

Summary
Endothelial dysfunction is a central hallmark of diabetes. The transcriptional coactivator PGC-1α is a powerful regulator of metabolism, but its role in endothelial cells remains poorly understood. We show here that endothelial PGC-1α expression is high in diabetic rodents and humans and that PGC-1α powerfully blocks endothelial migration in cell culture and vasculogenesis in vivo. Mechanistically, PGC-1α induces Notch signaling, blunts activation of Rac/Akt/eNOS signaling, and renders endothelial cells unresponsive to established angiogenic factors. Transgenic overexpression of PGC-1α in the endothelium mimics multiple diabetic phenotypes, including aberrant re-endothelialization after carotid injury, blunted wound healing, and reduced blood flow recovery after hindlimb ischemia. Conversely, deletion of endothelial PGC-1α rescues the blunted wound healing and recovery from hindlimb ischemia seen in type 1 and type 2 diabetes. Endothelial PGC-1α thus potently inhibits endothelial function and angiogenesis, and induction of endothelial PGC-1α contributes to multiple aspects of vascular dysfunction in diabetes.
Introduction
Vascular endothelial dysfunction predisposes diabetic patients to numerous cardiovascular complications, including critical limb ischemia, the leading cause of limb amputation worldwide (Hamilton et al., 2007; Rask-Madsen and King, 2007). Attenuated angiogenic response to tissue injury and hypoxia in diabetes likely contribute to the strong propensity to develop persistent decubitus and foot ulcers (Brem and Tomic-Canic, 2007; Falanga, 2005). The blunted angiogenesis may in part be caused by vascular endothelial growth factor (VEGF) resistance, in which there is reduced receptor signaling despite higher ligand expression, akin to insulin resistance (Sasso et al., 2005; Simons, 2005). Other signaling pathways have been also proposed to underlie diabetic endothelial dysfunction, including accelerated formation of advanced glycation endproducts, activation of protein kinase C, increased proinflammatory signaling, and impaired sensitivity of the phosphatidylinositol-3 kinase/Akt pathway (Potenza et al., 2009). However, the exact mechanisms by which angiogenic response is impaired in diabetes remain poorly understood.
PGC-1α is a transcriptional coactivator that regulates metabolism in numerous tissues. PGC-1α belongs to a small family of coactivators, comprised of PGC-1α, PGC-1β, and the more distant PRC (Rowe et al., 2010). Although originally named for its ability to coactivate PPARγ, it is now clear that PGC-1α coactivates a broad range of transcription factors, likely including most of the nuclear receptors. PGC-1α has widespread functions in different tissues. In hepatocytes, PGC-1α activates a gluconeogenic program in response to glucagon and low levels of insulin (Herzig et al., 2001; Yoon et al., 2001). In brown fat, PGC-1α responds to cold exposure and drives thermogenesis (Puigserver et al., 1998). In muscle and heart, PGC-1α powerfully activates a broad program of mitochondrial biogenesis (Lin et al., 2002; Russell et al., 2004). PGC-1α thus regulates metabolic programs in numerous cell types.
We have recently shown in muscle and heart that PGC-1α also regulates the formation of blood vessels (Arany et al., 2008; Patten et al., 2012), thus coordinating the control of fuel consumption in mitochondria with fuel delivery via blood vessels. PGC-1α regulates the expression of a number of angiogenic factors, including the canonical VEGF. Activation of VEGF is achieved by coactivation of ERRα, independently of hypoxia-inducible factor (HIF) activity. Transgenic expression of PGC-1α in myocytes dramatically increases capillary density in skeletal muscle and provides protection in an ischemic model (Arany et al., 2008). Conversely, deletion of PGC-1α in myocytes blocks the normal ability of mice to increase vascular content in response to exercise (Chinsomboon et al., 2009), and deletion of PGC-1α in cardiomyocytes leads to profound vascular defects and peripartum cardiomyopathy (Patten et al., 2012).
The above studies demonstrated that PGC-1α in myocytes critically regulates angiogenesis. The key cells that carry out angiogenesis, however, are the endothelial cells (ECs). The role of PGC-1α in ECs, if any, is much less studied. PGC-1α may have antiapoptotic properties in ECs (Won et al., 2010), as well as anti-ROS and anti-inflammatory properties (Borniquel et al., 2006; Kim et al., 2007; Valle et al., 2005). Studies of endothelial PGC-1α in intact animals are lacking. No studies have investigated a potential role of endothelial PGC-1α in diabetes. PGC-1α in the skeletal myocyte has been proposed to contribute to the etiology of insulin resistance (Mootha et al., 2003), although this remains controversial. Persistent upregulation of PGC-1α in the diabetic liver mediates hyperglycemia (Herzig et al., 2001; Yoon et al., 2001). Although these findings suggest broad implication of PGC-1α in diabetic pathophysiology, a role for PGC-1α in mediating or protecting against diabetic complications in the vasculature, if any, has not been investigated.
We show here, using both cell culture and a number of endothelial-specific genetic mouse models, that PGC-1α in ECs is induced by diabetes, strongly inhibits endothelial migration and angiogenesis in vivo, and mediates vascular dysfunction in diabetes.
Results
Diabetes Induces PGC-1α Expression in ECs
Diabetes is known to induce PGC-1α expression in the liver (Herzig et al., 2001; Yoon et al., 2001). To test the effects of diabetes on PGC-1α expression in ECs in vivo, mice were fed a high-fat diet (HFD) to induce type 2 diabetes and elevated glucose concentrations (Figure S1A available online). PGC-1α expression more than doubled in ECs isolated from either lung or heart tissue of these diabetic mice, compared to control animals (Figure 1A, left panels). PGC-1α was similarly induced in ECs in two genetic models of type 2 diabetes (ob/ob and db/db) and in streptozotocin-induced type 1diabetes (Figure 1A, right panels; Table S1). Human circulating CD34+ cells, which contain significant numbers of endothelial progenitor cells (EPCs) (Asahara et al., 1997; Fadini et al., 2006), isolated from patients with type 2 diabetes (see Table S2) as well as cultured EPCs established from the peripheral blood of diabetic patients also revealed elevated expression levels of PGC-1α, compared with control subjects (Figure 1B). Diabetes thus induces expression of PGC-1α in ECs in vivo in mice, and likely in humans as well.
Figure 1. Diabetes Induces PGC-1α Expression in ECs.
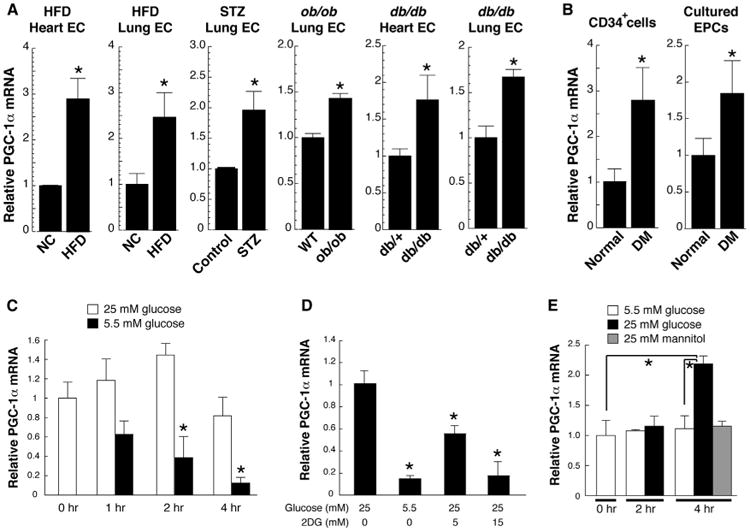
(A) Relative PGC-1α mRNA abundance in ECs freshly isolated from mouse models of type 1 (STZ) and type 2 (HFD, ob/ob, and db/db) diabetes. NC indicates normal chow.
(B) Relative PGC-1α mRNA abundance in vasculogenic circulating CD34+ cells and cultured endothelial progenitor cells (EPCs) isolated from patients with diabetes, versus matched normal subjects.
(C) Relative PGC-1α mRNA abundance in mouse muscle ECs (MMECs) at the indicated times after changing from hyperglycemia to normal glucose levels.
(D) Relative PGC-1α mRNA abundance in MMECs in absence or presence of 2-deoxyglucose (2DG) for 24 hr.
(E) Relative PGC-1α mRNA abundance in mouse heart ECs (MHECs) 2 and 4 hr after exposing cells to hyperglycemia or hyperosmolarity (mannitol). *p < 0.05. n = 3–5. Data are means ±SEM. See also Figure S1 and Tables S1 and S2.
The observation that both type 1 and type 2 diabetes induce PGC-1α expression in ECs suggested that hyperglycemia, rather than insulin resistance, stimulates endothelial PGC-1α. To test this, primary murine ECs were isolated from skeletal muscle, using standard techniques with CD31 antibody-coated bead cell capture. When these cells were cultured in high glucose (25 mM) and subsequently placed in physiological glucose concentrations (5.5 mM), the expression of PGC-1α rapidly declined by >90% within 4 hr (Figure 1C). This effect was mimicked by the addition of 2-deoxy glucose, an inhibitor of glycolysis (Figure 1D). Conversely, addition of high glucose to cells grown in low glucose induced PGC-1α mRNA expression (Figure 1E) and protein (Figure S1B). Treatment with mannitol had no effect, indicating that the induction of PGC-1α expression by high glucose is independent of osmotic effects (Figure 1E). Inhibition of reactive oxygen species (ROS) with N-acetyl cysteine (NAC) blocked the induction of PGC-1α (Figure S1C), while inhibition of O-GlcNAc transferase, the rate-limiting enzyme in glucose-mediated glycosylation pathways, did not (Figure S1C), suggesting that hyperglycemia-induced ROS production (Cosentino et al., 2003) (Figure S1D) mediates the induction of PGC-1α. Together, these data indicate that diabetes, likely at least in part via hyperglycemia, induces PGC-1α expression in ECs in vivo. Other pathways may also contribute.
PGC-1α Inhibits Endothelial Migration
Endothelial cell migration is a hallmark of endothelial function (Carmeliet and Jain, 2011) and is blunted in diabetes (Chen et al., 2007; Kränkel et al., 2005; Yan et al., 2009). Consistent with this, ECs isolated from streptozotocin (STZ)-treated animals, as well as from db/db animals, migrated significantly less in Transwell migration assays than did cells from control animals (Figures 2A, left panel, and 2B), as has been seen by others (Bae et al., 2013; Cui et al., 2011; Yan et al., 2009). EPCs isolated from diabetic patients revealed similarly blunted migration (Figure 2C). The blunting of migration coincided with induction of PGC-1α expression (Figures 1B and 2A, right panels), suggesting that PGC-1α may contribute to the decrease in cell migration. To test this notion, ECs were isolated from mice that overexpress PGC-1α specifically in ECs (see below for description of the mice). PGC-1α in these cells was induced 6-fold to 7-fold compared to cells from littermate controls (Figure 2D, right panel), a level of induction that is comparable to that seen in the STZ-treated wild-type animals (Figure 2A). ECs that overexpressed PGC-1α displayed significantly repressed migration, as measured in Transwell migration assays (Figure 2D, left). To validate this observation in cell-culture models, human umbilical vein ECs (HUVECs), primary mouse muscle ECs (MMECs), and primary human endothelial colony-forming cells (ECFCs) harvested from cord blood were transduced with retrovirus expressing PGC-1α, versus control. Endothelial migration was profoundly inhibited by expression of PGC-1α, as measured in scratch assays (Figure 2E) or in Transwell migration assays (Figures 2F–2G, S2A, and S2B). Overexpression of PGC-1α also inhibited the formation of tube-like structures by ECs (Figure 2H) while having little effect on cell viability (Figure S2C). Diverse promigratory stimuli, including VEGF (Figures 2F and S2B) or sphingosine 1-phosphate (S1P) (Figures 2G, S2A, and S2B), were blocked by PGC-1α. Interestingly, time-lapse videoscopy revealed that cells overexpressing PGC-1α remained capable of small-distance random vibration but failed to undergo directional cell movement (Movies S1 and S2). Consistent with this, the ability of VEGF and S1P to activate lamellipodia and stimulate assembly of subcortical F-actin was profoundly impaired (Figure 2I), mimicking defects seen in ECs isolated from db/db mice (Figure 2J), STZ-treated mice (Figure S2D), or diabetic patients (Figure S2E).
Figure 2. Endothelial PGC-1α Inhibits Cell Migration.
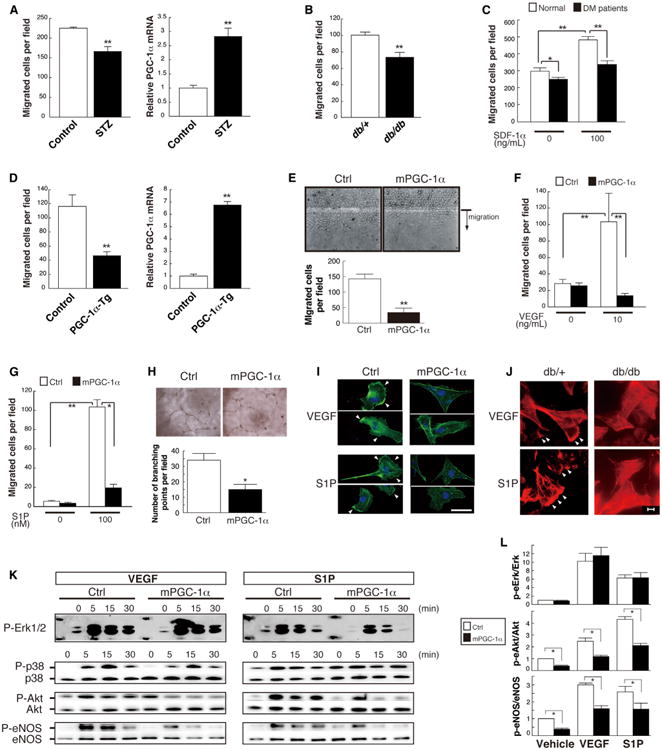
(A) Reduced migration of mouse heart ECs (MHECs) that were affinity-purified from STZ-treated mice and cultured to passage 2 (left), coincident with increased expression of PGC-1α mRNA (right).
(B and C) Reduced migration of mouse lung ECs (MLECs) isolated from db/db mice toward S1P (B) and cultured EPCs isolated from diabetic patients (C).
(D) Reduced migration (left) of MHECs isolated from mice that overexpress PGC-1α (right) in ECs.
(E–G) Stable expression of PGC-1α inhibits migration of HUVECs in scratch assays (E) and in Transwell assays stimulated by 10 ng/ml VEGF (F) or 100 nM S1P (G).
(H) Stable expression of PGC-1α inhibits the tube-forming activity of HUVECs.
(I–L) PGC-1α inhibits cortical F-actin (green) polymerization in response to VEGF and S1P (I), mimicking the impaired F-actin (red) accumulation to the same stimuli in db/db mouse lung ECs (J), and blocks activation of Akt and eNOS (but not Erk) by VEGF and S1P ([K], with densitometric quantification at 0 and 5 min in [L]) in human ECFCs. Arrowheads in (I) and (J) denote lamellipodia.*p < 0.05; **p < 0.01; n = 3–8. Data are means ±SEM. Scale bars are 50 μm in (I) and 20 μm in (J). See also Figure S2 and Movies S1 and S2.
VEGF and S1P act through distinct cell surface receptors (receptor tyrosine kinase versus G protein-coupled receptor, respectively) but converge on at least two intracellular signaling cascades: the ERK and Akt pathways. Endothelial cell cytoskeletal activation occurs in large part in response to Rac/Akt/eNOS signaling (Levine et al., 2007; Sawada et al., 2008). To test which signaling pathways are affected by PGC-1α, cells overexpressing PGC-1α were stimulated with VEGF or S1P, and activation of Akt, eNOS, and ERK was measured by phospho-protein western blotting. Overexpression of PGC-1α had little impact on activation of the ERK pathway by either VEGF or S1P (Figures 2K and 2L). By contrast, overexpression of PGC-1α strongly inhibited the activation of Akt (phosphorylation at Ser473) and eNOS (phosphorylation at Ser1177) by both VEGF and S1P (Figures 2K and 2L), as well as the production of NO (Figure S2F). PGC-1α thus inhibits the Akt/eNOS proangiogenic pathway. Interestingly, PGC-1α did not inhibit insulin-mediated Akt activation (Figure S2G), indicating that PGC-1α specifically suppresses only certain pathways.
PGC-1α Activates Notch Signaling to Inhibit Endothelial Migration
Notch signaling is increasingly recognized as a powerful inhibitor of endothelial migration and sprouting angiogenesis (Leslie et al., 2007; Lobov et al., 2007; Suchting et al., 2007; Williams et al., 2006). The Notch cell surface receptor is sequentially cleaved by matrix metalloproteases and γ-secretase, leading to nuclear localization of the Notch intracellular domain (NICD). NICD in turn induces the expression of key target genes including Hes1, Hey1, Nrarp, and RBPj, while repressing the expression of other genes, including Kdr (VEGFR2). Overexpression of PGC-1α in ECs increased the presence of the NICD (Figure 3A), and even mild induction of PGC-1α in cell culture (Figure S3A) or in vivo (Figure 3B) in ECs was sufficient to generate a gene expression signature of Notch activation that mimicked that seen in ECs treated with high glucose (Figure S3B) or harvested from STZ-treated mice (Figure S3C), from db/db mice (Figuer S3D), or from diabetic patients (Figure S3E). PGC-1α thus activates Notch signaling in ECs in culture and in vivo.
Figure 3. Endothelial PGC-1α Activates Notch to Inhibit Cell Migration.
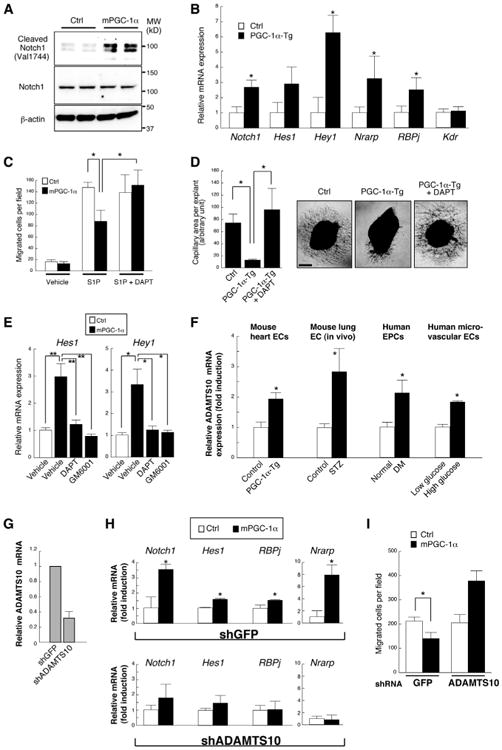
(A) Expression of PGC-1α in HUVECs activates Notch signaling, as assessed by western blotting of the Notch intracellular domain (NICD).
(B) mRNA expression of known Notch target genes in the endothelial compartment of the skeletal muscle from EC-specific PGC-1α-overexpressing mice, versus controls.
(C) Inhibition of Notch signaling with DAPT (2.5μM) rescues inhibition of migration in HUVECs retrovirally transduced with PGC-1α.
(D) Inhibition of Notch signaling with DAPT (2.5μM) rescues inhibition of capillary formation in mouse aortic explants from EC-specific PGC-1α-overexpressing mice.
(E) DAPT (2.5 μM) and pan-MMP inhibitor GM6001 (25 μM) equally inhibited the upregulation of Notch downstream gene mRNA expression in HUVECs retrovirally transduced with PGC-1α.
(F) Increased mRNA abundance of ADAMTS10 in the MHECs isolated from EC-specific PGC-1α transgenic mice, the endothelial compartment of STZ-induced diabetic mouse lung, cultured EPCs from diabetic patients, and human dermal microvascular ECs treated with high glucose for 24 hr.
(G) Gene knockdown efficiency by lentiviral transduction of HUVECs with short hairpin RNA targeting ADAMTS10.
(H and I) Knockdown of ADAMTS10 with shRNA abrogates the mRNA expression signature of Notch activation (H) and reduced Transwell migration toward S1P (I) by retroviral overexpression of PGC-1α in HUVECs. *p < 0.05 versus control. **p < 0.01. Data are means ±SEM. Scale bar indicates 500 μm. n = 3–18 for all groups. See also Figure S3.
Treating PGC-1α-overexpressing ECs with DAPT, a γ-secretase inhibitor, rescued the PGC-1α-mediated inhibition of migration (Figure 3C), indicating that the activation of Notch by PGC-1α is in part necessary for the inhibition of endothelial migration. Capillary sprouting from aortic explants, an ex vivo model of angiogenesis, was next used to test the effect of PGC-1α on capillary formation. Aortic explants were excised from mice overexpressing PGC-1α in ECs, versus littermate controls. Overexpression of PGC-1α strongly inhibited the formation of capillary sprouts in this model (Figure 3D). As with the inhibition of migration (Figure 3C), DAPT rescued the inhibition of capillary sprouting (Figure 3D). PGC-1α thus inhibits endothelial migration at least in part via activation of Notch signaling.
PGC-1α had no apparent impact on HIF-1α protein (Figure S3F) or on HIF-1α target genes (Figure S3G), echoing our previous results in skeletal muscle cells (Arany et al., 2008). PGC-1α also had only a mild effect on ROS-detoxifying enzymes (Figure S3H) and on the presence of ROS (Figure S3I), suggesting that the effect of PGC-1α on Notch likely involves other mechanisms. Inhibition of Notch signaling with either DAPT or GM6001, a pan-MMP inhibitor (Figure 3E), blocked the induction of Notch target genes by PGC-1α, indicating that PGC-1α likely acts upstream or at the level of the first Notch cleavage step by MMP/ADAM-family proteases. We next identified ADAMTS10, an ADAM-like MMP, as strongly induced in ECs from PGC-1α-overexpressing animals, as well as in ECs treated with high glucose and in ECs from diabetic animals or diabetic patients (Figure 3F). Knockdown of ADAMTS10 (Figure 3G) blocked the PGC-1α-mediated induction of Notch target genes (Figure 3H) as well as the PGC-1α-mediated suppression of endothelial migration (Figure 3I) (interestingly, migration is in fact increased by shADAMTS10). PGC-1α thus likely activates Notch signaling in part via the induction of ADAMTS10.
Loss of PGC-1α Activates EC Migration
The data above suggested that PGC-1α in ECs may act downstream of angiogenic stimuli. To test this, expression of PGC-1α was monitored after the addition of proangiogenic agents. As shown in Figure 4A, VEGF, S1P, and S-nitrosoglutathione (GSNO, an NO donor) rapidly inhibited the expression of PGC-1α in ECs—by as much as 90% within 4 hr. The suppression of PGC-1α expression was relieved by inhibition of PI-3K or Akt (Figure S4A). These data thus suggest that inhibition of PGC-1α may be necessary for EC activation and migration. To test directly the effects of loss of PGC-1α, ECs were isolated from PGC-1α-/- animals and littermate controls. The migratory capacity of the cells was then evaluated, using scratch and Transwell assays. In both cases, cells that lacked PGC-1α were markedly hypermigratory, to an extent comparable to control cells stimulated by VEGF (Figures 4B and 4C). Deletion of PGC-1α is therefore sufficient to mimic stimulation by VEGF. Capillary sprouting was also increased in aortic explants from PGC-1α-/- mice (Figure 4D), reflecting the key role of EC migration in capillary sprouting. The increased capillary-forming propensity of PGC-1α-/- vessels is reminiscent of the reported phenotypes of those deficient in Notch signaling (Leslie et al., 2007; Suchting et al., 2007). Indeed, the expression of Notch target genes was depressed in PGC-1α-/- cells (Figure 4E), mirroring the induction of the same genes in cells overexpressing PGC-1α (Figure 3B). Phosphorylation of Akt and eNOS was increased in PGC-1α-/- cells (Figure S4B); activation of Rac1 was constitutive in PGC-1α-/- ECs (Figure 4F), and inhibition of Rac (with NSC23766) or eNOS (with L-NAME or DPI) reversed the hypermigratory phenotype of PGC-1α-/- cells (Figure 4G), again indicating that PGC-1α inhibits the Rac/Akt/eNOS pathway. VEGF-induced ROS formation was not altered in PGC-1α-/- cells (Figure S4C), again showing the minimal contribution of ROS in mediating the antimigratory effect of PGC-1α. Together with the gain-of-function data shown in Figure 2, these data demonstrate that PGC-1α powerfully inhibits Rac/Akt/ eNOS signaling, and migration, in ECs.
Figure 4. Lack of PGC-1α Accelerates EC Migration and Decreases Notch Signaling.
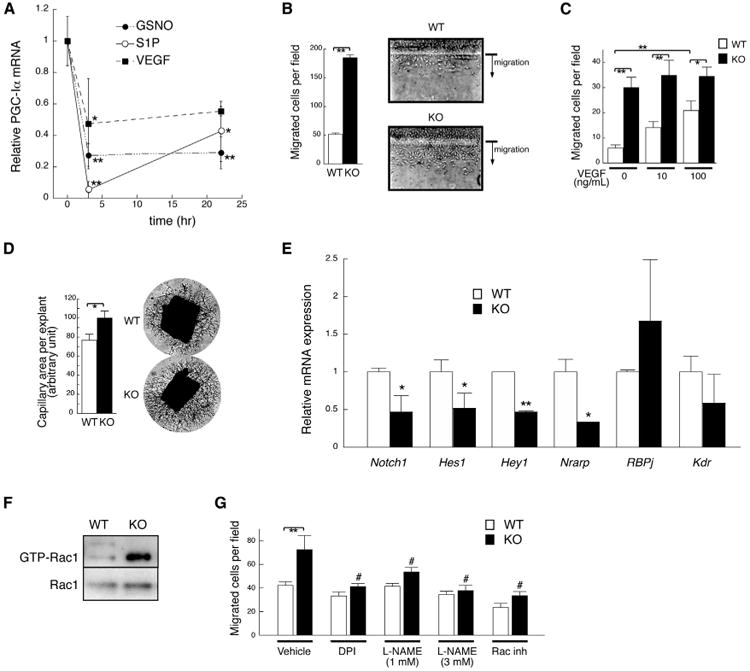
(A) Relative PGC-1α mRNA abundance in human dermal microvascular ECs stimulated with VEGF (10 ng/mL), S1P (100 nM), or S-nitrosoglutathione (GSNO, 100 μM).
(B and C) Isolated MHECs from PGC-1α-/- (KO) mice are hypermigratory in scratch assays (B) and Transwell migration assays (C).
(D) Capillary formation of KO (n = 31) and wild-type (WT) (n = 16) mouse aortic explants.
(E) mRNA expression of known Notch target genes in ECs isolated from KO mice, versus wild-type controls.
(F) GTP-Rac1 pull-down assay in WT and KO MHECs.
(G) VEGF (10 ng/ml)-induced Transwell migration of KO and WTMHECs, with or without DPI (diphenyleneiodonium chloride, 10 μM), L-NAME (L-NG-Nitroarginine Methyl Ester), or Rac inhibitor (NSC23766, 30 μM). *p< 0.05 versus control. **p < 0.01. The symbol # indicates p < 0.01 versus KO treated with vehicle. Data are means ±SEM. n = 3–18 for all groups. See also Figure S4.
Endothelial PGC-1α Inhibits Angiogenesis In Vivo
Endothelial cell migration is critical for the formation of new blood vessels. To test if PGC-1α inhibits the formation of blood vessels, we used an in vivo model of vasculogenesis in which ECFCs and mesenchymal progenitor cells are coinjected into nude mice (Yoder et al., 2007). Injection of ECFCs that overexpress PGC-1α significantly blunted the formation of new blood vessels in this model, compared to the use of control ECFCs (Figure 5A). To further test the role of endothelial PGC-1α in vivo, two double transgenic lines of mice with inducible expression of PGC-1α in ECs were generated, using either tetracycline-inhibited (“Tet-off”) or tetracycline-activated (“Tet-on”) versions (Figure 5B). Importantly, PGC-1α expression in the Tet-on mouse ECs in vivo was induced 5-fold to 7-fold (Figure 5C), roughly equivalent to the induction of PGC-1α seen with diabetes (Figure 1). Expression of PGC-1α in transgenic mouse endothelium did not affect baseline vascular density (Figure S5A) or systemic glucose handling (Figures S5B and S5C; Table S1), suggesting that endothelial PGC-1α does not play a prominent role in the systemic regulation of glucose levels.
Figure 5. Induced Expression of Endothelial PGC-1α In Vivo Phenocopies Diabetic Vascular Dysfunction.
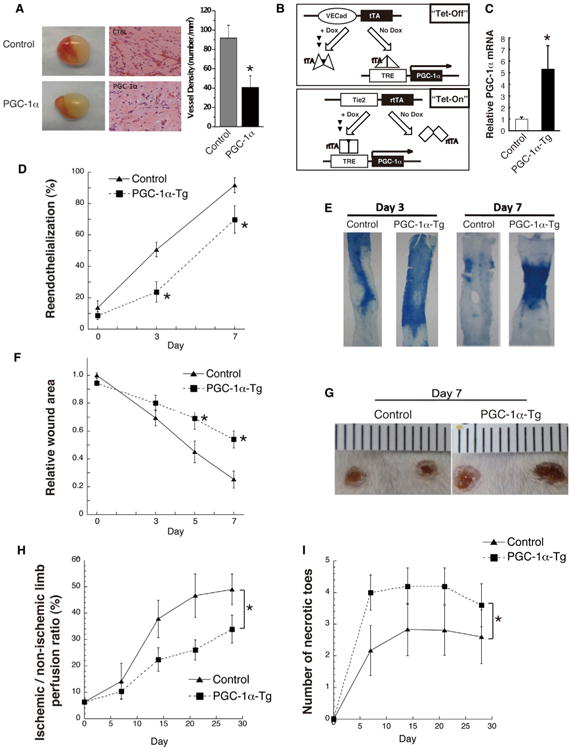
(A) In vivo vasculogenesis assay using mesenchymal progenitor cells and human ECFCs expressing PGC-1α versus control vector. Left, sample appearance of the Matrigel plugs. Middle, hematoxylin and eosin staining of plug sections. Right, quantification of vessel density.
(B) Schema of Tet-off and Tet-on tetracycline-inducible, endothelial-specific PGC-1α transgenic (Tg) mice.
(C) mRNA abundance of PGC-1α in freshly isolated lung ECs from transgenic animals versus control.
(D and E) Quantification (D) and representative images (E) of re-endothelialization following wire injury of mouse carotid arteries in Tet-off transgenic animals.
(F and G) Quantification (F) and representative images (G) of skin wound healing in Tet-off transgenic animals.
(H and I) Quantification of blood flow recovery (H) and necrotic toes (I) after induced hindlimb ischemia in Tet-on animals. n = 5–10 for all groups.*p < 0.05. Data are means ±SEM. See also Figure S5.
Endothelial cell migration is critical for re-endothelialization of vessel walls after wire injury, as occurs during percutaneous vascular interventions, and this process is dysfunctional in diabetes (Aronson et al., 1996; Mohan et al., 2008). Transgenic expression of PGC-1α in ECs strongly inhibited re-endothelialization in a carotid injury model (Figures 5D, 5E, and S5D). Endothelial cell migration and angiogenesis are also critical for various steps of wound healing, a process that is also dysfunctional in diabetes (Brem and Tomic-Canic, 2007; Falanga, 2005). Again, transgenic expression of PGC-1α, with both Tet-on and Tet-off lines, strongly inhibited the rate of wound healing in a dorsal skin punch injury model (Figures 5F, 5G, S5E, and S5F).
Normal EC function and NO production are also needed for vascular reactivity to endothelium-dependent vasodilatory agents such as acetylcholine. Impaired endothelial-dependent vasodilatory response is a hallmark of diabetic endothelial dysfunction (Rask-Madsen and King, 2007). Transgenic expression of PGC-1α had no effect on baseline blood pressure, systemic vascular resistance, or blood flow across the carotid artery (Figures S5G and S5H), but the ability to increase blood flow in response to acetylcholine challenge was significantly blunted (Figure S5H). Consistent with this, explanted vessels from mice expressing EC-PGC-1α revealed blunted vascular reactivity to carbachol compared to littermate controls (Figure S5I).
Finally, transgenic expression of PGC-1α also strongly inhibited the rate of recovery of blood flow in a murine hindlimb ischemia model of peripheral artery disease (Figures 5H and 5I), again a process that is dysfunctional in diabetes (Hazarika et al., 2007; Simons, 2005; Yan et al., 2009). These data thus demonstrate that expression of PGC-1α in ECs in vivo mimics multiple aspects of diabetic EC dysfunction. The effects are independent of tetracycline, because both “Tet-on” and “Tet-off” systems gave similar results. The effects are also almost certainly a consequence of expression of PGC-1α expression in the endothelium, because the effects are seen independently with transgenes driven by the divergent VE-cadherin and Tie2 promoters.
Loss of Endothelial PGC-1α Rescues Diabetic Endothelial Dysfunction
The above data suggested that vascular complications of diabetes are mediated, at least in part, via the induction of PGC-1α in the endothelium. To test this directly, mice lacking endothelial PGC-1α were generated via Cre/Lox recombination (Figure S6A). The PGC-1α locus was deleted with >80% efficiency, mRNA expression was repressed >70%, and PGC-1α protein was undetectable in isolated ECs from these animals (Figures S6B–S6D). The mice were then exposed to type 1 diabetes via STZ injection, or type 2 diabetes via high-fat feeding. Deletion of endothelial PGC-1α did not reduce the hyperglycemia seen with these models (EC-KO, 462.5 ± 55.2; control, 448.5 ± 42.7 mg/dl in type 1 diabetes model; see also Figures S6E and S6F), again suggesting that endothelial PGC-1α does not play a prominent role in the systemic regulation of glucose levels. Blood pressure and serum lipid profiles were also unaffected in EC-KO animals (Figure S6G; Table S1).
Wound healing is significantly retarded in the STZ model of type 1 diabetes (Seifter et al., 1981) and the high-fat fed model of type 2 diabetes (Nascimento and Costa, 2006; Seitz et al., 2010). As shown in Figures 6A and 6B, deletion of endothelial PGC-1α significantly accelerated wound healing in both of these diabetic models, rescuing the healing to nearly that seen in nondiabetic animals. The recovery of blood flow in murine hindlimbs after surgical induction of ischemia is also severely retarded in diabetic mice and often leads to necrotic limbs and autoamputation (Hazarika et al., 2007; Wang et al., 2009; Yan et al., 2009) (Figures 6C, 6E, 6F, and 6H). Again, deletion of PGC-1α in ECs reversed this defect in both type 1 and 2 diabetic models (Figures 6C–6H and S6H). Strikingly, the necrosis and loss of limbs that is frequently seen in diabetic animals after hindlimb ischemia was almost completely reversed in mice lacking endothelial PGC-1α (Figures 6E, 6F, and 6H). Deletion of PGC-1α in ECs also rescued the gene signature of Notch activation in ECs that is seen after STZ treatment (Figure S6I), indicating that PGC-1α is required for Notch activation by diabetes, a process implicated in reduced VEGF responsiveness of diabetic ECs (Cao et al., 2010). Together, these data indicate that deletion of PGC-1α in ECs in large part rescues numerous aspects of endothelial dysfunction in diabetes.
Figure 6. Ablation of Endothelial PGC-1α Rescues Diabetic Mice from Defective Wound Healing and Blood Flow Recovery in Ischemic Limbs.
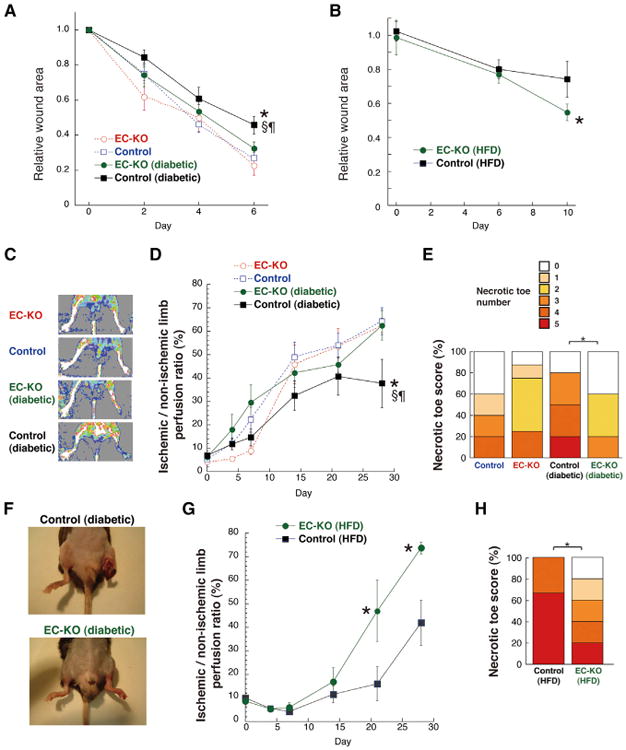
(A) Skin wound healing in endothelial-specific PGC-1α knockout (EC-KO) and control mice,either nondiabetic or rendered diabetic by treatment with STZ.
(B) Skin wound healing in EC-KO and control mice that were fed HFD for 12 months.
(C–F) Recovery of blood flow after surgical induction of hindlimb ischemia in type 1 diabetic EC-KO animals: (C) representative day 14 laser Doppler perfusion images, (D) quantification of blood perfusion recovery in the ischemic limbs, (E) number of necrotic toes in the ischemic foot at day 28, and (F) representative appearance of diabetic mouse ischemic limbs at day 14.
(G and H) Recovery of blood flow after hindlimb ischemia in type 2 diabetic EC-KO animals: (G) quantification of blood perfusion recovery in the ischemic limbs and (H) number of necrotic toes in the ischemic foot at day 21. n = 6–14. *p < 0.05 versus EC-KO; the symbol § indicates p < 0.05 versus control; the symbol ¶ indicates p < 0.05 versus EC-KO (diabetic). Data are means ±SEM. See also Figure S6.
Discussion
Diabetes and hyperglycemia are associated with significant microvascular dysfunction, leading to a large burden of disease worldwide. The data presented here indicate that PGC-1α is elevated in ECs from diabetic organisms, that elevated endothelial PGC-1α inhibits proper endothelial migration, and that deletion of endothelial PGC-1α rescues multiple aspects of vascular dysfunction in diabetes. We suggest that PGC-1α in ECs acts as a brake on endothelial activation that is normally overcome by stimulation with VEGF, S1P, and other angiogenic ligands. Hyperglycemia induces PGC-1α in ECs and accentuates the brakes on the system to a point that can no longer be overcome by angiogenic stimuli. As a result, PGC-1α inhibits vessel re-endothelialization, blunts wound healing, and worsens foot necrosis, all of which are characteristic features of diabetic vascular complications. Conversely, deletion of PGC-1α in ECs rescues these defects in diabetic models. These data thus indicate that PGC-1α mediates in part the vascular dysfunction caused by diabetes.
The data presented here identify PGC-1α as a potent negative regulator of endothelial migration, likely affecting both angiogenesis and vasculogenesis, and functioning at least in part via induction of Notch signaling. Notch signaling plays a central role in maintaining ECs in a relatively nonactivated, stalk-like phenotype during sprouting angiogenesis (Leslie et al., 2007; Roca and Adams, 2007; Suchting et al., 2007). The activation of Notch by PGC-1α is thus consistent with the induction of a quiescent state in ECs, resistant to migration or angiogenesis. Indeed, Notch inhibition has been shown to rescue VEGF resistance in diabetic ECs and to improve blood flow recovery in the murine hindlimb ischemia model (Cao et al., 2010). Activation of Notch by hyperglycemia and diabetes has also been described in renal podocytes (Ahn and Susztak, 2010; Lin et al., 2010; Niranjan et al., 2008) and in hepatocytes (Pajvani et al., 2011). The mechanism by which Notch is activated by hyperglycemia is not known, although interestingly, O-glucosylation of the extracellular domain of Notch is required for Notch activity (Acar et al., 2008; Rana and Haltiwanger, 2011). The data shown here indicate that PGC-1α is required for Notch activation in diabetic ECs. It will be of future interest to deconvolute how PGC-1α induces Notch activity and inhibits Akt/eNOS. It will also be of interest to investigate if a hyperglycemia/PGC-1α/Notch axis is also at play in kidney and liver cells.
The data here also reveal an important relationship between angiogenesis and a central regulator of metabolism. Metabolic regulation of ECs is increasingly garnering interest, especially in the context of cancer (Harjes et al., 2012). It will thus be of interest to understand the role of PGC-1α in modulating EC metabolism and understanding the role of metabolic perturbations, if any, in EC and Notch activation. Cellular replication and anabolism, such as occurs during angiogenesis, is generally thought to require an increase in glycolytic flux at the expense of fuel consumption in mitochondria. PGC-1α may inhibit this process and thus may contribute to EC quiescence in this manner as well.
Interestingly, baseline vascular density and development did not appear to be affected by either deletion or overexpression of endothelial PGC-1α in our mouse models. This suggests that PGC-1α primarily mediates postnatal physiological and pathological responses but has little role during development. This is consistent with observations in other tissues: absence of PGC-1α often has little effect at baseline but blunts responses to physiological stimuli (Arany et al., 2005; Chinsomboon et al., 2009; Lin et al., 2004).
Physiological endothelial activation is blunted in diabetes, but pathological endothelial activation contributes to multiple pathologies, including tumor angiogenesis and choroidal neovascularization in age-related macular degeneration. It will therefore be of interest to test if activation of endothelial PGC-1α can inhibit these processes. Similarly, endothelial activation and angiogenesis have also been proposed to contribute to atherosclerosis, although this remains controversial (Moulton et al., 2003). PGC-1α-/- mice are hyperactive and protected from diet-induced obesity and diabetes (Lin et al., 2004), both of which would be predicted to be antiatherosclerotic. Despite this fact, total-body deletion of PGC-1α had a net-neutral effect on atheromatous burden in ApoE-/- mice (Stein et al., 2010). Deletion of PGC-1α must thus also have proatherosclerotic effects, which may stem from deletion of PGC-1α in the endothelial compartment. It will therefore be of interest to test if endothelial PGC-1α modulates atherosclerosis.
In summary, the work presented here supports the notion that induction of PGC-1α in ECs mediates endothelial dysfunction and vascular complications in diabetes. The data thus in principle identify endothelial PGC-1α as a potential new therapeutic target. We have shown previously, however, that in cardiomyocytes and in skeletal myotubes, PGC-1α is proangiogenic by virtue of stimulating the secretion of numerous proangiogenic factors (Arany et al., 2008; Patten et al., 2012). Modulation of PGC-1α in different cells would thus have opposing effects. Similarly, activation of PGC-1α has been suggested as a therapeutic approach to improve insulin resistance, but the current findings suggest that this might be achieved at the expense of worsening diabetic complications. This striking observation stresses the need for caution in therapeutic endeavors aimed at PGC-1α in this and other contexts.
Experimental Procedures
Mouse and Human ECs
All procedures were performed using protocols approved at the relevant institution by Institutional Review Boards for human studies and Institutional Animal Care and Use Committees. Mouse ECs were isolated as described (Sawada et al., 2008, 2009). Isolated ECs were either immediately harvested to represent the endothelial compartment of mouse organs or cultured to passage 2 or 3 for cell migration and other assays. Human umbilical vein (HUVECs) and dermal microvascular ECs (HDMVECs) were purchased from Lonza. The preparation of CD34+ cells was modified from our previous reports for the preparation of CD133+ cells (Masuda et al., 2007, 2011; Nakamura et al., 2012). Human ECFCs were isolated as previously described (Melero-Martin et al., 2008).
Mouse Models
PGC-1α knockout mice are described (Lin et al., 2004). PGC-1α conditional allele knockin mice (Handschin et al., 2007) were backcrossed at least eight times onto the C57BL/6 strain. Two strains of Tie2-Cre transgenic mice were used for the study. The first line (Flavell) (Koni et al., 2001) was obtained from Jackson Laboratory (Bar Harbor). The second line (Yanagisawa)(Kisanuki et al., 2001) was obtained from Dr. Thomas N. Sato. Two independent lines of tetracycline-inducible, endothelial-specific PGC-1α transgenic mice were generated. The first line (“Tet-off” mice) was created as double Tg mice by crossing VE-cadherin promoter-driven tetracycline transactivator (tTA) Tg mice (VECad-tTA) (Sun et al., 2005) with tetracycline response element-driven PGC-1α Tg mice (TRE-PGC-Iα) (Russell et al., 2004), a generous gift from Dr. Dan Kelly. The second line (“Tet-on” mice) was similarly generated by crossing Tie2-promoter/enhancer-driven reverse tetracycline transactivator (rtTA) Tg mice (Tie2-rtTA) (Teng et al., 2002), purchased from the Jackson Laboratory, with TRE-PGC-1α mice. We used four diabetic mouse models: (i) STZ administration, (ii) high-fat feeding (HFD) using Rodent Diet with 60 kcal% fat (cat# D12492, Research Diets Inc., New Brunswick), (iii) ob/ob mice (Charles River, Yokohama), and (iv) db/db mice (CLEA Japan, Tokyo). Endothelial denudation of the common carotid artery was performed with five passes of an 0.14 in guide wire, which was inserted through an arteriotomy in external carotid artery. Carotid artery blood flow was recorded using an ultrasonic flow probe placed on the artery and sampled at 0.1 kHz using a transit-time perivascular flow meter (Transonic, MA0.5PSB and TS420, respectively). Wound healing assay in vivo was performed according to a published method (Dioufa et al., 2010). Hindlimb ischemia was performed as described previously (Sawada et al., 2008).
Reagents and Cellular Assays
Anti-PGC-1α (H300) antibody was from Santa Cruz. Lentivirus gene knockdown constructs were from RNAi Consortium (Broad Institute, Cambridge). Tube formation assays (Jiang et al., 2009) and capillary sprouting assays (Sawada etal., 2008, 2009) were performed as described previously. The intracellular ROS levels of cells were determined using CM-H2DCFDA (Molecular Probe). NO measurements were performed with 10 μM diaminofluorescein-2 (DAF-2) (Cayman Chemical, Ann Arbor), an NO-sensitive fluorescent dye. Vascular reactivity of carotid artery explants to carbachol was performed as described (Kanda et al., 2009).
Statistics
All data are expressed as means ±SEM. One-way or two-way ANOVA, followed by post hoc Fisher's test, was used for comparison between groups. Values of p of <0.05 were considered significant. See Supplemental Experimental Procedures for full description of the materials and methods.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
N.S. was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology; the Takeda Science Foundation; the Japan Vascular Disease Research Foundation; the Astra Zeneca Research Grant; and the Suzuken Memorial Foundation. H.K. and A.J. were supported by postdoctoral fellowships from the AHA. J.B. was supported by HL094262. A.M. was supported by the CAPES foundation. C.W.K. was supported by HL076136. F.A.J. was supported by HL108229. T.M. was supported by grants from the NHLBI and NIGMS, and Z.A. was supported by the ADA, the Ellison Medical Foundation, and the NHLBI.
Footnotes
Supplemental Information: Supplemental Information includes six figures, two tables, two movies, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2013.12.014.
References
- Acar M, Jafar-Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA, Pan H, Haltiwanger RS, Bellen HJ. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell. 2008;132:247–258. doi: 10.1016/j.cell.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SH, Susztak K. Getting a notch closer to understanding diabetic kidney disease. Diabetes. 2010;59:1865–1867. doi: 10.2337/db10-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun GD, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- Aronson D, Bloomgarden Z, Rayfield EJ. Potential mechanisms promoting restenosis in diabetic patients. J Am Coll Cardiol. 1996;27:528–535. doi: 10.1016/0735-1097(95)00496-3. [DOI] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Bae ON, Wang JM, Baek SH, Wang Q, Yuan H, Chen AF. Oxidative stress-mediated thrombospondin-2 upregulation impairs bone marrow-derived angiogenic cell function in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2013;33:1920–1927. doi: 10.1161/ATVBAHA.113.301609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 2006;20:1889–1891. doi: 10.1096/fj.05-5189fje. [DOI] [PubMed] [Google Scholar]
- Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest. 2007;117:1219–1222. doi: 10.1172/JCI32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Arany PR, Kim J, Rivera-Feliciano J, Wang YS, He Z, Rask-Madsen C, King GL, Mooney DJ. Modulating Notch signaling to enhance neovascularization and reperfusion in diabetic mice. Biomaterials. 2010;31:9048–9056. doi: 10.1016/j.biomaterials.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Lin SJ, Lin FY, Wu TC, Tsao CR, Huang PH, Liu PL, Chen YL, Chen JW. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes. 2007;56:1559–1568. doi: 10.2337/db06-1103. [DOI] [PubMed] [Google Scholar]
- Chinsomboon J, Ruas J, Gupta RK, Thom R, Shoag J, Rowe GC, Sawada N, Raghuram S, Arany Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc Natl Acad Sci USA. 2009;106:21401–21406. doi: 10.1073/pnas.0909131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino F, Eto M, De Paolis P, van der Loo B, Bachschmid M, Ullrich V, Kouroedov A, Delli Gatti C, Joch H, Volpe M, Lüscher TF. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation. 2003;107:1017–1023. doi: 10.1161/01.cir.0000051367.92927.07. [DOI] [PubMed] [Google Scholar]
- Cui X, Chopp M, Zacharek A, Ye X, Roberts C, Chen J. Angiopoietin/Tie2 pathway mediates type 2 diabetes induced vascular damage after cerebral stroke. Neurobiol Dis. 2011;43:285–292. doi: 10.1016/j.nbd.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dioufa N, Schally AV, Chatzistamou I, Moustou E, Block NL, Owens GK, Papavassiliou AG, Kiaris H. Acceleration of wound healing by growth hormone-releasing hormone and its agonists. Proc Natl Acad Sci USA. 2010;107:18611–18615. doi: 10.1073/pnas.1013942107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini GP, Sartore S, Albiero M, Baesso I, Murphy E, Menegolo M, Grego F, Vigili de Kreutzenberg S, Tiengo A, Agostini C, Avogaro A. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol. 2006;26:2140–2146. doi: 10.1161/01.ATV.0000237750.44469.88. [DOI] [PubMed] [Google Scholar]
- Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–1743. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- Hamilton SJ, Chew GT, Watts GF. Therapeutic regulation of endothelial dysfunction in type 2 diabetes mellitus. Diab Vasc Dis Res. 2007;4:89–102. doi: 10.3132/dvdr.2007.026. [DOI] [PubMed] [Google Scholar]
- Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, Neubauer N, Hu J, Mootha VK, Kim YB, et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J Clin Invest. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjes U, Bensaad K, Harris AL. Endothelial cell metabolism and implications for cancer therapy. Br J Cancer. 2012;107:1207–1212. doi: 10.1038/bjc.2012.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazarika S, Dokun AO, Li Y, Popel AS, Kontos CD, Annex BH. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ Res. 2007;101:948–956. doi: 10.1161/CIRCRESAHA.107.160630. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Jiang A, Hu W, Meng H, Gao H, Qiao X. Loss of VLDL receptor activates retinal vascular endothelial cells and promotes angiogenesis. Invest Ophthalmol Vis Sci. 2009;50:844–850. doi: 10.1167/iovs.08-2447. [DOI] [PubMed] [Google Scholar]
- Kanda T, Brown JD, Orasanu G, Vogel S, Gonzalez FJ, Sartoretto J, Michel T, Plutzky J. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J Clin Invest. 2009;119:110–124. doi: 10.1172/JCI36233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Park KG, Yoo EK, Kim YH, Kim YN, Kim HS, Kim HT, Park JY, Lee KU, Jang WG, et al. Effects of PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid Redox Signal. 2007;9:301–307. doi: 10.1089/ars.2006.1456. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kränkel N, Adams V, Linke A, Gielen S, Erbs S, Lenk K, Schuler G, Hambrecht R. Hyperglycemia reduces survival and impairs function of circulating blood-derived progenitor cells. Arterioscler Thromb Vasc Biol. 2005;25:698–703. doi: 10.1161/01.ATV.0000156401.04325.8f. [DOI] [PubMed] [Google Scholar]
- Leslie JD, Ariza-McNaughton L, Bermange AL, McAdow R, Johnson SL, Lewis J. Endothelial signalling by the Notch ligand Deltalike 4 restricts angiogenesis. Development. 2007;134:839–844. doi: 10.1242/dev.003244. [DOI] [PubMed] [Google Scholar]
- Levine YC, Li GK, Michel T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK → Rac1 → Akt → endothelial nitric-oxide synthase pathway. J Biol Chem. 2007;282:20351–20364. doi: 10.1074/jbc.M702182200. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, et al. Transcriptional coactivator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jäger S, Vianna CR, Reznick RM, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin CL, Wang FS, Hsu YC, Chen CN, Tseng MJ, Saleem MA, Chang PJ, Wang JY. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes. 2010;59:1915–1925. doi: 10.2337/db09-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA. 2007;104:3219–3224. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H, Kalka C, Takahashi T, Yoshida M, Wada M, Kobori M, Itoh R, Iwaguro H, Eguchi M, Iwami Y, et al. Estrogen-mediated endothelial progenitor cell biology and kinetics for physiological postnatal vasculogenesis. Circ Res. 2007;101:598–606. doi: 10.1161/CIRCRESAHA.106.144006. [DOI] [PubMed] [Google Scholar]
- Masuda H, Alev C, Akimaru H, Ito R, Shizuno T, Kobori M, Horii M, Ishihara T, Isobe K, Isozaki M, et al. Methodological development of a clonogenic assay to determine endothelial progenitor cell potential. Circ Res. 2011;109:20–37. doi: 10.1161/CIRCRESAHA.110.231837. [DOI] [PubMed] [Google Scholar]
- Melero-Martin JM, De Obaldia ME, Kang SY, Khan ZA, Yuan L, Oettgen P, Bischoff J. Engineering robust and functional vascular networks in vivo with human adult and cord blood-derived progenitor cells. Circ Res. 2008;103:194–202. doi: 10.1161/CIRCRESAHA.108.178590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan S, Reddick RL, Musi N, Horn DA, Yan B, Prihoda TJ, Natarajan M, Abboud-Werner SL. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Lab Invest. 2008;88:515–528. doi: 10.1038/labinvest.2008.23. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, Lo KM, Gillies S, Javaherian K, Folkman J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci USA. 2003;100:4736–4741. doi: 10.1073/pnas.0730843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Tsutsumi V, Torimura T, Naitou M, Iwamoto H, Masuda H, Hashimoto O, Koga H, Abe M, Ii M, et al. Human peripheral blood CD34-positive cells enhance therapeutic regeneration of chronically injured liver in nude rats. J Cell Physiol. 2012;227:1538–1552. doi: 10.1002/jcp.22873. [DOI] [PubMed] [Google Scholar]
- Nascimento AP, Costa AM. Overweight induced by high-fat diet delays rat cutaneous wound healing. Br J Nutr. 2006;96:1069–1077. doi: 10.1017/bjn20061955. [DOI] [PubMed] [Google Scholar]
- Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, Susztak K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14:290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- Pajvani UB, Shawber CJ, Samuel VT, Birkenfeld AL, Shulman GI, Kitajewski J, Accili D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat Med. 2011;17:961–967. doi: 10.1038/nm.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L, Hacker MR, Rhee JS, Mitchell J, Mahmood F, et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012;485:333–338. doi: 10.1038/nature11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenza MA, Gagliardi S, Nacci C, Carratu' MR, Montagnani M. Endothelial dysfunction in diabetes: from mechanisms to therapeutic targets. Curr Med Chem. 2009;16:94–112. doi: 10.2174/092986709787002853. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Rana NA, Haltiwanger RS. Fringe benefits: functional and structural impacts of O-glycosylation on the extracellular domain of Notch receptors. Curr Opin Struct Biol. 2011;21:583–589. doi: 10.1016/j.sbi.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Madsen C, King GL. Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21:2511–2524. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res. 2010;107:825–838. doi: 10.1161/CIRCRESAHA.110.223818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M, Scardone M, Marra C, Nasti R, Marfella R, Cozzolino D, et al. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J Am Coll Cardiol. 2005;46:827–834. doi: 10.1016/j.jacc.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Sawada N, Salomone S, Kim HH, Kwiatkowski DJ, Liao JK. Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ Res. 2008;103:360–368. doi: 10.1161/CIRCRESAHA.108.178897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada N, Kim HH, Moskowitz MA, Liao JK. Rac1 is a critical mediator of endothelium-derived neurotrophic activity. Sci Signal. 2009;2:ra10. doi: 10.1126/scisignal.2000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifter E, Rettura G, Padawer J, Stratford F, Kambosos D, Levenson SM. Impaired wound healing in streptozotocin diabetes. Prevention by supplemental vitamin A. Ann Surg. 1981;194:42–50. doi: 10.1097/00000658-198107000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz O, Schürmann C, Hermes N, Müller E, Pfeilschifter J, Frank S, Goren I. Wound healing in mice with high-fat diet- or ob gene-induced diabetes-obesity syndromes: a comparative study. Exp Diabetes Res. 2010;2010:476969. doi: 10.1155/2010/476969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M. Angiogenesis, arteriogenesis, and diabetes: paradigm reassessed? J Am Coll Cardiol. 2005;46:835–837. doi: 10.1016/j.jacc.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Stein S, Lohmann C, Handschin C, Stenfeldt E, Borén J, Lüscher TF, Matter CM. ApoE-/- PGC-1α-/- mice display reduced IL-18 levels and do not develop enhanced atherosclerosis. PLoS ONE. 2010;5:e13539. doi: 10.1371/journal.pone.0013539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchting S, Freitas C, le Noble F, Benedito R, Bréant C, Duarte A, Eichmann A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA. 2007;104:3225–3230. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, Benjamin LE. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci USA. 2005;102:128–133. doi: 10.1073/pnas.0403198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng PI, Dichiara MR, Kömüves LG, Abe K, Quertermous T, Topper JN. Inducible and selective transgene expression in murine vascular endothelium. Physiol Genomics. 2002;11:99–107. doi: 10.1152/physiolgenomics.00059.2002. [DOI] [PubMed] [Google Scholar]
- Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Wang CY, Kim HH, Hiroi Y, Sawada N, Salomone S, Benjamin LE, Walsh K, Moskowitz MA, Liao JK. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci Signal. 2009;2:ra11. doi: 10.1126/scisignal.2000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CK, Li JL, Murga M, Harris AL, Tosato G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood. 2006;107:931–939. doi: 10.1182/blood-2005-03-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won JC, Park JY, Kim YM, Koh EH, Seol S, Jeon BH, Han J, Kim JR, Park TS, Choi CS, et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha overexpression prevents endothelial apoptosis by increasing ATP/ADP translocase activity. Arterioscler Thromb Vasc Biol. 2010;30:290–297. doi: 10.1161/ATVBAHA.109.198721. [DOI] [PubMed] [Google Scholar]
- Yan J, Tie G, Park B, Yan Y, Nowicki PT, Messina LM. Recovery from hind limb ischemia is less effective in type 2 than in type 1 diabetic mice: roles of endothelial nitric oxide synthase and endothelial progenitor cells. J Vasc Surg. 2009;50:1412–1422. doi: 10.1016/j.jvs.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R, Temm CJ, Prchal JT, Ingram DA. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.