and transporters to fit diverse physiological roles that range from controlling acidification in intracellular compartments to muscle contraction and salt reabsorption in the kidney. Further work is needed to elucidate the coupling mechanism between the inner and outer gates in the CLC transporters.
A transport model for the CLC exchangers
Our findings, together with previous work, allow us to propose a novel model describing the CLC exchange cycle (Fig. 7). The experimental observations built into our proposal are: (i) the transport stoichiometry is 2 Cl–:1 H+3; (ii) Glu148 adopts three distinct conformations9,11,12; (iii) Glu148 and Tyr445 respectively form the external12 and internal gates (Fig. 5); (iv) opening of the two gates is coupled through helix O (Fig. 6); (v) Cl– and H+ binding is synergistic38; (vi) proton exchange with the intracellular solution occurs via Glu20351. State (1) represents the transporter in its apo and occluded state. A conformational change opens the inner gate (2) allowing 2 Cl– ions to bind from the intracellular side displacing Glu148 (3), allowing its protonation from the outside and triggering the closure of the inner gate. This gives rise to a fully loaded state of the transporter in its outward-facing conformation (4). A closed intracellular gate guarantees that the Cl– ions will exit to the extracellular solution. The still-protonated Glu148 then re-enters the ion transport pathway (5) and the proton from Glu148 is –somehow– transferred to Glu203 (6) to be released towards the intracellular solution (a). Every step in the proposed cycle is reversible and does not require additional ad-hoc assumptions to guarantee its proper function. The synergistic binding of the substrates gives rise to a fully loaded state in which 2 Cl– and 1 H+ are simultaneously bound to the transporter38 (Fig, 7d), a state that is explicitly forbidden for classical “ping-pong” alternating access exchange mechanisms. Our data suggests that helix O is part of the coupling machinery between the two gates and we hypothesize that this allows the simultaneous binding of substrates to occur while preventing ion slippage. Finally, the mechanism leading to opening of the inner gate in the apo state (Fig. 7a-b) cannot be inferred from known data and the structure of the ion-free CLC-ec1 is similar to that of the ion-bound form52. It is possible that the two states might exist in thermal equilibrium that is shifted towards state (3) by Cl– binding. Based on our finding that the A399C A432CHg crosslink inhibits Cl–/Cl– exchange as well as net Cl– transport (Fig. 2, 5), we hypothesize that it either prevents the transition between states (1) and (2) or between (3) and (4). This proposed cycle also explain other, more unusual, characteristics of the CLC transporters. For example, the direct correlation between the size of the side chain at the inner-gate position 445 and the stoichiometry of transport39,40 is easily rationalized as smaller residues do not efficiently seal the intracellular access of the pathway, so that state (4) becomes leaky to Cl–, therefore degrading coupling. Similarly, the presence of a pH-independent inner gate enables the CLC exchangers to sustain tight coupling even at acidic pHs, a necessity in light of their role in survival of enteric bacteria to the extreme acid challenges presented by the host's stomach53. The CLC channels might have originated because of mutations that led to the disruption of the seal formed by the inner gate. This predicts that remnants of the CLC exchange cycle should remain in the gating of the CLC channels. Indeed, the key role of Gluex in CLC channel gating is well documented12,54 and, in at least one case, channel gating entails transmembrane H+ transport55. Finally, in the CLC-0 and CLC-1 channels56 Thr471 and Ser537, which correspond to Ile402 in CLC-ec1, selectively affect the apparent affinity of blockers for the closed but not for the open state, suggesting that gating in these homologues entails a conformational change likely involving their intracellular vestibules56. Our results suggest that the conformational rearrangements of helix O might be evolutionarily conserved in the gating processes of both CLC channels and transporters. Further work is needed to elucidate how these movements lead to the coordinated opening and closing of the inner and outer gates of the CLC transporters, while in the CLC channels movement of the outer gate is sufficient to allow for ion permeation. Elucidating the conformational changes underlying gating of the CLC channels and transporters will allow us to understand how disease-causing mutations disrupt the function of these proteins and may enable design of novel therapies for these devastating conditions.
Figure 7. Transport cycle for Cl–H+ exchange in the CLC transporters.

The details of the transport model are given in the main text. The states are (1) Apo state with the inner (Tyr445) and outer (Glu148) gates closed. The inner gate opens (2) thereby allowing Cl– binding to CLC-ec1 (3). (4) The inner gate closes, the external gate opens and becomes protonated. (5) Cl– ions can move to the extracellular solution and the external gate closes while still protonated. (6) The proton goes to the intracellular proton acceptor, Glu203 from which it diffuses into the intracellular solution returning the transporter to the apo state.
Online Methods
Protein Purification
Expression and purification of WT and mutant CLC-ec1 was performed according to the published protocols39,51,57. In some preparations the detergent was exchanged from 5 mM DM (n-Decyl-β-D-Maltopyranoside) to 1 mM DMNG (2,2-dioctylpropane-1,3-bis-β-D-maltopyranoside). The protein remains stable in both detergents. The eluted protein was run on a Superdex 200 column (GE Healthcare) pre-equilibrated in 100 mM NaCl, 20 mM Hepes, 5 mM DM or 50 μM DMNG, pH 7.5 (buffer B) for liposome reconstitution and crystallography.
Hg-induced crosslink and PEGylation assay
After purification the protein sample reacted with a 4-fold molar ratio excess of HgCl2 for 1 hr at room temperature. Excess Hg2+ was removed by addition of 1mM EDTA and/or gel filtration. Successive re-runs of the purified protein showed that it remained stable.
To determine whether the introduced cysteines become crosslinked after incubation with Hg2+, we used a gel-shift assay (Supplementary Fig. 2). The purified protein was concentrated to ~1 mg/ml and 3 μl of 40 mM maleimide 5,000 polyethylene-glycol (mPEG5K; Sigma-Aldrich) were added to 20 μl of Hg-reacted and unreacted protein samples in the presence of 0.5 % Sodium Dodecyl Sulfate (SDS), to unfold the protein. Over time single cysteine mutants shift from their unreacted position to a single higher molecular weight band (MW) (Supplementary Fig. 2a) while the double mutants eventually convert to a second band of higher MW that corresponds to a doubly reacted protein (Supplementary Fig. 2b). The reaction of the protein with mPEG5K is completely prevented by incubation with a 4-fold molar excess of Hg2+ for 60 min (Supplementary Fig. 2c), indicating that Hg2+ oxidizes the cysteines. Furthermore, a 1:1 molar ratio of Hg2+ to protein is sufficient to protect both cysteines present in two different double mutants, A399C/A432C and A392C/T428C (Supplementary Fig. 2d, e), suggesting that Hg2+ induces the formation a crosslink. In all cases the proteins ran mostly as monodisperse peaks on gel filtration after crosslink formation (Supplementary Fig. 2f) indicating that the Hg-treatment was well tolerated. A 60 min incubation with Hg2+ was sufficient to completely crosslink all 7 double mutants (Supplementary Fig. 3).
Liposome Reconstitution
E. coli polar lipids (Avanti lipids) were dried under N2 and resuspended in pentane and dried again. The dry lipids were suspended to a final concentration of 20 mg/ml in reconstitution buffer (Buffer R) 300mM KCl, 25 mM citric acid, adjusted to pH 7.0 with NaOH, to which 35 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) was added. The suspension was bath sonicated to clarity, and after a 2 hrs incubation the purified protein was added to the desired concentration: 0.2–1 ug protein/mg lipid for proteoliposomes reconstitution and 1.5 μg/mg for 36Cl– uptake. Detergent was dialyzed out overnight in Buffer R, and the resulting liposomes were fast frozen in ethanol/dry ice and stored at –80°C.
Measurement of Cl– and H+ fluxes and rate determination
H+ and Cl– efflux measurements were carried out as described3,40. Briefly, H+ influx and/or Cl– efflux was initiated by the addition of 1 μl of 1 mg/ml of the K+ ionophore valinomycin, which plays a dual role: it shunts the voltage established by the Cl– gradient and sets the membrane potential to the K+ equilibrium potential. The time courses of H+ and Cl– fluxes were respectively monitored using a pH meter or an Ag:AgCl electrode. H+ influx was terminated by the addition of 1 μl of 1 mg/ml of the H+ ionophore Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) while Cl– efflux was terminated by the addition of 40 μl of 1.5 M n-octyl-β-d-glucopyranoside (Affymetrix) to dissolve liposomes. In most cases the time course of Cl– release was fit to a single exponential with a leak term, as previously described40. However, for slow mutants the time course of efflux was better approximated by a linear fit, as the time constant became very large (>>100 s). In these cases efflux was fit to:
Where Cl(t) is the [Cl] at time t, v is the velocity of efflux mediated by CLC-ec1 and vL is the velocity of the leak, which was experimentally determined from 6+ independent sets of protein-free liposomes v =1.24 ± 0.27 × 10–3 nMole Cl–.s–1. The turnover rate, γ, was calculated from the velocity as described40. As expected, for fast mutants the two approaches gave comparable estimate of γ.
Crystallization
The A399C/A432C mutant of CLC-ec1 was crystallized as a complex with a FAB fragment as previously described12,39. Briefly, protein was incubated with a 3-fold excess of FAB fragment and then purified over gel filtration pre-equilibrated in Buffer B with 50 μM DMNG. The complex, concentrated to 5–12 mg/mL, was mixed with an equal volume of 100 mM NaCl, 20–30% (w/v) PEG400, 50 mM ADA pH 6.5/ Hepes pH 7.5/ Tris pH 8.5. Crystals grew by vapor diffusion in sitting drop trays for 2–10 weeks at 20 °C and were directly harvested from the drop, flash-frozen and stored in liquid N2. Diffraction data were collected to 3.5 Å at the X25 beamline (Brookhaven NSLS), and were processed by HKL2000 (Table 2). Phases were obtained by molecular replacement using the WT protein in complex with FAB (PDB ID: 1OTS) 12 as the search model with ions, waters, and CLC-ec1 residues 397-401 and 429-434 removed12,58. Refinement was done by Phenix with initial rigid body refinement followed by several rounds of minimization rebuilding the region of the Cys399 and Cys432 crosslink59.
Binding measurements with Isothermal Titration Calorimetry
Cl– binding to the A399C/A432C mutant of CLC–ec1 was carried out as described36,38 using a nanoITC instrument (TA Instruments). For these experiments, the final purification step of the protein was purified over a gel filtration column pre-equilibrated in 100 mM Na-K-Tartrate, 20 mM Hepes, 50 μM DMNG, pH 7.5 (Buffer B0) and concentrated to 50-195 μM. The A399C/A432C mutant was not stable in the absence of Cl– upon incubation with Hg2+. Therefore, in this case the Hg2+ reaction was carried out in Buffer B. Excess Cl– and Hg2+ were successively removed by running the protein over a gel filtration column pre-equilibrated in buffer B0. The injection syringe was filled with buffer B0 with 50 mM KCl added, to achieve final Molar Ratios of 100-250. Each experiment consisted of 30-48 injections of 1 μl of the ligand solution at 3-4 min intervals into the experimental chamber kept under constant stirring at 350 rpm and at 25.0±0.1 °C. All solutions were filtered and degassed prior to use. The ITC data was fit to a single site Wiseman isotherm as described36,38 using the NanoAnalyze program from TA instruments.
Cl–/Cl– exchange
Cl– uptake measurements were carried out as previously described57,60. The averaged time course was fit to a saturating single exponential function of the form C(t)= aM*(1– exp(–t/τ)) for A399C/A432C and to a line C(t)= a0+K*t for A399C/A432CHg. The initial rate of uptake, K, was calculated as aM/τ for A399C/A432C and determined from the fit for A399C/A432CHg.
Mutagenesis
All mutagenesis is carried out with the Quickchange method (Stratagene) and the mutated genes fully sequenced.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors wish to thank C. Miller, Brandeis University, for the generous gift of 36Cl, O. Boudker, C. Nimigean, O. Andersen and members of the Accardi lab for helpful discussions and comments on the manuscript. This work was supported by US National Institutes of Health grant GM085232 and an Irma T. Hirschl- Monique Weill-Caulier Scholar Award (to A.A.).
Footnotes
Author Contributions
D.B., K.N. and A.P. performed experiments, D.B. and A.A. analyzed the data; A.A. designed research and wrote the paper; all authors contributed to the editing of the manuscript.
Accession Codes
The structure of the Hg2+-treated A399C A432C mutant of CLC-ec1 was deposited in the PDB database with accession number 4MQX.
References
- 1.Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit. Rev. Biochem. Mol. Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- 2.Accardi A, Picollo A. CLC channels and transporters: proteins with borderline personalities. Biochim Biophys Acta. 2010;1798:1457–1464. doi: 10.1016/j.bbamem.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Accardi A, Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- 4.Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- 5.Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- 6.Graves AR, Curran PK, Smith CL, Mindell JA. The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:5. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- 7.De Angeli A, et al. The nitrate/proton antiporter AtCLCa mediates nitrate accumulation in plant vacuoles. Nature. 2006;442:939–942. doi: 10.1038/nature05013. [DOI] [PubMed] [Google Scholar]
- 8.Stockbridge RB, et al. Fluoride resistance and transport by riboswitch-controlled CLC antiporters. Proc Natl Acad Sci U S A. 2012;109:15289–15294. doi: 10.1073/pnas.1210896109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 10.Jayaram H, Robertson JL, Wu F, Williams C, Miller C. Structure of a slow CLC Cl−/H+ antiporter from a cyanobacterium. Biochemistry. 2011;50:788–794. doi: 10.1021/bi1019258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng L, Campbell EB, Hsiung Y, MacKinnon R. Structure of a Eukaryotic CLC Transporter Defines an Intermediate State in the Transport Cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 13.Chen TY, Chen MF, Lin CW. Electrostatic control and chloride regulation of the fast gating of ClC-0 chloride channels. J. Gen. Physiol. 2003;122:641–651. doi: 10.1085/jgp.200308846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zúñiga L, et al. The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J. Physiol. 2004;555:671–682. doi: 10.1113/jphysiol.2003.060046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engh AM, Faraldo-Gomez JD, Maduke M. The mechanism of fast-gate opening in ClC-0. J. Gen. Physiol. 2007;130:335–349. doi: 10.1085/jgp.200709759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng L, Campbell EB, MacKinnon R. Molecular mechanism of proton transport in CLC Cl−/H+ exchange transporters. Proc Natl Acad Sci U S A. 2012;109:11699–11704. doi: 10.1073/pnas.1205764109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguitragool W, Miller C. CLC Cl−/H+ transporters constrained by covalent cross-linking. Proc. Natl. Acad. Sci. U. S. A. 2007;104:20659–20665. doi: 10.1073/pnas.0708639104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zdebik AA, et al. Determinants of anion-proton coupling in mammalian endosomal CLC proteins. J. Biol. Chem. 2008;283:4219–4227. doi: 10.1074/jbc.M708368200. [DOI] [PubMed] [Google Scholar]
- 19.Robertson JL, Kolmakova-Partensky L, Miller C. Design, function and structure of a monomeric ClC transporter. Nature. 2010;468:844–847. doi: 10.1038/nature09556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 21.Morth JP, et al. Crystal structure of the sodium-potassium pump. Nature. 2007;450:1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- 22.Olesen C, et al. The structural basis of calcium transport by the calcium pump. Nature. 2007;450:1036–1042. doi: 10.1038/nature06418. [DOI] [PubMed] [Google Scholar]
- 23.Reyes N, Ginter C, Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaback HR, Smirnova I, Kasho V, Nie Y, Zhou Y. The alternating access transport mechanism in LacY. J Membr Biol. 2011;239:85–93. doi: 10.1007/s00232-010-9327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forrest LR, Krämer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochem Biophys Acta. 2011;1807 doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 26.Perez C, Koshy C, Yildiz O, Ziegler C. Alternating-access mechanism in conformationally asymmetric trimers of the betaine transporter BetP. Nature. 2012;490:126–130. doi: 10.1038/nature11403. [DOI] [PubMed] [Google Scholar]
- 27.Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi Y. Common folds and transport mechanisms of secondary active transporters. Annu Rev Biophys. 2013;42:51–72. doi: 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- 29.Artigas P, Gadsby DC. Na+/K+-pump ligands modulate gating of palytoxin-induced ion channels. Proc Natl Acad Sci U S A. 2003;100:501–505. doi: 10.1073/pnas.0135849100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ludewig U, Jentsch TJ, Pusch M. Analysis of a protein region involved in permeation and gating of the voltage-gated Torpedo chloride channel ClC-0. J. Physiol. 1997;498:691–702. doi: 10.1113/jphysiol.1997.sp021893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pusch M, Steinmeyer K, Koch MC, Jentsch TJ. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the CIC-1 chloride channel. Neuron. 1995;15:1455–1463. doi: 10.1016/0896-6273(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 32.Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 2002;19:423–434. doi: 10.1002/humu.10063. [DOI] [PubMed] [Google Scholar]
- 33.Bell SP, Curran PK, Choi S, Mindell JA. Site-directed fluorescence studies of a prokaryotic ClC antiporter. Biochemistry. 2006;45:6773–6782. doi: 10.1021/bi0523815. [DOI] [PubMed] [Google Scholar]
- 34.Elvington S, Liu C, Maduke M. Substrate-driven conformational changes in ClC-ec1 observed by fluorine NMR. EMBO J. 2009;28:3090–3102. doi: 10.1038/emboj.2009.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osteen J, Mindell JA. Zn2+ inhibition of CLC-4. Biophysical journal. 2008;95:4668–4675. doi: 10.1529/biophysj.108.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picollo A, Malvezzi M, Houtman JC, Accardi A. Basis of substrate binding and conservation of selectivity in the CLC family of channels and transporters. Nat Struct Mol Biol. 2009;16:1294–1301. doi: 10.1038/nsmb.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jayaram H, Accardi A, Wu F, Williams C, Miller C. Ion permeation through a Cl- -selective channel designed from a CLC Cl−/H+ exchanger. Proc Natl Acad Sci U S A. 2008;105:6. doi: 10.1073/pnas.0804503105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Picollo A, Xu Y, Johner N, Bernèche S, Accardi A. Synergistic substrate binding determines the stoichiometry of transport of a prokaryotic H(+)/Cl(-) exchanger. Nat Struct Mol Biol. 2012;19:525–531. doi: 10.1038/nsmb.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Accardi A, Lobet S, Williams C, Miller C, Dutzler R. Synergism between halide binding and proton transport in a CLC-type exchanger. J. Mol. Biol. 2006;362:691–699. doi: 10.1016/j.jmb.2006.07.081. [DOI] [PubMed] [Google Scholar]
- 40.Walden M, et al. Uncoupling and turnover in a Cl−/H+ exchange transporter. J. Gen. Physiol. 2007;129:317–329. doi: 10.1085/jgp.200709756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bykova EA, Zhang XD, Chen TY, Zheng J. Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nat. Struct. Mol. Biol. 2006;13:1115–1119. doi: 10.1038/nsmb1176. [DOI] [PubMed] [Google Scholar]
- 42.Zifarelli G, Pusch M. Intracellular regulation of human ClC-5 by adenine nucleotides. Embo Rep. 2009;10:1111–1116. doi: 10.1038/embor.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Angeli A, et al. ATP binding to the C terminus of the Arabidopsis thaliana nitrate/proton antiporter, AtCLCa, regulates nitrate transport into plant vacuoles. J Biol Chem. 2009;284:26526–26532. doi: 10.1074/jbc.M109.005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orhan G, Fahlke C, Alekov AK. Anion- and proton-dependent gating of ClC-4 anion/proton transporter under uncoupling conditions. Biophys J. 2011;100:1233–1241. doi: 10.1016/j.bpj.2011.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leisle L, Ludwig CF, Wagner FA, Jentsch TJ, Stauber T. ClC-7 is a slowly voltage-gated 2Cl(−)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J. 2011;30:2140–2152. doi: 10.1038/emboj.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bennetts B, Parker MW. Molecular determinants of common gating of a ClC chloride channel. Nat Commun. 2013;4 doi: 10.1038/ncomms3507. [DOI] [PubMed] [Google Scholar]
- 47.Nguitragool W, Miller C. Uncoupling of a CLC Cl−/H+ exchange transporter by polyatomic anions. J. Mol. Biol. 2006;362:682–690. doi: 10.1016/j.jmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 48.Ludewig U, Jentsch TJ, Pusch M. Inward rectification in ClC-0 chloride channels caused by mutations in several protein regions. J. Gen. Physiol. 1997;110:165–171. doi: 10.1085/jgp.110.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wollnik B, Kubisch C, Steinmeyer K, Pusch M. Identification of functionally important regions of the muscular chloride channel CIC-1 by analysis of recessive and dominant myotonic mutations. Hum. Mol. Genet. 1997;6:805–811. doi: 10.1093/hmg/6.5.805. [DOI] [PubMed] [Google Scholar]
- 50.Saviane C, Conti F, Pusch M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 1999;113:457–468. doi: 10.1085/jgp.113.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Accardi A, et al. Separate ion pathways in a Cl−/H+ exchanger. J. Gen. Physiol. 2005;126:563–570. doi: 10.1085/jgp.200509417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lobet S, Dutzler R. Ion-binding properties of the ClC chloride selectivity filter. Embo J. 2006;25:24–33. doi: 10.1038/sj.emboj.7600909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iyer R, Iverson TM, Accardi A, Miller C. A biological role for prokaryotic ClC chloride channels. Nature. 2002;419:715–718. doi: 10.1038/nature01000. [DOI] [PubMed] [Google Scholar]
- 54.Traverso S, Elia L, Pusch M. Gating competence of constitutively open CLC-0 mutants revealed by the interaction with a small organic Inhibitor. J. Gen. Physiol. 2003;122:295–306. doi: 10.1085/jgp.200308784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lísal J, Maduke M. The ClC-0 chloride channel is a ‘broken’ Cl−/H+ antiporter. Nat Struct Mol Biol. 2008;15:805–810. doi: 10.1038/nsmb.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Accardi A, Pusch M. Conformational changes in the pore of CLC-0. J. Gen. Physiol. 2003;122:277–293. doi: 10.1085/jgp.200308834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Accardi A, Kolmakova-Partensky L, Williams C, Miller C. Ionic currents mediated by a prokaryotic homologue of CLC Cl− channels. J. Gen. Physiol. 2004;123:109–119. doi: 10.1085/jgp.200308935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maduke M, Pheasant DJ, Miller C. High-level expression, functional reconstitution, and quaternary structure of a prokaryotic ClC-type chloride channel. J. Gen. Physiol. 1999;114:713–722. doi: 10.1085/jgp.114.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.