

Abstract
The effects of valvular endothelial cell (VlvEC) paracrine signaling on VIC phenotype and nodule formation were tested using a co-culture platform with physiologically relevant matrix elasticities and diffusion distance. 100μm thin poly(ethylene glycol) (PEG) hydrogels of 3 to 27 kPa Young’s moduli were fabricated in transwell inserts. VICs were cultured on the gels, as VIC phenotype is known to change significantly within this range, while VlvECs lined the underside of the membrane. Co-culture with VlvECs significantly reduced VIC activation to the myofibroblast phenotype on all gels with the largest percent decrease on the 3 kPa gels (~70%), while stiffer gels resulted in approximately 20–30% decrease. Additionally, VlvECs significantly reduced αSMA protein expression (~2 fold lower) on both 3 and 27 kPa gels, as well as the number (~2 fold lower) of nodules formed on the 27kPa gels. Effects of VlvECs were prevented when nitric oxide (NO) release was inhibited with L-NAME, suggesting that VlvEC produced NO inhibits VIC activation. Withdrawal of L-NAME after 3, 5, and 7 days with restoration of VlvEC NO production for 2 additional days led to a partial reversal of VIC activation (~25% decrease). A potential mechanism by which VlvEC produced NO reduced VIC activation was studied by inhibiting initial and mid-stage cGMP pathway molecules. Inhibition of soluble guanylyl cyclase (sGC) with ODQ or protein kinase G (PKG) with RBrcGMP or stimulation of Rho kinase (ROCK) with LPA, abolished VlvEC effects on VIC activation. This work contributes substantially to the understanding of the valve endothelium’s role in preventing VIC functions associated with aortic valve stenosis initiation and progression.
1. Introduction
Fibrocalcific aortic valve disease (FCAVD) is typically classified by fibrosis and calcification of the aortic heart valve. This disease affects a surprisingly large portion of the US population, with 2–4% of adults over the age of 65 diagnosed with FCAVD and 70,000 aortic valve replacement surgeries occurring annually [1]–[3]. While CAVD was initially thought to result from a progressive wearing out of the valve tissue over time, it is now understood to be a dynamic process, which involves resident valvular interstitial cells (VICs) [1]. VICs are critical in maintaining aortic valve homeostasis and function through many actions, such as proliferation [4]–[6], secretion of matrix metalloproteinases (MMPs) [4], [5], [7], [8], and de novo extracellular matrix (ECM) molecules [4]–[6], [9]–[11]. However, when inflammatory signaling is prolonged with repeated valve injury, as in hypertension or diabetes [12], [13], regulation of VIC phenotype can be lost. If myofibroblastic VICs persist, VICs can increase valve stiffness through excess remodeling of the valve tissue (e.g., excess collagen deposition), and cause VICs to express genes associated with osteogenesis [14], [15]. This process of pathological VIC activation to secretory myofibroblasts and eventually osteoblast-like cells is facilitated by several factors, including the potent cytokine transforming growth factor-β1 (TGF-β1) [4], [7]. Unfortunately, very few cues are known to prevent VIC activation, like fibroblast growth factor (FGF) [16] and no small molecule or drug has been found that reverses VIC activation in vivo or in vitro, once it has occurred.
VICs reside in the intersitium of the valve tissue, and this tissue is lined with valvular endothelial cells (VlvECs) [17], [18] that provide a protective and selective barrier between blood and VICs [19], [20]. Interestingly, initiation of aortic sclerosis (i.e., an early stage of FCAVD) has been linked to endothelial cell dysfunction [17], [21]. Endothelial cells appear to be a critical modulator of the progression of CAVD by regulation of VIC activation and calcification [17], [22], [23], [24], [25]. This regulation of VIC phenotype has been correlated with VlvEC produced nitric oxide (NO), as synthetically generated NO can significantly decrease calcific nodule formation in VIC cultures on treated tissue culture plastic [26]. But tissue culture plastic has an elastic modulus of ~1 GPa, which is known to cause high levels of VIC myofibroblast activation [14]. Co-culture of VICs with vascular endothelial cells (VECs) isolated from the aorta, or addition of synthetically generated NO, also decrease activation of VICs cultured in osteogenic media (supplemented with dexamethasone) [24]. However, endothelial cells likely secrete multiple beneficial signaling factors (e.g., fibroblast growth factor (FGF) [27] and prostaglandins [28]), which might also suppress VIC activation.
Previous studies have yielded valuable insight into potential signaling pathways associated with endothelial regulation of VIC phenotype, although activation of VICs has been induced primarily with biochemical factors (e.g., dexamethasone). Additionally, less is known about the role of microenvironmental elasticities, which is an important aspect of valve disease progression toward FCAVD (i.e., tissue stiffening). Thus, we sought to use a simple co-culture system that would allow one to study the role of microenvironmental mechanical cues on VIC activation in the presence of VlvECs. We tested the hypothesis that matrix elasticity may modulate the interaction of VICs and VlvECs, as the ability of biochemical cues to reverse or inhibit VIC activation may vary with the local microenvironmental stiffness.
Culture substrate elasticity has been shown to direct the activation of VICs to myofibroblasts [10], [15], [29]. Specifically, Kloxin et al. demonstrated that culturing VICs on a substrate with a Young’s modulus (E) of 33 kPa, VICs differentiated into a predominately myofibroblastic population (~80%) [29]. Conversely, when VICs were cultured on a softer substrate (E~7 kPa), the population was mainly fibroblastic. Clearly, biophysical cues are an important aspect of VIC phenotype regulation, and having adept control of the culture substrate elasticity provides a useful tool for studying the role of mechanical signals in FCAVD initiation and progression. We have used peptide-functionalized poly(ethylene glycol) (PEG) hydrogels as a VIC culture platform that facilitates control of both biophysical and biochemical microenvironmental cues [9], [14], [29], [30], [31]. Part of the motivation for using PEG-based materials relates to their high water content (similar to that of many soft tissues), minimal nonspecific adsorption of protein, and mechanical moduli of physiological relevance to valve tissue [10], [32], [33].
In this study, VICs were cultured on ~100 μm thin PEG hydrogels to allow for a physiologically relevant diffusion distance between VlvECs and VICs, while the crosslinking density of the gels was controlled to yield materials of varying Young’s moduli. This range of elasticity was selected to direct VIC activation from mostly quiescent fibroblasts to activated myofibroblasts, allowing control of the percentage of myofibroblasts in the VIC population, which increases during valve tissue stiffening associated with FCAVD progression [34], [35]. Then, the effect of VlvECs on VIC phenotype and nodule formation were assessed by lining the underside of the insert membrane with VlvECs and analyzing αSMA immunostaining or bright field images of formed nodules, respectively. The key paracrine signaling molecule and its mechanism were studied with small molecule inhibititors and assessment of VIC activation.
2. Materials and Methods
2.1. Materials
Eight-armed poly(ethylene glycol) (PEG, Mn: 20 & 40 kDa) was purchased from JenKem. All amino acids and resin for solid phase peptide synthesis were purchased from Chem-Impex and Novabiochem, respectively. Porcine hearts were obtained from Hormel Inc. for VIC and VlvEC isolation, and M199 and porcine endothelial media were purchased from Life Technologies and Genlantis, respectively. All other chemicals were purchased from Sigma-Aldrich, unless otherwise specified.
2.2. Monomer Synthesis
Eight-armed PEG-norbornene (PEG-N) (Mn: 20 & 40 kDa) was synthesized as previously described by Fairbanks et al. [32]. Briefly, the reaction was carried out under anhydrous conditions in the organic solvent dichloromethane (DCM), where a PEG solution was added drop-wise to a stirred solution of N,N′-dicyclohexylcarbodiimide (DCC) and norbornene acid, and allowed to react overnight at room temperature. The norbornene functionalized PEG in this solution was then precipitated in ice-cold ethyl ether, filtered, and re-dissolved in chloroform. This chloroform PEG solution was then washed with a glycine buffer and brine before being precipitated in ice-cold ethyl ether and filtered again. The filtered PEG was then placed in a vacuum chamber to remove excess ether. The percent functionalization of PEG arms with norbornene groups was determined using 1H-NMR by comparing the hydrogen peaks associated with the carbon adjacent to the ester linkage (~4.2ppm) to the hydrogen peaks associated with the PEG molecule (~3.6ppm). Only synthesis products with greater than 95% functionalization were used in subsequent experiments.
Two peptides were included in the thiol-ene formulation, one derived from fibronectin (CGRGDS) [36] to promote cell adhesion and a non-degradable di-thiol linker peptide (KCGGPQGIdWGQGCK) [32], [37] to facilitate crosslinking of the polymer network, where d denotes a reversed d chirality of the I amino acid. These peptides were synthesized using solid phase peptide synthesis (SPPS) on a Protein Technologies Tribute peptide synthesizer. After a 5 wt% phenol trifluoroacetic acid (TFA) cleavage and ice cold ether precipitation, if the purity was found to be less than 95% via high performance liquid chromatography (HPLC), then large scale HPLC purification was performed. The correct eluate fraction, based on MW, was determined through matrix assisted laser desorption ionization (MALDI). The HPLC buffer was removed from the peptide in solution via lyophilization.
2.3. Hydrogel Fabrication
In a sterile cell culture hood, eight-armed PEG-N, non-degradable dithiol linker peptide, CGRGDS, and 0.2 mM lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) [38] were dissolved in sterile PBS to yield the final monomer solution concentrations indicated in Table 1. As drawn schematically in Figure 2a, these monomer solutions were transferred onto a Teflon pedestal, with enough volume (10 μl) to create a ~100 μm thin gel (Fig. 2 b & c), and a 12-well insert (0.4 μm pore size) was then pressed on top. The Teflon pedestal, monomer solution, and insert sandwich were then placed under 365 nm UV light at 3 mW/cm2 for 2 minutes [38]. The resulting gels were allowed to swell overnight in serum free, low glucose DMEM media at 37° C before cell seeding.
Table 1.
Monomer concentrations and PEG-N molecular weights for each of the final Young’s modulus materials used for cell culture.
| PEG-N Mn (kDa) | [PEG-N] (mM) | Thiol to Ene Ratio | [Dithiol Linker Peptide] (mM) | [CGRGDS] (mM) | Young’s Modulus (kPa) |
|---|---|---|---|---|---|
| 40 | 2.5 | 2 to 3 | 6.0 | 2.0 | 3 |
| 40 | 2.5 | 1 to 1 | 9.0 | 2.0 | 10 |
| 20 | 5.0 | 3 to 4 | 14.3 | 2.0 | 15 |
| 20 | 5.0 | 1 to 1 | 19.0 | 2.0 | 27 |
Figure 2.

(a) Schematic of transwell insert PEG hydrogel fabrication material platform used to coculture VICs and VlvECs. (b) Representative fluorescent gel tagging confocal microscopy z-stack image and (c) quantification. (d) Young’s modulus as a function of crosslinking density for each of the four hydrogel formulations used for cell culture.
For formed gel thickness characterization, a fluorescent CGRGDS peptide was covalently tethered in the transwell gel and allowed to swell overnight in PBS. The gel was then imaged on a Ziess 2-photon confocal microscope, taking a z-stack in 10 micron increments from 50 microns below the membrane to 150 microns above (Fig. 2b). Significant fluorescence persisted from the membrane to approximately 105 microns above (i.e., the actual gel thickness) (Fig. 2c).
Final gel Young’s modulus (E) was determined by measuring the shear modulus (G) of swollen hydrogels via parallel plate rheometry and converting to Young’s modulus via rubber elasticity theory (E=2*(1+ν)*G=3*G), which assumes a Poisson’s ratio (ν) of 0.5 for an incompressible material (water) [32].
2.4. VIC and VlvEC Isolation and Culture
Primary VICs and VlvECs were isolated from aortic leaflets, which were excised from fresh porcine hearts acquired from Hormel within 24 hours of slaughter via a sequential collagenase digestion as previously described [39], [40] and aliquots were frozen until needed. Briefly, the leaflets were incubated in Earle’s balanced salt solution containing 250 U/mL collagenase for 10 minutes to remove the VlvECs, followed by an additional 20 minutes of collagenase digestion to remove the remaining endothelium. VICs were then removed by continuing incubation in fresh collagenase solution for 60 minutes.
The VlvEC suspension was pelleted and resuspended in porcine endothelial media supplemented with 15 v/v% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomyocin, and 1 μg/mL fungizone; plated on TCPS dishes; and cultured to 100% confluency at 37°C and 5% CO2. The VlvECs were then trypsinized, pelleted, and resuspended in 45 v/v% porcine endothelial media, 50 v/v% FBS, and 5 v/v% dimethyl-sulfoxide (DMSO) to 1,000,000 cells/mL. This suspension was transferred to cryovials, which were then placed in a slow temperature gradient freezing box at −70°C overnight. Collected VlvECs were stained with Von Willebrand factor (VWF) and CD31 (abcam) to ensure that a pure population of endothelial cells was obtained (Supplemental Figure 1).
The VIC suspension was filtered through a 100 μm strainer to remove the degraded leaflets and pelleted. The pellet was resuspended in Media 199 supplemented with 15 v/v% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomyocin, and 1 μg/mL fungizone; plated on TCPS dishes; and cultured to 50% confluency at 37°C and 5% CO2. VICs were then trypsinized, pelleted, and resuspended in 45 v/v% M199 media, 50 v/v% FBS, and 5 v/v% dimethyl-sulfoxide (DMSO) to 1,000,000 cells/mL. This suspension was transferred to cryovials, which were then placed in a slow temperature gradient freezing box at −70°C overnight.
Frozen vials were stored in a liquid nitrogen cooled tank until needed, and then thawed and expanded on tissue culture polystyrene (TCPS) in porcine endothelial media or Media M199 supplemented with 15 v/v% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomyocin, and 1 μg/mL fungizone in an incubator set to 37°C and 5% CO2 for VlvECs and VICs, respectively. Only VICs and VlvECs of the third passage were used for experiments.
For direct co-culture studies presented in Figure 1, VlvECs were grown up to 90% confluency, trypsinized, pelleted, and resuspended in 1 mL of serum and phenol red free RPMI media (Life Technologies) with 10 μM red cell tracker dye (Life Technologies) and incubated at 37°C for 15 minutes to allow for discrimination between VlvECs and VICs during image analysis. In parallel, VICs were grown up to 80% confluency, trypsinized, pelleted, and resuspended in 1 v/v% FBS low glucose DMEM media (Life Technologies). VlvECs were then diluted to the correct concentration in 1% FBS low glucose DMEM media and seeded on treated TCPS at 50,000 cells/cm2 [22]. Then, VICs were seeded on top at 20,000 cells/cm2 to avoid significant cell-cell contacts. The cells were then placed in an incubator at 37 C and 5% CO2 for 3 days.
Figure 1.
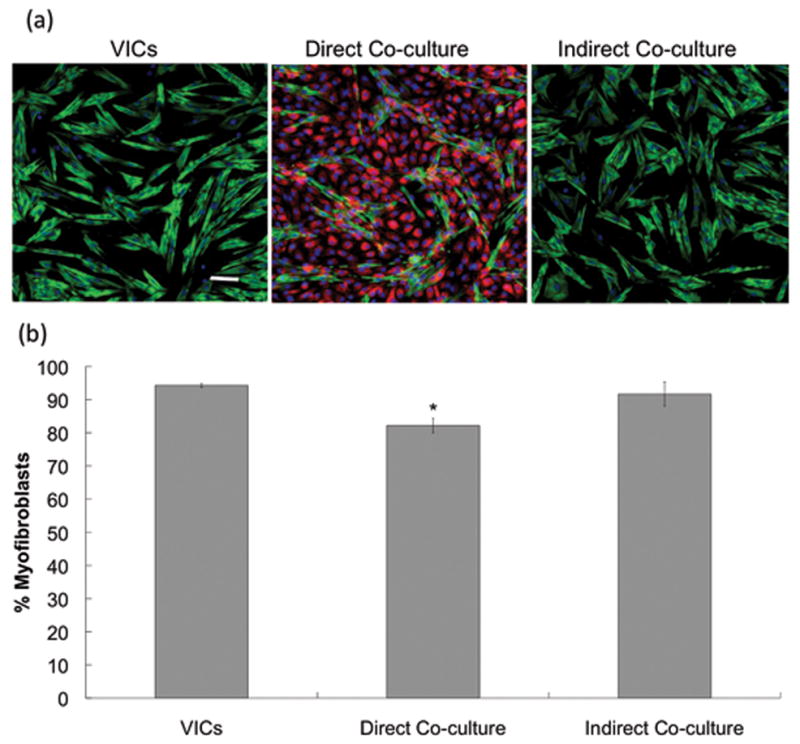
(a) Example immunostaining images for VICs cultured alone or in direct or indirect contact with VlvECs on TCPS. Green is αSMA, red is cell tracker transfected into VlvECs, blue is the cell nuclei, and the scale bar is 100 μm. (b) Quantification of the percentage of VICs activated to the myofibroblast phenotype from immunostaining images for VICs cultured alone or co-cultured in director indirect contact with VlvECs on TCPS. VICs and VlvECs were co-cultured for 3 days in 1% FBS low glucose DMEM media at 10,000 cells/cm2 and confluence, respectively, before fixation and immunostaining. * p<0.05 vs. VICs.
For indirect co-culture presented in Figure 1, VlvECs were seeded at 50,000 cells/cm2 in a transwell insert [22], and VICs were seeded on the TCPS below at 20,000 cells/cm2 in 1 v/v% FBS low glucose DMEM media (Life Technologies) and cultured for up to 3 days before fixation and immunostaining.
For gel-transwell experiments, VlvECs were seeded at 50,000 cells/cm2 on the underside of the inverted 12-well insert membranes and allowed to attach for 24 hours, before turning the insert right-side up and culturing to confluency (~2 days) (Supplemental Figure 2) [22]. Afterward, VICs were seeded on top the gel inside the insert at a density specified for each assay below in 1 v/v% FBS low glucose DMEM media (Life Technologies) and cultured for up to 6 days. Small molecule inhibitors of nitric oxide synthases (NOS), soluble guanylyl cyclase (sGC), protein kinase G (PKG), including NG-nitro-L-arginine methyl ester (100 μM L-NAME) [41], 1H-[1,2,4] Oxadiazolo [4,3-a] quinoxalin-1-one (3 μM ODQ) [42], and Rp-8-Bromo-β-phenyl-1,N2-ethenoguanosine 3′,5′-cyclic monophosphorothioate sodium salt (0.5 mM RBrcGMP) [25], respectively, or a Rho kinase (ROCK) activator Oleoyl-L-α-lysophosphatidic acid sodium salt (20 μM LPA) [43] were added to the culture media.
2.5. VIC Activation and αSMA Immunostaining
VICs were seeded at 10,000 cells/cm2 and cultured for up to 9 days in an incubator at 37 C and 5% CO2 before fixing the cells in 10 v/v% buffered formalin overnight at 4°C. The next day, VICs were stained with mouse anti-αSMA primary (abcam), goat anti-mouse FITC-488 secondary (Life Technologies), and DAPI (Life Technologies) at previously determined dilutions. Samples were imaged on a Zeiss microscope and manually analyzed in ImageJ software to determine the percentage of the VIC population with polymerized αSMA within the cell body [5], [6], [39], [44].
2.6. α-SMA Western Blot
VICs were seeded at 30,000 cells/cm2 and cultured for 3 days. Afterward, VICs were trypsinized off of the gels, pelleted, and resuspended in a 1X RIPA lysis buffer (Upstate Cell Signaling Solutions) supplemented with phosphotase and protease inhibitors (Calbiochem). VICs were lysed for 20 minutes at 4°C before removing cell debris via centrifugation at 4°C and 10,000 g for 15 minutes. Total protein concentrations were determined via a microBCA assay (Thermo Scientific). Samples were then diluted to the lowest measured concentration, supplemented with an SDS-β-mercaptoethanol solution, and heated to 95°C for 5 minutes to denature proteins. The samples were then cooled to room temperature, and 10 μg of total protein was loaded into a 10 wt% gel, and resolved with a BioRad system. The separated proteins in the gel were then transferred to a PVDF membrane via a wet transfer method. The membrane was washed in 0.05 wt% Tween-20 PBS (PBST) and blocked in a 2 wt% BSA, 4 wt% dry milk PBST solution for 1 hr at room temperature. The membrane was then incubated overnight at 4°C with αSMA (abcam) or GAPDH (Cell Signaling Technology) primary antibodies at previously determined dilutions. Afterward, the membrane was washed in PBST and incubated at room temperature for 1 hour with an HRP secondary antibody. The membrane was washed again before addition of the HRP luminescent substrate (GE), film exposure, and development. Afterward, the developed film was scanned and analyzed in ImageJ software to determine the ratio of αSMA to GAPDH for each sample.
2.7. Nodule Formation
VICs were seeded at confluency on top of the gel formed inside of the transwell insert of 3 or 27 kPa and with or without VlvECs present. The VICs or co-culture were grown for 6 days in 1% FBS M199 media to allow for VIC contraction into multicellular nodules. The number of nodules/mm2 was determined by manually counting nodules from bright field images obtained on day 6 ImageJ software and dividing by the field of view area.
2.8. Statistics
Data are presented as mean ± standard error with three experimental replicates and three biological (i.e., isolation) replicates. Data were compared using an ANOVA, and significance was established for p < 0.05.
3. Results
3.1. VIC Response to VlvECs on TCPS
To investigate the effect of VlvEC co-culture paracrine signaling on VIC phenotype and function, VICs were first studied on untreated TCPS with VlvECs in direct or indirect contact with transwell inserts. Figure 1 shows representative immunostaining images of VICs for αSMA fiber formation for each of these culture conditions. Interestingly, direct co-culture with VlvECs minimally, yet significantly decreased the percentage of myofibroblastic VICs as compared to VICs culture alone (Fig. 1b), while no decrease in myofibroblasts was observed in the indirect co-culture. This initial study suggested that both matrix elasticity and diffusion length scale were both important factors in understanding how VlvECs regulate VIC phenotype and function in vivo. Thus, we developed a new co-culture platform to control of both matrix elasticity and diffusion distance between VICs and VlvECs.
3.2. New Co-culture Gel Platform
The new method modifies a transwell insert with a material based substrate to control cell-matrix interactions, local elasticity, and spatially proximity of co-culture cells, as shown schematically in Figure 2a. Specifically, 8-armed PEG-N monomers of 20 or 40 kDa molecular weight and small peptides were combined at ratios specified in Table 1 and pipetted onto a Teflon insert of the same diameter as the transwell insert membrane that was then pressed on top. The Teflon and transwell insert sandwich was then placed under a 365 nm UV light of 3 mW/cm2 for 2 minutes to photopolymerize the monomer solution into a hydrogel with the desired mechanical properties and height (100 μm). The thickness of the hydrogels was determined by fluorescently labeling the hydrogel and imaging with confocal microscopy. Z-stack images, like the representative image in Figure 2b, were analyzed and quantified (panel c) and demonstrate the thickness of formed gels was approximately 105 μm (5% error).
The hydrogel substrates were further characterized by measuring the Young’s modulus as a function of crosslinking density (Fig. 2d). The range of Young’s moduli implemented in this study for VIC-VlvEC co-cultures (E ~ 3–27 kPa) included a range of elasticities that should direct both low and high activation levels and that fall within the range of normal and sclerotic valve tissue [45]. In general, the gel elasticity is readily tunable and directly proportional to the crosslinking density of the final material. Importantly, as the crosslinking density and moduli of the culture substrate were increased, a shift in VIC phenotype was observed from mostly quiescent fibroblasts (αSMA−) to primarily activated myofibroblasts (αSMA+). As expected, when VICs were cultured alone on hydrogels of increasing Young’s modulus (3, 10, 15, and 27 kPa), a significant increase in the percentage of myofibroblasts was observed as defined by the presence of αSMA organized in fibers (36 ± 3%, 48 ± 2%, 63 ± 2%, and 72 ± 2%, respectively) (Fig. 3) [29]. However, when VICs were cultured in the presence of VlvECs, VIC activation was significantly reduced and each modulus had significantly lower VIC activation when cultured in the presence of VlvECs, with 3, 10, 15, and 27 kPa gels yielding only 13 ± 3%, 35 ± 4%, 48 ± 4%, and 52 ± 8% VIC activation, respectively. Further, the percentage reduction of VIC activation to the myofibroblast phenotype was significantly larger on the 3 kPa gels (Fig. 3c).
Figure 3.

(a) Representative immunostaining images of VICs cultured on gels of increasing modulus, and in the absence of VlvECs, where green and blue are αSMA and nuclei, respectively. Scale bar = 100 μm. (b) Quantification of the percentage of VICs activated to the myofibroblast phenotype from immunostaining images for VICs cultured alone (diamond) or VICs co-cultured with VlvECs (square) as a function of gel modulus. VICs and VlvECs were co-cultured for 3 days in 1% FBS low glucose DMEM media at 10,000 cells/cm2 and confluence, respectively, before fixation and immunostaining. (c) Percent decrease in myofibroblast activation with the addition of VlvECs.
3.3. Role of Gel Elasticity on VIC Response
As 3 and 27 kPa represented the lowest and highest levels of VIC activation and percentage reduction examined, these conditions were used to further probe the potential influence of substrate elasticity on the response of VICs to signals from VlvECs. When VICs were co-cultured with VlvECs in the presence of a small molecule inhibitor of endothelial nitric oxide synthase (eNOS), L-NG-nitroarginine methyl ester (L-NAME at 100 μM), VIC myofibroblast activation was restored to levels observed with VICs alone, from 30 to 47% on 27 kPa gels and from 8 to 26% on 3 kPa gels (Fig. 4a).
Figure 4.

(a) Percentage of myofibroblasts on low (E~3 kPa) and high (E~27 kPa) activating gel formulations and (b) Western blot bands and quantification of αSMA expression normalized to GAPDH ratio for VICs alone (white), coculture (grey), and coculture with 100 μM L-NAME (black) after 3 days in culture. VICs and VlvECs were co-cultured in 1% FBS low glucose DMEM media.
VlvEC paracrine signaling not only influenced the assembly of αSMA into cytoskeletal fibers, but also the amount of αSMA protein expressed. Figure 4b demonstrates that as the culture substrate elasticity was increased, the relative amount of αSMA expression normalized to GAPDH increased from ~1.2 to ~1.9 for VICs cultured in the absence of VlvECs. However, when VlvECs were added to VIC cultures, the ratio of αSMA to GAPDH decreased to ~0.6 and ~0.9 on 3 and 27 kPa gels, respectively. Interestingly, the level of αSMA protein expressed by VICs on the 27 kPa gels in the presence of VlvECs was no longer significantly different from VICs cultured alone on the non-activating, 3 kPa substrates. Further, when L-NAME was added to the co-cultures, αSMA expression returned to levels similar to that of VICs cultured alone on both the 3 and 27 kPa gels (~1.6 and 2.0, respectively).
Organization of αSMA into stress fibers is one of the key markers for VIC activation to the myofibroblast phenotype, but persistent and elevated αSMA expression has also been shown to be directly linked to VIC nodule formation [43], which is an important aspect of progression of aortic stenosis. When VICs were cultured alone on 3 and 27 kPa substrates for 6 days, the number of nodules formed per area significantly increased from ~0.1 to ~0.6 nodules/mm2 (Fig. 5b). However, when VICs were co-cultured with VlvECs, the number of nodules significantly decreased to 0.3 nodules/mm2 on the 27 kPa hydrogels, while no differences were observed on the softest substrate, E ~3 kPa. Interestingly, the VlvEC-mediated reduction in nodule formation on the 27 kPa gels was prevented when L-NAME was added to the co-cultures and the level returned to that of VICs cultured alone. Further, the size of formed nodules in the presence of VlvECs on the 27 kPa gels was significantly less than that of VICs cultured alone and was prevented by L-NAME (Supplemental Figure 3). In contrast, no significant differences were observed on the non-activating 3 kPa gels.
Figure 5.

(a) Representative bright field images of VIC nodule formation on 27 kPa gels and (b) quantification of the number of the nodules on 3 and 27 kPa gels for VICs alone without (white) or with (light grey) 100 μM L-NAME and co-culture without (dark grey) or with 100 μM L-NAME (black) after 6 days in culture. VICs and VlvECs were co-cultured in 1% FBS low glucose DMEM media at confluence. Scale bar = 100μm.
3.4. VlvECs Deactivate Myofibroblastic VICs
An important, unanswered question is whether restoration of NO, in the presence of activated VICs would deactivate them back to fibroblasts. Thus, we hypothesized that NO paracrine signaling of VlvECs might not only suppress VIC activation but possibly reverse it as well. To test this hypothesis, VICs were cultured alone or with VlvECs on activating 27 kPa gels in the presence of 100 μM L-NAME for 3, 5, or 7 days, where relatively high levels of VIC activation (~46/51, 43/42, and 50/62%, respectively) were observed for both conditions (Fig. 6). Then, L-NAME was removed from the media and VIC activation was assessed two days later (5, 7, or 9 days of total culture). A major finding is that VIC myofibroblast activation was significantly reduced in the co-cultures to ~24, 22, and 18% (p < 0.01) on days 5, 7, and 9, respectively, and in fact, was similar to that of the co-cultures initially studied without L-NAME (Fig 6).
Figure 6.

The percentage of VICs activated to the myofibroblast phenotype as a function of culture time with 100 μM L-NAME exposure for 3, 5, or 7 days (black) and two additional days (i.e., 5, 7, or 9 total) without L-NAME (white) for VICs cultured alone (a) or in the presence of VlvECs (b). Lines were added to show trends. VICs and VlvECs were co-cultured in 1% FBS low glucose DMEM media at 10,000 cells/cm2 and confluence, respectively, before immunostaining. * p<0.05.
3.5. Mechanism of VIC Activation Reduction
To examine the mechanism by which VlvECs might influence VIC phenotype, we investigated effects of small molecule inhibitors or activators of NO intracellular signaling molecules in the cGMP pathway. These pathways have been implicated in relaxation of smooth muscle cells (SMCs) through down regulation of RhoA and myosin light chain-1 (MLC-1) [46], [47]. Because VICs and SMCs have similar pathways of regulation of αSMA expression [48], we hypothesized that the cGMP pathway may also facilitate NO reduction of VIC activation. Specifically, soluble guanylyl cyclase (sGC) and protein kinase G (PKG) functions were inhibited in co-cultures on activating 27 kPa gels by the addition of 3 μM 1H-[1,2,4] Oxadiazolo [4,3-a] quinoxalin-1-one (ODQ) and 0.5 mM Rp-8-Bromo-β-phenyl-1,N2-ethenoguanosine 3′,5′-cyclic monophosphorothioate sodium salt (RBrCGMP), respectively. Additionally, the activity of Rho kinase (ROCK), which is regulated by RhoA, was stimulated by addition of 20 μM Oleoyl-L-α-lysophosphatidic acid sodium salt (LPA). Figure 7 a and b report the percent myofibroblasts when sGC or PKG was inhibited and show that beneficial effect of VlvECs on suppressing substrate elasticity induced VIC activation was abolished (~40 to 60% myofibroblasts). Interestingly, when ROCK activity was stimulated by LPA addition, protective effects of VlvECs on VIC activation was again blocked as activation significantly increased from ~40 to 60% (Fig. 7c). Taken together, these data suggest that release of NO by VlvECs may act through the cGMP pathway to inhibit ROCK activity (Fig. 7d), VIC αSMA expression, and subsequent activation to the myofibroblast phenotype.
Figure 7.

Percentage of VICs activated to the myofibroblast phenotype as a function of culture conditions after 3 days (white) and after exposure to small molecule inhibitors of sGC (3 μM ODQ (a)) or PKG (0.5 mM RBrcGMP (b)), or the ROCK activator (20 μM LPA (c)) (black). VICs and VlvECs were co-cultured in 1% FBS low glucose DMEM media at 10,000 cells/cm2 and confluence, respectively for 3 days before immunostaining. (d) Model for the intersection of stiffness and NO signaling. The increased prevalence of stabilized focal adhesions of VICs attached to stiff substrates increases myofibrotic pathways (red). NO secreted by local VECs acts to increase anti-fibrotic pathways (blue).
4. Discussion
Activated VICs are a key mediator in progression toward aortic valve stenosis (AS) through elevated levels of collagen deposition and matrix remodeling [4], [5], [49], both of which cause valve tissue stiffening. Recent evidence suggests that VlvECs may regulate VIC phenotype and perhaps aid in prevention of AS initiation [17], [24]. Endothelial cell function can be inhibited by many factors, including hypercholesterolemia and diabetes [21], [50], [51], which may thereby lead to dysregulation of the VIC phenotype and ultimately AS. However, isolating the effect of endothelium on VIC phenotype and function is difficult to assess in vivo because many factors, both biophysical and biochemical, likely affect VIC function and phenotype. Studying VIC and VlvEC paracrine signaling dynamics in vitro allows testing of specific hypotheses concerning this essential paracrine signaling event and associated mechanisms.
Stiffening of heart valve tissue may have profound consequences at the cellular level, and aspects of this can be tested in vitro through advances in biomaterials used for cell culture. Here, VlvECs were shown to have a beneficial effect on VIC phenotype and function when physiologically relevant elasticity levels were used, and the effect was difficult to discern when TCPS direct and indirect co-culture methods were used. The small effects are likely due to the extreme stiffness of TCPS, which produces activation of essentially all of the VIC population to myofibroblasts [14], and may impair the response of VICs to VlvECs even when in direct contact. Further, if the distance of diffusion between the cells is increased to the mm scale (i.e., a typical distance between transwell membranes and the plate below), the VlvECs are no longer able to significantly impact VIC phenotype. This motivated the development of an alternative co-culture method to study the dynamics of VlvEC paracrine signaling on VIC phenotype by introducing more physiological elasticities and spatial distances between cells.
A range of elastic moduli were selected and characterized to reduce or enhance VIC activation to the myofibroblast phenotype to study how the cellular microenvironmental stiffness may affect VIC response to VlvEC paracrine signaling. Additionally, gels were fabricated in transwell inserts, commonly used for co-cultures, and were approximately 100 μm thin to provide a controlled distance between cell types relevant to the valve structure [52], while still allowing for control of the cell culture substrate elasticity. VIC activation to the secretory myofibroblast phenotype was significantly elevated with increasing Young’s modulus of the underlying substrate (Fig. 3). This result is comparable to the previous finding that reported a 15 kPa threshold for significant VIC activation [29]. Interestingly, co-culture with VlvECs significantly reduced VIC activation to the αSMA+ myofibroblasts on all matrix elasticities studied, which suggests that the valve endothelium is able to suppress AS progression even in fibrotic tissue stiffnesses. However, VICs cultured on the lowest elasticity (3 kPa) were significantly more responsive than those on higher elasticities. This result suggests that VIC populations with a lower percentage of activated myofibroblasts are able to respond more robustly to VlvEC paracrine signaling. This responsiveness may occur because RhoA is less activated in cells on the softer substrates and/or that NO paracrine signaling through the cGMP pathway dominates regulation of RhoA, and thus αSMA expression. Clearly, future studies would be warranted to confirm this hypothesis. In particular, if verified, these results might have significant implications for the treatment of aortic valve stenosis (AS), if by improving endothelial cell function, or dosing with exogenous NO, VIC function and potentially valve function could be improved.
VIC nodule formation can also be modulated by culture substrate modulus, and αSMA is necessary for nodule formation [14]. Similarly, we found that the number of nodules formed significantly increased when the culture substrate elasticity was increased from 3 to 27 kPa, and importantly, that the co-culture significantly reduced the number and size of nodules induced on the 27 kPa substrates. This result is also consistent with previous studies using synthetically generated NO that blocked VIC contraction mediated nodule formation [26].
Previous work also demonstrated that exogenous NO dosage or co-culture with aortic/vascular endothelial cells (VECs) could prevent VIC activation induced by dexamethasone [24]. While this work was amongst the first to show the beneficial influences of endothelium on VIC phenotype, it should be noted that VECs have unique gene expression profiles as compared to VlvECs [53], and dexamethasone is a synthetic corticosteroid that is not typically present in vivo during AS initiation or progression. In our results, we also observe that NO is an important VlvEC paracrine signaling molecule that reduces disease-like VIC functions, as effects were blocked with the addition of the eNOS inhibitor, L-NAME. Specifically, we observed that VIC activation, αSMA protein expression, and nodule formation levels in the presence of both VlvECs and L-NAME return to those of VICs cultured alone. However, other molecules secreted by VlvECs, like c-type natriuretic peptide (CNP) or prostaglandins may also play important roles in regulating VIC phenotype. Additionally, activated VICs may also paracrine signal VlvECs and alter their gene expression and secreted proteins, such as PDGF and S1P1.
While this work and previous studies highlight the importance of VlvECs in preventing disease-like VIC functions (e.g., activation), aortic valve disease is generally not detected until valve disease is advanced. At that point, the valve tissue have irreversibly stiffened, calcified, and must be replaced. It would be most useful to develop a treatment that would reverse the progression of AS, instead of solely preventing it. Here, we demonstrate that VlvEC secreted NO is also able to reverse VIC activation even after one week of L-NAME treatment. This result suggests that if endothelial dysfunction could be detected early and restored to normal, then proper VIC and valve function may potentially be restored. Fortunately, many studies have shown that endothelial cell function can be improved through altered diet or medication, resulting in delaying of atherosclerosis progression and increased NO levels to pre-dysfunction levels [54].
Clearly, VlvECs have a protective effect on VIC phenotype and function, and NO appears to be a major player in this process. We speculate that understanding mechanisms by which NO suppresses VIC activation and nodule formation might aid in drug development to target specific signaling pathways. Nitric oxide is a potent vasodilator and has been well characterized in relation to regulation of SMCs and in preventing atherosclerosis. In SMCs, NO acts through the cGMP signaling pathway, which ultimately down regulates Rho Kinase (ROCK) [47] and myosin light chain-1 (MLC-1) [46]. ROCK also regulates αSMA expression in VICs [43], which led us to hypothesize that NO acts through the cGMP pathway in VICs to ultimately regulate αSMA expression. Indeed, co-cultures with VlvECs in the presence of the small molecule inhibitors ODQ and RBrcGMP for sGC and PKG, respectively, (i.e., initial and mid-stage molecules in the cGMP pathway) did not reduce VIC activation (Fig. 7 a & b). Further, the co-culture’s beneficial effect was prevented by addition of LPA, which stimulates ROCK activity (Fig. 7c). Thus, regulation of ROCK may also be involved in NO mediated reduction of disease-like VIC functions. These results suggest that NO is likely acting through the cGMP pathway to regulate VIC αSMA expression, which in turn reduces VIC activation to the myofibroblast phenotype and contraction into nodules (Fig. 7d).
It is also probable that NO affects expression of other proteins related to activation of VICs to the myofibroblast phenotype. One possibility is that MLC-1 is also down-regulated, reducing cytoskeletal tension, which is a prerequisite for αSMA fiber formation [55]. Additionally, it is likely that VICs are able to secrete soluble signaling molecules to influence protein expression of VlvECs (e.g., NO, FGF, CNP), and the paracrine signaling molecules secreted by VICs is likely to be greatly influenced by their phenotype (quiescent fibroblast versus activated myofibroblast). Further, there is likely a complex and multifaceted interplay between VIC intracellular signaling pathways like the cGMP pathway studied here and previously implicated like Notch1 [25] that work together to prevent FCAVD progression when initiated by VlvEC paracrine signaling molecules. Further studies with this co-culture platform are on-going and motivated to better understand the complex and dynamic paracrine signaling between VICs and VlvECs for the prevention and treatment of AS.
5. Conclusions
A co-culture platform was developed that allows for the control of microenvironmental matrix cues and spatial proximity of cell types. Here, this system facilitated the study of effects of both NO and matrix elasticity on VIC activation and subsequent intracellular signaling pathways when co-cultured with VlvECs. VICs responded to changes in microenvironmental stiffness and activated to a myofibroblast phenotype; however, these changes were highly dependent on signaling from VlvECs. VlvECs suppress substrate elasticity-induced VIC activation, αSMA protein expression, and nodule formation, and reverse VIC activation even after one week of L-NAME treatment in a manner that relates to VlvEC secreted NO paracrine signaling. Results suggest that NO may act primarily through the cGMP intracellular signaling pathway and ultimately down regulate ROCK activity and αSMA gene expression. Additionally, the co-culture platform presented herein is a simplified modification to traditional methods, but affords facile user defined control of cell culture substrate elasticity and cell proximity (i.e., gel thickness), allowing one to simultaneously examine the effects of microenvironmental elasticity and dynamic paracrine signaling between cells.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Huan Wang for providing the western blot protocol and our funding sources, including the National Institute of Health (NIH R01 HL089260 and HL 62984) and Howard Hughes Medical Institute (HHMI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease pathogenesis, disease progression, and treatment strategies. Circulation. 2005;111:3316–3326. doi: 10.1161/CIRCULATIONAHA.104.486738. [DOI] [PubMed] [Google Scholar]
- 2.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 3.Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of sortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. New Eng J Med. 1999;341:142–147. doi: 10.1056/NEJM199907153410302. [DOI] [PubMed] [Google Scholar]
- 4.Chester AH, Taylor PM. Molecular and functional characteristics of heart-valve interstitial cells. Philos Trans R Soc Lond, B, Biol Sci. 2007;362:1437–1443. doi: 10.1098/rstb.2007.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chester AH, Latif N, Yacoub MH, Taylor PM. Vascular complications in human disease, Chapter 18: Aortic Valve: From Function to Tissue Engineering. Springer; London: 2008. [Google Scholar]
- 6.Cushing MC, Jaeggli MP, Masters KS, Leinwand LA, Anseth KS. Serum deprivation improves seeding and repopulation of acellular matrices with valvular interstitial cells. J Biomed Mater Res A. 2005;75:232–241. doi: 10.1002/jbm.a.30412. [DOI] [PubMed] [Google Scholar]
- 7.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253–260. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 8.Adelöw C, Segura T, Hubbell JA, Frey P. The effect of enzymatically degradable poly(ethylene glycol) hydrogels on smooth muscle cell phenotype. Biomaterials. 2008;29:314–326. doi: 10.1016/j.biomaterials.2007.09.036. [DOI] [PubMed] [Google Scholar]
- 9.Benton JA, Fairbanks BD, Anseth KS. Characterization of valvular interstitial cell function in three dimensional matrix metalloproteinase degradable PEG hydrogels. Biomaterials. 2009;30:6593–6603. doi: 10.1016/j.biomaterials.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gould ST, Darling NJ, Anseth KS. Small peptide functionalized thiol–ene hydrogels as culture substrates for understanding valvular interstitial cell activation and de novo tissue deposition. Acta Biomater. 2012;8:3201–3209. doi: 10.1016/j.actbio.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 12.Weber KT, Janicki JS, Pick R, Capasso J, Anversa P. Myocardial fibrosis and pathologic hypertrophy in the rat with renovascular hypertension. Am J Cardiol. 1990;65:1–7. doi: 10.1016/0002-9149(90)90952-w. [DOI] [PubMed] [Google Scholar]
- 13.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 14.Benton JA, Kern HB, Anseth KS. Substrate properties influence calcification in valvular interstitial cell culture. J Heart Valve Dis. 2008;17:689–699. [PMC free article] [PubMed] [Google Scholar]
- 15.Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–42. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 16.Cushing MC, Liao JT, Jaeggli MP, Anseth KS. Material-based regulation of the myofibroblast phenotype. Biomaterials. 2007;28:3378–3387. doi: 10.1016/j.biomaterials.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Simmons CA. Aortic valve mechanics: an emerging role for the endothelium. J Am Coll Cardiol. 2009;53:1456–1458. doi: 10.1016/j.jacc.2008.12.052. [DOI] [PubMed] [Google Scholar]
- 18.Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510–1524. doi: 10.1161/CIRCRESAHA.110.234237. [DOI] [PubMed] [Google Scholar]
- 19.Butcher JT, Nerem RM. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos Trans R Soc Lond, B, Biol Sci. 2007;362:1445–1457. doi: 10.1098/rstb.2007.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell and Reece, Biology, Seventh. San Francisco, CA: Pearson Benjamin Cummings; 2005. [Google Scholar]
- 21.Poggianti E, Venneri L, Chubuchny V, Jambrik Z, Baroncini LA, Picano E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. J Am Coll Cardiol. 2003;41:136–141. doi: 10.1016/s0735-1097(02)02622-0. [DOI] [PubMed] [Google Scholar]
- 22.Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng. 2006;12:905–915. doi: 10.1089/ten.2006.12.905. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Janssens SP, Wingler K, Schmidt HHHW, Moens AL. Modulating endothelial nitric oxide synthase: a new cardiovascular therapeutic strategy. Am J Physiol Heart Circ Physiol. 2011;301:H634–646. doi: 10.1152/ajpheart.01315.2010. [DOI] [PubMed] [Google Scholar]
- 24.Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, et al. Side-specific endothelial-dependent regulation of aortic valve calcification: interplay of hemodynamics and nitric oxide signaling. Am J Pathol. 2013;182:1922–1931. doi: 10.1016/j.ajpath.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol. 2013;60:27–35. doi: 10.1016/j.yjmcc.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kennedy JA, Hua X, Mishra K, Murphy GA, Rosenkranz AC, Horowitz JD. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur J Pharmacol. 2009;602:28–35. doi: 10.1016/j.ejphar.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 27.Tazawa S, Hayakawa Y, Ishikawa T, Niiya K, Sakuragawa N. Heparin stimulates the proliferation of bovine aortic endothelial cells probably through activation of endogenous basic fibroblast growth factor. Thromb Res. 1993;72:431–439. doi: 10.1016/0049-3848(93)90243-h. [DOI] [PubMed] [Google Scholar]
- 28.Toniolo A, Buccellati C, Pinna C, Gaion RM, Sala A, Bolego C. Cyclooxygenase-1 and prostacyclin production by endothelial cells in the presence of mild oxidative stress. PLoS ONE. 2013;8:e56683. doi: 10.1371/journal.pone.0056683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kloxin AM, Benton JA, Anseth KS. In situ elasticity modulation with dynamic substrates to direct cell phenotype. Biomaterials. 2010;31:1–8. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Haeger SM, Kloxin AM, Leinwand LA, Anseth KS. Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS ONE. 2012;7:e39969. doi: 10.1371/journal.pone.0039969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Tibbit M, Langer SJ, Leinwand LA, Anseth KS. Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway. PNAS. 2013;110:19336–19341. doi: 10.1073/pnas.1306369110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hern DL, Hubbell JA. Incorporation of adhesion peptides into nonadhesive hydrogels useful for tissue resurfacing. J Biomed Mater Res. 1998;39:266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Xu S, Gotlieb AI. The progression of calcific aortic valve disease through injury, cell dysfunction, and disruptive biologic and physical force feedback loops. Cardiovasc Pathol. 2013;22:1–8. doi: 10.1016/j.carpath.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Yip CYY, Simmons CA. The aortic valve microenvironment and its role in calcific aortic valve disease. Cardiovasc Pathol. 2011;20:177–182. doi: 10.1016/j.carpath.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 37.Patterson J, Hubbell JA. Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials. 2010;31:7836–7845. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 38.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials. 2009;30:6702–6707. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filip DA, Radu A, Simionescu M. Interstitial cells of the heart valves possess characteristics similar to smooth muscle cells. Circ Res. 1986;59:310–320. doi: 10.1161/01.res.59.3.310. [DOI] [PubMed] [Google Scholar]
- 40.Brody S, McMahon J, Yao L, O’Brien M, Dockery P, Pandit A. The effect of cholecyst-derived extracellular matrix on the phenotypic behaviour of valvular endothelial and valvular interstitial cells. Biomaterials. 2007;28:1461–1469. doi: 10.1016/j.biomaterials.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 41.El-Hamamsy I, Balachandran K, Yacoub MH, Stevens LM, Sarathchandra P, Taylor PM, et al. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. J Am Coll Cardiol. 2009;53:1448–1455. doi: 10.1016/j.jacc.2008.11.056. [DOI] [PubMed] [Google Scholar]
- 42.Feelisch M, Kotsonis P, Siebe J, Clement B, Schmidt HH. The soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one is a nonselective heme protein inhibitor of nitric oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol Pharmacol. 1999;56:243–253. doi: 10.1124/mol.56.2.243. [DOI] [PubMed] [Google Scholar]
- 43.Gu X, Masters KS. Role of the Rho pathway in regulating valvular interstitial cell phenotype and nodule formation. Am J Physiol Heart Circ Physiol. 2011;300:H448–H458. doi: 10.1152/ajpheart.01178.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen JH, Chen WLK, Sider KL, Yip CYY, Simmons CA. β-catenin mediates mechanically regulated, transforming growth factor-β1-induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol. 2011;31:590–597. doi: 10.1161/ATVBAHA.110.220061. [DOI] [PubMed] [Google Scholar]
- 46.Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. PNAS. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato M, Blanton R, Wang GR, Judson TJ, Abe Y, Myoishi M, et al. Direct binding and regulation of RhoA protein by cyclic GMP-dependent protein kinase Iα. J Biol Chem. 2012;287:41342–41351. doi: 10.1074/jbc.M112.421040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harhun MI, Szewczyk K, Laux H, Prestwich SA, Gordienko DV, Moss RF, Bolton TB. Interstitial cells from rat middle cerebral artery belong to smooth muscle cell type. J Cell Mol Med. 2009;13:4532–4539. doi: 10.1111/j.1582-4934.2008.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thayer P, Balachandran K, Rathan S, Yap CH, Arjunon S, Jo H, Yoganathan AP. The effects of combined cyclic stretch and pressure on the aortic valve interstitial cell phenotype. Ann Biomed Eng. 2011;39:1654–1667. doi: 10.1007/s10439-011-0273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abebe W, Mozaffari M. Endothelial dysfunction in diabetes: potential application of circulating markers as advanced diagnostic and prognostic tools. EPMA J. 2010;1:32–45. doi: 10.1007/s13167-010-0012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guerraty MA, Grant GR, Karanian JW, Chiesa OA, Pritchard WF, Davies PF. Hypercholesterolemia induces side-specific phenotypic changes and peroxisome proliferator-activated receptor-gamma pathway activation in swine aortic valve endothelium. Arterioscler Thromb Vasc Biol. 2010;30:225–231. doi: 10.1161/ATVBAHA.109.198549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, Sridhar B, Leinwand LA, Anseth KS. Characterization of Cell Subpopulations expressing progenitor cell markers in porcine cardiac valves. PLoS ONE. 2013;8:e69667. doi: 10.1371/journal.pone.0069667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farivar RS, Cohn LH, Soltesz EG, Mihaljevic T, Rawn JD, Byrne JG. Transcriptional profiling and growth kinetics of endothelium reveals differences between cells derived from porcine aorta versus aortic valve. Eur J Cardiothorac Surg. 2003;24:527–534. doi: 10.1016/s1010-7940(03)00408-1. [DOI] [PubMed] [Google Scholar]
- 54.Harrison DG, Armstrong ML, Freiman PC, Heistad DD. Restoration of endothelium-dependent relaxation by dietary treatment of atherosclerosis. J Clin Invest. 1987;80:1808–1811. doi: 10.1172/JCI113276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hinz B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol. 2006;3–4:175–181. doi: 10.1016/j.ejcb.2005.09.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.