

Abstract
Regulatory T cells (Treg cells), which maintain immune homeostasis and self-tolerance, form an immunological synapse (IS) with antigen-presenting cells (APCs). However, signaling events at the Treg IS remain unknown. Here we show that protein kinase C-η (PKC-η) associated with CTLA-4 and was recruited to the Treg IS. PKC-η-deficient Treg cells displayed defective suppressive activity, including suppression of tumor immunity but not autoimmune colitis. Phosphoproteomic analysis revealed an association between CTLA-4-PKC-η and the GIT-PIX-PAK complex, an IS-localized focal adhesion complex. Defective activation of this complex in PKC-η-deficient Treg cells was associated with reduced CD86 depletion from APCs by Treg cells. These results reveal a novel CTLA-4-PKC-η signaling axis required for contact-dependent suppression, implicating this pathway as a potential cancer immunotherapy target.
The discovery and recognition of CD4+Foxp3+ Treg cells as a distinct subset of T cells with immunoregulatory function represents a major advance in our understanding of the immune system1-3. Treg cells actively maintain immune homeostasis and self-tolerance, and one prominent Treg cell-mediated suppressive mechanism is dependent upon contact with antigen presenting cells (APCs)4. This physical contact promotes the formation of a specialized signaling platform, known as the immunological synapse (IS), at the Treg cell-APC interface.
CTLA-4 is a potent negative regulator of T cell-mediated immune responses through its actions in both Teff and Treg cells. CTLA-4 is highly expressed on Treg cells3, and this high expression, as well as the higher affinity of CTLA-4 for its CD80 (B7-1) and CD86 (B7-2) ligands by comparison with CD285 is associated with predominant localization of CTLA-4 at the Treg cell IS and, consequently, displacement of CD28 from the IS6. However, despite extensive studies on CTLA-4, little is known about the intracellular signaling pathways initiated upon CTLA-4 engagement by its ligands. The SHP1, SHP2 and PP2A phosphatases, which represent binding partners of CTLA-47, may account for the intrinsic inhibitory activity of CTLA-4 in Teff cells, but a recent study demonstrated that these phosphatases are not recruited to the Treg cell IS together with CTLA-46. Thus, how CTLA-4 exerts its signaling effects at the Treg cell IS remains unknown.
The Treg cell IS is distinguishable from the “conventional” IS formed between naïve or effector T (Teff) cells and APCs in several respects. First, although the TCR is present in the central supramolecular activation cluster (cSMAC) in both types of IS, the costimulatory CD28 receptor is recruited to the Teff IS, whereas CTLA-4 is present at the T IS6, 8. Second, PKC-θ is absent from the Treg cell IS and, moreover, in contrast to Teff cells, it negatively regulates the function of Treg cells4. Physical association of PKC-θ, mediated by its V3 domain, with the costimulatory CD28 receptor underlies its cSMAC recruitment and essential functions in driving the activation, proliferation and differentiation of Teff cells9. Hence, the absence of PKC-θ in the Treg cell IS suggests that TCR signaling events in these cells could differ significantly from those of Teff cells. Nevertheless, proximal TCR signaling appears intact in Treg cells, as indicated by the phosphorylation and activation of TCR, Lck10, PDK111, LAT and PLCγ112, all of which have been implicated in the in vitro suppressive function of Treg cells. Because of these findings and, in particular, the importance of the association between LAT and activated PLCγ112, which is required for the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) and generation of diacylglycerol (DAG), the PKC-activating second messenger, we hypothesized that DAG should be produced locally13 upon IS formation in Treg cells and, furthermore, that this would lead to the IS recruitment and activation of a PKC family member other than PKCθ, which may positively regulate the function of Treg cells.
Here we show that, by analogy with the PKC-θ-CD28 interaction in Teff cells, which promotes their activation and function9, the CD28-related receptor CTLA-4, which is highly expressed on Treg cells and is required for their suppressive function14, 15, physically recruits another member of the novel PKC (nPKC) subfamily, PKC-η, which localizes at the Treg cell IS following stimulation. This association required phosphorylated serine residues in PKC-η and a conserved, membrane-proximal motif in the cytoplasmic tail of CTLA-4, respectively. Although Treg cell development and the expression of typical Treg cell markers were normal in PKC-η-deficient (Prkch−/−) Treg cells, these cells displayed a grossly impaired contact-dependent suppressive activity in vitro and in vivo, which was associated with a grossly defective activation of NFAT and NF-κB. Lastly, we show that defective activation of a focal adhesion complex consisting of p21-activated kinase (PAK) and two additional proteins, PIX and GIT2, which was physically associated with CTLA-4 and PKC-η, was associated with reduced ability of Prkch−/− Treg cells to serially engage APCs, thereby providing a potential mechanistic basis for the impaired suppressive activity of these cells. Therefore, strategies that interfere with this novel signaling pathway may be beneficial for inhibiting Treg cell function in cancer and, hence, boosting the immune response against tumors.
RESULTS
Phospho-PKC-η interacts with CTLA-4 and is recruited to the Treg cell IS
By analogy to the interaction of PKC-θ with the CTLA-4-related costimulatory receptor, CD28, which is critical for the activation of Teff cells9, we first tested if any PKC isoform could physically interact with CTLA-4. Using T hybridoma cells, we found that CTLA-4 coimmunoprecipitated with a higher than normal molecular weight (MW) species of PKC-η (Fig. 1a), but not with any other T cell-expressed PKC isoforms (Supplementary Fig. 1a). We reasoned that the MW shift of PKC-η might be a result of its phosphorylation. Indeed, alkaline phosphatase treatment partially reversed the shift of the higher MW species of PKC-η and resulted in the appearance of a lower MW species (Fig. 1b), indicating that CTLA-4 interacts predominantly with phosphorylated PKC-η. Consistently, phospho-PKC-η was found in unstimulated Foxp3+ Treg cells, but not in naïve T cells (Fig. 1c), despite the fact that PKC-η protein expression was not significantly different between Teff and Treg cells (Supplementary Fig. 1b). Using a Treg cell-APC conjugation system and confocal microscopy, we observed that, relative to PKC-θ, PKC-η was preferentially localized in the Treg cell IS, where it was partially colocalized with the TCR (Fig. 1d). We analyzed the relative intensity of the PKC-η or PKC-θ fluorescence signals in the Treg cell IS by calculating the intensity ratio in the IS vs. the opposite T cell pole. About 15% (3/20) of imaged cells displayed preferential localization of PKC-θ in the IS (ratio ≥ 5), consistent with the observation that a similar percentage of cells in this population were FoxP3− (by intracellular staining), representing contaminating activated Teff cells in the iTreg culture. Among the remaining cells, the intensity ratios for PKC-η and PKCθ were 2.84 ± 0.22 and 1.51 ± 0.18 (P < 0.0001), respectively (data not shown). Taken together, these results indicate that phospho-PKC-η associates with CTLA-4 in Treg cells and, furthermore, that PKC-η preferentially colocalizes with CTLA-4 in the IS.
Figure 1.
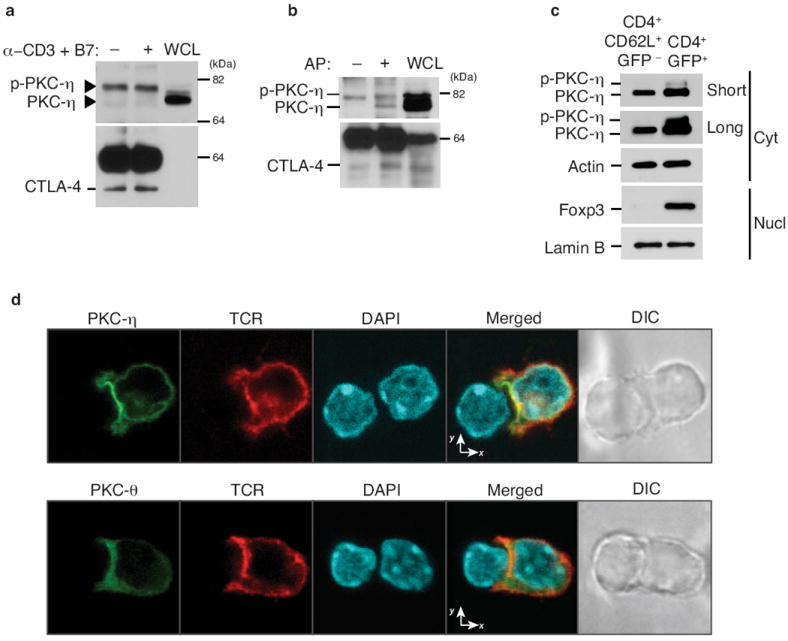
IS recruitment and CTLA-4 interaction of PKC-η in Treg cells. (a) Immunoblot analysis of T hybridoma cells left unstimulated (-) or stimulated (+) with anti-CD3 plus CD86-Fc for 5 min. CTLA-4 IPs (left and center lanes) or whole cell lysates (WCL) were immunoblotted with the indicated Abs. Arrowheads mark the two PKC-η species. (b) Immunoblot analysis of IPs (left and center lanes) or WCL (right lane) from T hybridoma cells stimulated with anti-CD3 plus CD86-Fc for 5 min. CTLA-4 IPs were left untreated (-) or treated with alkaline phosphatase (AP; +) prior to immunoblotting. (c) Immunoblot analysis of cytosol or nuclear fractions from sorted CD4+CD62L+GFP- (naïve) and CD4+GFP+ (Treg) cells (2 × 106 each) from wild-type FIG mice. Data in (a-c) are representative of at least four experiments. (d) Confocal imaging of PKC-η (top row) or PKC-θ (bottom row) and TCRβ localization in in vitro differentiated iTreg cells from AND TCR-Tg Rag2−/− mice, which were retrovirally transduced with eGFP-tagged mouse PKC-θ or PKC-η 1 d after anti-CD3 and -CD28 stimulation. Sorted GFP+ T cells (~90% FoxP3+ by intracellular staining; not shown) obtained on d 4 and stimulated for 5-10 min by conjugation with MCC-pulsed LPS-stimulated B cells were fixed and analyzed. eGFP-tagged PKC, TCR and nuclear DAPI staining are shown in green, red and blue, respectively. Images are representative of at least 100 cells collected from three independent experiments.
Development of Foxp3+ Treg cells is independent of PKC-η
Given the critical role of CTLA-4 in contact-dependent Treg cell suppressive function, which involves depletion of CD80 and CD86 from APCs14, 15, we next examined the CD4+Foxp3+ Treg cell population in the lymphoid organs of Prkch−/− mice by intracellular Foxp3 staining. The number and frequency of CD4+Foxp3+ cells was not significantly altered in the thymi and spleens of Prkch−/− mice compared to wild-type mice (Fig. 2a, b and Supplementary Fig. 2a). However, there was a consistent increase in the numbers of CD4+Foxp3+ Treg cells in the peripheral (pLN) and mesenteric (mLN) lymph nodes of these mice (Fig. 2c, d), suggesting that PKCη is dispensable for the in vivo development of CD4+Foxp3+ T cells. Phenotypically, cells that have been “licensed” to become Foxp3+ expressed similar levels of typical Treg cell markers, including Foxp3, TCR-β chain, CTLA-4, CD25, glucocorticoid-induced tumor necrosis factor receptor (GITR) and CD44 (Fig. 2e-j). Thus, PKC-η is dispensable for nTreg cell development in vivo, and for iTreg cell differentiation in vitro as assessed by expression of typical Treg cell markers.
Figure 2.
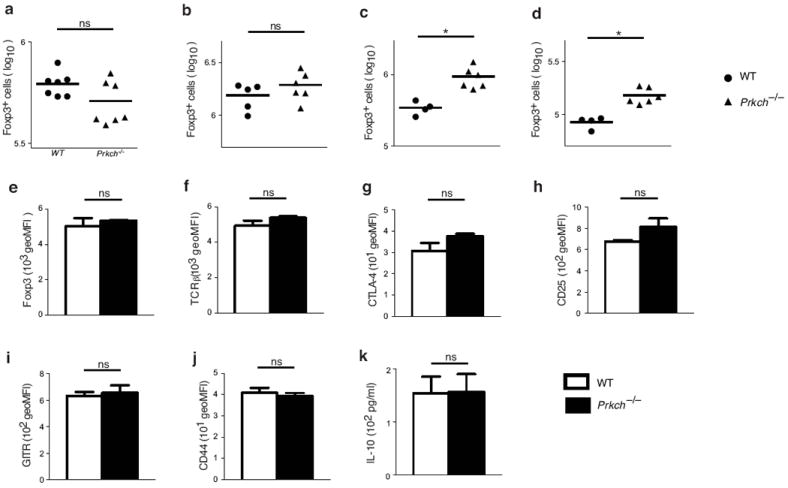
Development of Foxp3+ Treg cells is independent of PKC-η. (a-d) Cell counts (Log10) of CD4+Foxp3+ cells from thymi (a), spleens (b), peripheral lymph nodes (pLN; c) and mesenteric lymph nodes (mLN; d) of 8-12 week-old wild-type or Prkch−/− mice determined by intracellular Foxp3 staining. Each data point represents a single mouse. ns, non significant; *P < 0.05. (e-j) Mean fluorescence intensity (MFI) of Foxp3 (e), TCRβ (f), CTLA-4 (g), CD25 (h), GITR (i) and CD44 (j) expression determined on gated CD4+Foxp3+-cells from wild-type or Prkch−/− mice. (k) IL-10 production (measured by ELISA) by CD4+GFP+ Treg cells from wild-type and Prkch−/− FIG mice, which were stimulated with plate-bound anti-CD3 mAb and CD86-Fc in the presence of IL-2 overnight. For description of FIG mice, see Fig. 3 and the corresponding text.
PKC-η is required for Foxp3+ Treg cell suppressive function
We next explored the possibility that PKC-η might be required for the suppressive function of Foxp3+ Treg cells. To enable definitive identification of Treg cells, we crossed Prkch−/− mice with mice coexpressing Foxp3 and enhanced GFP under the control of the endogenous Foxp3 promoter (Foxp3-IRES-eGFP, hereafter called FIG)16 to generate Prkch−/−-FIG mice. We sorted CD4+GFP+ Treg cells from wild-type and Prkch−/− FIG mice, and found that, upon stimulation, they produced similar amount of interleukin 10 (IL-10) (Fig. 2k), a cytokine that can mediate the suppressive activity of Treg cells17, 18.
We next considered the possibility that, despite the presence of an apparently normal Treg cell population in Prkch−/− mice, the suppressive activity of these cells may be defective. We first used a conventional in vitro Treg cell suppression assay, which assessed the proliferation of naïve T cells cocultured with Treg cells and stimulated with anti-CD3 plus splenic dendritic cells (DCs) as an APC source. The percentage of dividing Teff cells was consistently higher in cultures with Prkch−/− Treg cells compared to Teff cells cocultured with wild-type Treg cells (Fig. 3a). The Prkch−/−Treg cells were substantially impaired in their ability to suppress the proliferation of Teff cells even at high Treg/Teff cell ratios, indicating that PKC-η expression by Treg cells is important for their function.
Figure 3.
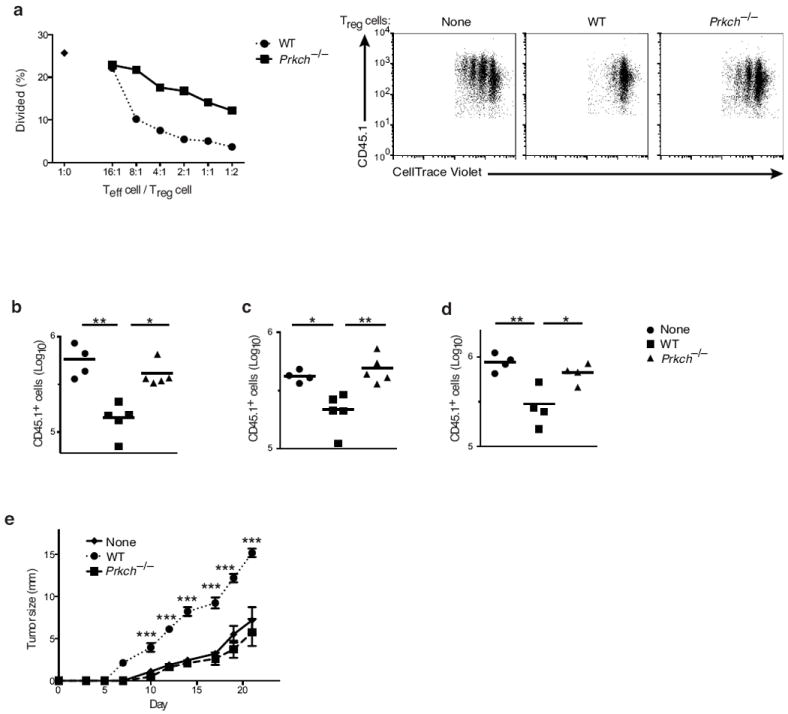
Contact-dependent suppression by Treg cells depends on PKC-η. (a) In vitro suppression assay measuring the proliferation of CellTrace Violet-labeled naïve B6 CD4+CD25- Teff cells cocultured in the absence (1:0) or presence of Foxp3+ Treg cells from wild-type or Prkch−/−FIG mice at the indicated Teff/Treg cell ratios, and stimulated with anti-CD3 mAb and splenic DCs for 3 d. Percentages of CellTrace Violet-diluting Teff cells were calculated (left panel). The three right panels show representative FACS dot plots of dye-diluting Teff cells cultured with Treg cells at a 1:1 ratio. Data are representative of at least five independent experiments. (b-d) In vivo homeostatic proliferation assay showing the number (Log10) of B6.SJL CD45.1+ naïve T cells recovered from spleens (b), pLN (c) or mLN (d) of recipient Rag1−/− mice 7 d after i.v. transfer of naïve cells (2 × 106) in the absence (none) or presence of FACS-sorted CD4+GFP+ Treg cells (.5 × 106) from FIG or Prkch−/−-FIG mice. Each data point represents a single mouse. (e) Sequential measurements of B16-F10 melanoma growth in groups of Rag1-/- mice (n ≥ 5), which received CD25-depleted B6 splenic cells (1.5 × 107; a source of Teff cells) without or with 0.5 × 106 CD4+GFP+ Treg cells from wild-type or Prkch−/− FIG mice, and were inoculated one day later with tumor cells (2 × 105). Tumor diameters along two perpendicular axes were measured 2-3 times/week. *P < 0.05; **P < 0.01; ***P < 0.001. This experiment is representative of at least three independent experiments.
To demonstrate the importance of PKC-η in Treg cell function in vivo, we used two distinct experimental models, namely, homeostatic T cell expansion and tumor growth. Transfer of naïve CD4+ CD62L+ T cells into immune deficient mice leads to their massive proliferation, a process generally referred to as homeostatic expansion, although it is likely that some of this proliferation is antigen driven19. Treg cells control the expansion of Teff cells in a lymphopenic environment20. Purified naïve CD45.1+CD4+ T cells, either alone or in the presence of sorted wild-type or Prkch−/− CD45.2+CD4+GFP+ Treg cells, were adoptively transferred into Rag1−/− mice, and T cell numbers were determined one week post-transfer. In the presence of wild-type Treg cells, CD45.1+ Teff cell expansion was significantly reduced in all secondary lymphoid organs examined; in contrast, minimal or no reduction in Teff cell expansion was observed in the presence of Prkch−/− Treg cells (Fig. 3b-d). Similar numbers of wild-type and Prkch−/− Treg cells were present in these tissues (Supplementary Fig. 2b).
We next investigated the ability of Prkch−/− Treg cells to inhibit the immune response against a growing tumor. Splenocytes depleted of CD25+ T cells were adoptively transferred into Rag1−/− mice as a source of Teff cells in the absence or presence of Treg cells one day prior to inoculation of B16-F10 melanoma cells14, 21. Transfer of CD25+-depleted splenocytes alone resulted in relatively small skin tumors, whereas mice receiving Teff cells together with wild-type Treg cells developed massive B16 tumors, reflecting inhibition of the Teff cell anti-tumor response by the cotransferred Treg cells (Fig. 3e). Cotransfer of Prkch−/− Treg cells resulted in substantially reduced tumor growth similar to that seen in mice receiving only Teff cells (Fig. 3e). Taken together, these results indicate that in the absence of PKC-η, the in vivo suppressive function of Treg cells is attenuated, leading to enhanced homeostatic proliferation and anti-tumor immunity.
Consistent with the importance of PKC-η in Treg cell suppressive functions, Prkch−/− mice exhibit lymphadenopathy, reflected by increased T and B cell numbers, as well as a higher proportion of CD44hi T cells, characteristic of an activated phenotype22. Indeed, Prkch−/− CD44hi T cells secreted significantly elevated amounts of effector cytokines, including IL-2, IFN-γ, IL-4 and IL-17A (Fig. 4a-d), upon in vitro stimulation with anti-CD3 plus -CD28 monoclonal antibodies (mAbs). Consistent with this hyperactive phenotype, Prkch−/− mice displayed elevated serum levels of IgE (Fig. 4e) and autoantibodies against double-stranded DNA and histone (Fig. 4f, g, respectively) at 8-12 weeks of age, implying a deregulated, hyperactive immune system in the absence of PKC-η.
Figure 4. Cytokine production and serum antibody levels of WT vs. Prkch−/− mice.
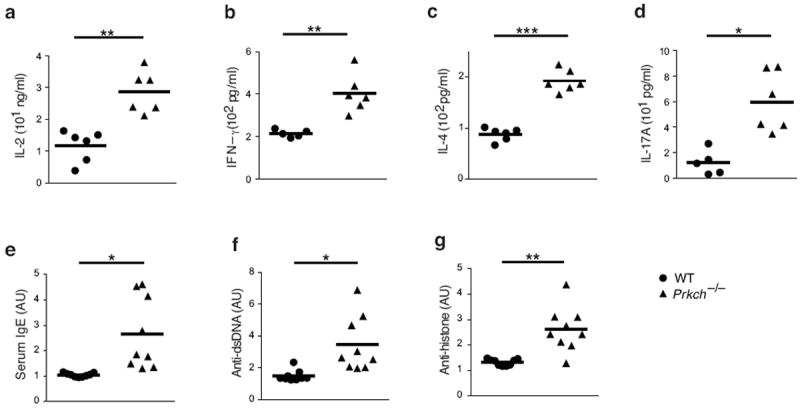
(a-d) Cytokine expression in culture supernatants of WT or Prkch−/− FACS-sorted CD44hi T cells, which were stimulated with anti-CD3 plus -CD28 mAbs for 24 h and assayed by an ELISA for levels of IL-2 (a), IFNγ (b), IL-4 (c) and IL-17A (d). (e-g) Serum levels of IgE (e), anti-double stranded DNA (f) or anti-histone (g) antibodies in 8-12 week-old wild-type and Prkch−/− mice determined by an ELISA. Each data point represents a single mouse from two pooled experiments. *P < 0.01 **P < 0.001; ***P < 0.0001.
We also assessed the ability of wild-type and Prkch−/− Treg cells to inhibit the development of autoimmune colitis in an established in a T cell transfer model. While the transfer of naïve wild-type T cells alone into Rag1−/− recipient mice induced weight loss, indicative of the development of chronic inflammatory bowel disease, cotransfer of either wild-type or Prkch−/− GFP+ Treg cells protected the recipients against weight loss (Supplementary Fig. 3a) and inhibited the expansion of Teff (CD45.1+) cells (Supplementary Fig. 3b). Thus, despite the in vitro (Fig. 3a) and in vivo (Fig. 3b-e) severe defects in their suppressive function, Prkch−/− Treg cells were still able to protect, albeit perhaps incompletely, recipient mice against the development of colitis. This finding suggests that in this particular disease model, Prkch−/− - cells employ an alternative, PKCη-independent suppressive mechanism(s), e.g., IL-10-mediated suppression (Fig. 2k). Furthermore, increased proliferation (or localization) of Prkch−/− Treg cells in the inflammatory bowel environment may compensate for their defective intrinsic suppressive function. Indeed, greater numbers of cotransferred Prkch−/− Treg cells relative to wild-type Treg cells were found in the secondary lymphoid organs of the recipient mice, and this effect tended to be more pronounced in the mLNs, which drain the site of inflammation (Supplementary Fig. 3c). Together, these findings suggest that PKC-η is not globally required for all forms of Treg cell-induced suppression, and that it is dispensable for Treg-mediated inhibition of colitis
Mapping and biological significance of CTLA-4-PKC-η interaction sites
The predominant association of phospho-PKC-η with CTLA-4 in Treg cells suggested that phosphorylated residues mediate this interaction. To pinpoint potential phosphorylation site(s) in PKC-η required for its interaction with CTLA-4, we mutated six predicted PKC-η phosphorylation sites, transfected these mutants into JTAg cells, a Jurkat cell derivative that does not express CD28 but expresses CTLA-4 (data not shown), and immunoprecipitated the transfected CTLA-4 proteins. Upon costimulation with anti-CD3 mAb and CD86-Fc recombinant protein, we found that mutations of amino acid residues S28 and S32 in the C2 domain, or S317 in the V3 domain of PKC-η abolished the interaction with CTLA-4 (Fig. 5a); as a control, mutation of three other phosphorylation sites (S327, T656 or S675) did not affect this interaction (Fig. 5a). Thus, phosphorylation of S28 and S32 or S317 in PKC-η is critical for the interaction with CTLA-4.
Figure 5.
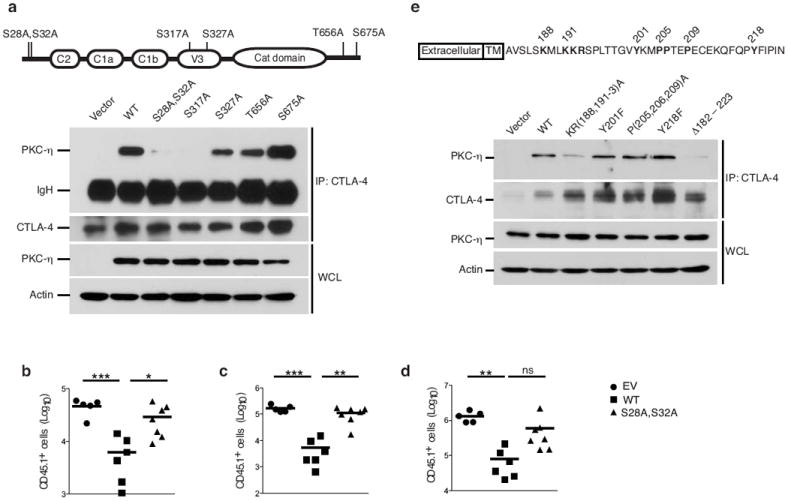
Mapping and biological significance of the CTLA-4-PKC-η interaction sites. (a) Immunoblot analysis of CTLA-4 IPs or WCL from JTAg cells, which were cotransfected with the indicated PKC-η vectors plus wild-type CTLA-4 and stimulated as in Fig. 1a. IgH, heavy chain of the precipitating antibody. Schematic representation of human PKC-η and its phosphorylation sites is shown on top. Data are representative of at leas five independent experiments. (b-d) In vivo homeostatic proliferation assay performed as described in Fig. 3b-d showing the number (Log10) of B6.SJL CD45.1+ naïve T cells, which were recovered from spleens (b), pLN (c) or mLN (d) of recipient Rag1−/− mice 10 d post i.v. transfer of naive cells (2 × 106) in the absence (vector) or presence of FACS-sorted rCD2+CD4+GFP+ Treg cells (.5 × 106). Treg cells were derived from Prkch−/− BM mouse chimeras reconstituted with wild-type PKC-η or a CTLA-4 non-interacting PKC-η-S28A, S32A using retroviral pMIG-IRES-rCD2 vector. Each data point represents a single mouse. ns, non significant; *P < 0.05; **P < 0.01; ***P < 0.001. (e) Immunoblot analysis of CTLA-4 IPs or WCL from JTAg cells cotransfected with the indicated CTLA-4 vectors plus wild-type PKC-η, and processed as in (a). Schematic representation of mouse CTLA-4 is shown on top.
In order to determine whether the association between PKC-η and CTLA-4 is required for the suppressive function of Treg cells, we generated bone marrow (BM) chimeric mice by retrovirally reconstituting Prkch−/−-FIG BM cells with wild-type PKC-η or a mutant (PKC-η-S28A, S32A) that does not interact with CTLA-4. The retroviral vector used coexpressed a non-signaling rat CD2 to allow isolation of transduced T cells. Transduced Treg cells (rCD2+GFP+CD45.2+) from chimeric mice were sorted and assayed for their ability to suppress the in vivo homeostatic proliferation of cotransferred naïve CD45.1+ T cells. Unlike Treg cells expressing wild-type PKC-η, Treg cells reconstituted with PKC-η-S28A, S32A were incapable of suppressing naïve T cell proliferation in the spleens and lymph nodes of adoptively transferred recipients (Fig. 5b-d).
To map the critical motif within the cytoplasmic tail of CTLA-4 that is required for the interaction with PKC-η, we mutated four conserved tail residues, i.e., a membrane proximal positively charged motif (K188XXKKR193), a proline-rich motif (P205, 206, 209) and two tyrosine residues (Y201 or Y218). We found that mutation of the positively charged K188XXKKR193 motif, as well as complete deletion of the CTLA-4 cytoplasmic tail (Δ183-223) greatly reduced the association with PKC-η; in contrast, mutations of the conserved tyrosine residues or the proline-rich motif did not affect the interaction (Fig. 5e). This membrane proximal motif is highly conserved throughout evolution from fish to primates (Supplementary Fig. 4a). Partial truncation of the cytoplasmic tail of CTLA-4 (Δ192-223), which left intact nine membrane proximal residues, including K188 and K191, resulted in a residual association with PKC-η, which, however, was weaker than that displayed by full-length CTLA-4 (Supplementary Fig. 4b). Interestingly, Treg cells expressing CTLA-4 with a similar incomplete truncation of the cytoplasmic tail were reported to retain suppressive activity23, 24. We also found that the interaction between CTLA-4 and PKC-η was not affected by PP2, an inhibitor of Src-family kinases (data not shown), consistent with the lack of effect of Y201 or Y218 CTLA-4 tail mutations on this interaction. Taken together, these results reveal that the CTLA-4-PKC-η interaction is necessary for the suppressive function of Treg cells, thereby implicating PKC-η in a signaling axis linking CTLA-4 to Treg-cell mediated suppression.
Impaired APC dissociation and CD86 depletion by Prkch−/− Treg cells
In comparison to Teff cells, Treg cells preferentially form aggregates with APCs4, 25, and this is due in part to the higher expression of adhesion molecules such as LFA-126 and neuropilin-127 on Treg cells. Such Treg cell-APC engagement has been implicated as a potential suppression mechanism, by allowing Treg cells to effectively compete with Teff cells in engaging APCs and, thus, inhibiting Teff cell activation25. However, activation of LFA-1 and its conversion to a high affinity state, as measured by adhesion to its ligand, ICAM-1, was intact in Prkch−/− Treg cells (Supplementary Fig. 5). Similarly, wild-type and Prkch−/− Treg cells expressed similar levels of neuropilin-1, CD39 and CD73 (data not shown), the latter two representing cell surface molecules that have been implicated in Treg cell-mediated suppression through an adenosine-dependent action28.
To elucidate the signaling mechanism potentially responsible for the PKC-η-mediated suppression, we performed a phosphoproteomic analysis of wild-type vs. Prkch−/− Treg cells and found that PAK2 and GIT2, two components of a focal adhesion complex that promotes focal adhesion disassembly and, hence, cellular motility29, 30, were significantly hypophosphorylated in Prkch−/− Treg cells (Supplementary Fig. 6a). A complex of these two proteins together with the guanine nucleotide exchange factor αPIX was found to translocate to the T cell IS and to be required for optimal Teff cell activation31. Moreover, CTLA-4 immunoprecipitates from anti-CD3- plus -CTLA-4-costimulated wild-type Treg cells contained not only PKC-η, but also GIT2, αPIX and PAK kinase (Fig. 6a). These proteins were also present in PKC-η immunoprecipitates (data not shown). Of note, recruitment of this complex was unique to CTLA-4 costimulation, because the association of the GIT-PIX-PAK complex with CTLA-4 was barely above background level when the cells were costimulated with anti-CD3 plus - CD28 mAbs (Supplementary Fig. 6b). Thus, this particular signaling event is not shared between CTLA-4 and CD28. Furthermore, the activating phosphorylation of PAK kinases was dramatically reduced in Prkch−/− Treg cells (Fig. 6b), indicating impaired activation of this complex.
Figure 6.
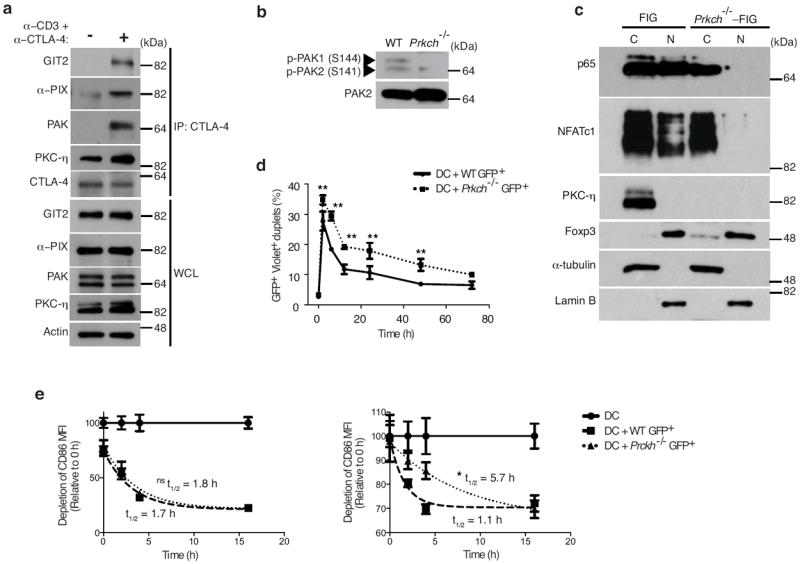
CTLA-4-PKC-η recruits GIT-PIX-PAK complex and modulates Treg cell-APC interaction. (a) Immunoblot analysis of cytosolic (C) and nuclear (N) fractions of FACS-sorted wild-type or Prkch−/− FIG CD4+GFP+ Treg cells, which were stimulated with anti-CD3ε plus -CTLA-4 mAbs for 5 min. (b) Immunoblot analysis of CTLA-4 IPs or WCL of FACS-sorted GFP+ Treg cells derived from FIG mice and left unstimulated (-) or stimulated (+) with ani-CD3 plus anti-CTLA-4 antibodies for 5 min. (c) Expression of phospho-PAK and total PAK2 in lysates of CD4+GFP+ Treg cells from wild-type or Prkch−/− FIG mice determined by immunoblotting with the corresponding antibodies. (d) Conjugation assay measuring formation of cell doublets between FACS-sorted CD4+GFP+ wild-type or Prkch−/− FIG Treg and CellTrace Violet-labeled splenic DCs at different times during a 3 d coculture period in the presence of anti-CD3 mAb and IL-2. Percentages of GFP+ and Violet+ double-positive doublets are shown. *P < 0.05; **P <0.001. (e) CD86 depletion from APCs cocultured in the absence (circles) or presence of FACS-sorted CD4+GFP+ wild-type (squares) or Prkch−/− (triangles) FIG Treg cells. A first set of CD45.2+ splenic DCs was cultured for 9 h in the presence of anti-CD3 mAb and IL-2 prior to the addition of a second set of CD45.1+ splenic DCs. The geometric mean fluorescence intensity of CD86 was enumerated on gated CD11c+ Annexin V− CD45.2+ and CD11c+ Annexin V− CD45.1+ cells, respectively. The t1/2 values of CD86 decay curves were calculated using the GraphPad Prism™ program. This experiment is representative of three independent experiments. ns, non significant; *P < 0.05.
Given the impaired PAK activation, and because PAK kinase was found to be required for TCR-induced transcriptional activation of NFAT and the CD28 response element (which includes an NF-κB-binding site) in Jurkat T cells31, 32, we also examined whether Prkch−/− Treg cells display impaired activation of these transcription factors following costimulation with anti-CD3ε plus -CTLA-4 mAbs. In these conditions we observed a severe defect in NFATc1 and NF-κB activation in the Prkch−/− Treg cells (Fig. 6c).
Because the PAK2-GIT2 complex promotes cellular motility through focal adhesion disassembly29, 30, we investigated if the defective activation of the GIT-PIX-PAK complex in Prkch−/− Treg cells might result in a more stable conjugation of Prkch−/− Treg cells with APCs. We noticed that the efficiency of conjugation between Prkch−/− Treg cells and APCs was significantly higher by comparison to APC conjugates of wild-type Treg cells (Fig. 6d), suggesting that in Prkch−/− Treg cells, impaired activation of the GIT-PIX-PAK complex leads to defective breaking of Treg cell-APC contacts.
If Prkch−/− Treg cells display impaired dissociation from engaged APCs due to defective activation of the GIT-PIX-PAK complex, this defect may be expected to translate into reduced ability of Prkch−/− Treg cells to serially engage new APCs, which, in turn, could result in reduced suppressive activity. This prediction is based on findings that Treg cells can capture CD80 and CD86 molecule from APCs, a process that depletes the ligands required for CD28 costimulation of Teff cells15 and, thus, considered as a potential mechanism of contact-dependent Treg cell-mediated suppression. To address this possibility, we tested the ability of wild-type vs. Prkch−/− Treg cells to deplete CD86 from cocultured CD45.2+CD11c+ APCs as an indirect measure of APC engagement by the Treg cells. wild-type and Prkch−/− Treg cells were equally capable of depleting CD86 from these APCs (Fig. 6e, left panel). However, upon subsequent addition of a second pool of APCs, distinguished from the first APC population by their CD45.1 expression, Prkch−/− Treg cells had a significant delay in their ability to deplete CD86 from these newly introduced APCs, as indicated by the fact that it took them ~4-fold longer time (~16 vs. ~4 h) to execute the same level of CD86 depletion as that accomplished by wild-type Treg cells (Fig. 6e, right panel). This observation supports the notion that the relative inefficiency of Prkch−/−Treg cells to serially engage new APCs and, hence, to effectively deplete APC-expressed CD86, could account, at least in part, for their reduced suppressive activity. Importantly, our findings imply that a Treg cell-intrinsic, CTLA-4-PKC-η-dependent signaling mechanism is required in order to manifest the cell-extrinsic suppressive function of CTLA-4 toward Teff cells.
DISCUSSION
We report a novel interaction of CTLA-4 with PKC-η and preferential IS localization of PKC-η in Treg cells upon their activation. Moreover, PKC-η and its CTLA-4 association were required for the contact-dependent suppressive function of Treg cells, albeit not for their development. Although Prkch−/− Treg cells displayed defective suppressive activity in a conventional in vitro suppression assay and in in vivo homeostatic proliferation and anti-tumor immunity models, they could still inhibit the development of autoimmune colitis. Our findings provide two potential explanations for these apparently discrepant observations. First, enhanced expansion of Prkch−/− Treg cells in the inflammatory bowel environment, which was particularly noticeable in the mLN of recipient mice, could compensate for their reduced intrinsic suppressive function. Second, Treg cells use various mechanisms of suppression and the relative importance of each mechanism depends on the environment and the context of the immune response. For example, similar to Prkch−/− Treg cells, Ctla4−/− Treg cells suppress the development of colitis through intact compensatory expression of IL-1033, and we found that Prkch−/− Treg cells expressed normal levels of IL-10. Thus, the CTLA-4-associated PKC-η-dependent suppressive pathway that we describe here may be less relevant in Treg suppressive mechanisms that do not depend on Treg cell-APC contact but, rather, on soluble mediators. If our observations of a dispensable role of PKC-η in Treg cell protection against colitis can be extended to other autoimmune disease models, this would suggest that strategies that selectively inhibit PKC-η or its association with CTLA-4 may disable Treg cells from inhibiting anti-tumor immunity without affecting their ability to protect against autoimmune diseases, a notion with important potential translational implications.
The Treg cell defect revealed by our study may explain the increased cytokine production and higher amounts of serum IgE and autoantibodies in Prkch−/− mice, although intrinsic hyperactivity of the Teff cells in these mice could also contribute to this increase. Nevertheless, it is interesting to note that mice with a Treg-specific deletion of Ctla4 display similar increases in cytokine, IgE and autoantibody amounts14, reinforcing the notion of a functionally relevant link between CTLA-4 and PKC-η at the level of Treg cell-mediated suppression.
The recruitment of a GIT-PIX-PAK complex to CTLA-4 (and PKC-η) upon TCR plus CTLA-4 (but not CD28) costimulation of Treg and its impaired activation in Prkch−/− Treg cells suggest a potential mechanism underlying their impaired suppressive activity, based on previous findings that this macromolecular complex is required for the disassembly of focal adhesions in neurons and epithelial cells29, 30 Given the reported localization of this complex to the T cell IS and its importance for T cell activation31, it is possible that the TCR-activated GIT-PIX-PAK complex destabilizes T cell-APC contacts, thereby promoting conversion of the mature, concentric IS into an unstable IS (i.e., kinapse), which is important for the motility of T cells and their ability to serially engage new APCs34. In the context of contact-dependent, Treg cell-mediated suppression, increased stability of Treg cell-APC conjugates and the concomitant reduction in their ability to serially engage new APCs would be translated to impaired overall suppressive activity. Consistent with this notion, we found that Prkch−/− Treg cells form more efficient contacts with APCs in comparison with their wild-type counterparts and, furthermore, these Prkch−/− Treg cells displayed a reduced ability to deplete CD86 from a second set of added APCs, likely a reflection of impairment in their disengagement from the first set of APCs. When considered at the population level and the relatively long time during which Treg cells have the opportunity to engage APCs, this effect could translate into an overall reduction in CD86 depletion and, consequently, reduced Treg cell suppression. Hence, our findings identify a Treg cell-intrinsic signaling mechanism consisting of a CTLA-4-bound PKC-η-GIT-PIX-PAK complex, which plays a role in promoting the cell-extrinsic suppressive function of Treg cell-expressed CTLA-4 on Teff cells.
The biology of CTLA-4 is complex and its mechanisms of action in T cells are still incompletely understood. This complexity reflects, to a large extent, the fact that CTLA-4 is expressed both in Teff cells where it functions in a cell-autonomous manner in cis to inhibit their activation, and in Treg cells where it operates in a cell-non-autonomous manner in trans to dampen Teff cell responses17, 18, 35. Earlier studies did not distinguish between these functions, but with the conditional deletion of Ctla4 in Treg cells only, it became evident that CTLA-4 expression by Treg cells is in most (but not all) cases important for their suppressive function through depletion of CD80 and CD86 from APCs14, 15, 36.
Ctla4−/− mice display fatal lymphoproliferative disorder characterized by the systemic infiltration of pathogenic self-reactive T cells37, 38. Although Prkch−/− mice displayed moderate lymphadenopathy, increase in memory-activated T cells and cytokine expression, and elevated IgE and autoantibodies at a relatively young age, they lived into adulthood (~1 year) with no gross signs of pathology (data not shown). Several functional disparities could account for these different phenotypes. First, CTLA-4 inhibits thymocyte negative selection39 and, as a result, Ctla4−/− mice harbor autoreactive T cells that cause tissue damage40. However, thymic selection is largely intact in the absence of PKC-η22, suggesting that the lack of overt autoreactivity and lymphoproliferation in Prkch−/− mice might limit the self-destructive nature of the hyperactive Prkch−/− Teff cells. Second, the inhibitory effect of CTLA-4 is mediated by a combination of non-mutually exclusive mechanisms consisting of Treg cell-intrinsic as well as Teff cell-intrinsic mechanisms, and both mechanisms can cooperate to dampen Teff cell responses. Third, our findings strongly suggest that the intrinsic ability Prkch−/− Treg cells to deplete CD86 from APCs by transendocytosis (which depends on the CTLA-4extracellular domain) is intact, while only the newly described signaling function of CTLA-4, i.e., the dissociation of Treg cells from APCs mediated by the PKC-η-GIT-PIX-PAK complex (which depends on the CTLA-4 cytoplasmic domain), is impaired. In contrast, Treg cells from germline Ctla4−/− mice or Foxp3-Cre conditional Ctla4−/− mice lack both of these CTLA-4 functions.
Our findings of altered APC engagement and presumed reduced motility of Prkch−/− Treg cells may be related to the reported ability of CTLA-4 to reverse the TCR stop signal and promote the motility of Teff cells, possibly via LFA-1 activation41, 42. In contrast, Treg cells were found to be resistant to the CTLA-4-induced reversal of the TCR stop signal, resulting in longer contact times with APCs43, a scenario that would lead to lower overall Treg cell motility and less frequent serial encounters with new APCs. However, we did not observe a defect in LFA-1 expression or activation in Prkch−/− Treg cells and, furthermore, while LFA-1 promotes the formation of a stable IS, our findings point to a defect at a later stage, i.e., disassembly of the IS in the Prkch−/− Treg cells.
The successful clinical use of anti-CTLA-4 heralded a new era of cancer immunotherapy by targeting immunosuppression44. In many tumor tissues, infiltrating Treg cells restrict the function of Teff cells and, therefore, inhibiting critical Treg cell signaling molecules that are important for their function could lead to enhanced antitumor responses45. Here, we showed that phosphorylated PKC-η is recruited to the Treg cell IS and is associated with the cytoplasmic tail of CTLA-4, and demonstrated the critical importance of this association for the contact-dependent suppressive activity of Treg cells. Hence, the CTLA-4-PKC-η axis could represent a key therapeutic target for Treg cell-dependent suppression in controlling cancer.
METHODS
Antibodies (Abs) and reagents
mAbs specific for mouse CD3 (clone 145-2C11), CD28 (clone 37.51), or CTLA-4 (clone UC10-4B9) were purchased from Biolegend, as were flourophore-conjugated anti-CD4 (Clone GK1.5), -CD8 (53-6.7), -Foxp3 (Clone FJK-16s), -CD25 (Clone PC61), -CD44 (Clone IM7) and –GITR (Clone DTA-1) mAbs. Anti-human CD3 mAb (OKT3) was purified in-house. Polyclonal anti-PKC-θ (sc-212), anti-PKC-η (C-15 and sc-215), anti-PAK (N-20 and sc-882), anti-NFATc1 (7A6), anti-lamin B (M-20), and anti-α-tubulin (TU-02) Abs were obtained from Santa Cruz Biotechnology. Anti-p65 (NF-κB), anti-GIT2 and -αPIX Abs were obtained from Cell Signaling Technology. Anti-Foxp3 Abs (clone 150D/E4 for immunoblotting and clone FJK-16s for flow cytometry) were purchased from eBiosciences. Alexa-647-conjugated anti-mouse Ig and Alexa-555-conjugated anti-rabbit Ig were obtained from Molecular Probes. Digitonin was obtained from EMD Chemicals. Calf intestinal alkaline phosphatase was purchased from New England Biolabs. Recombinant CD86-Fc was previously described46.
Plasmids
Plasmids of full-length human Prkch and mouse Ctla4 were generated via PCR amplification and cloned into the pEF4/HisC expression vector or pMIG retroviral vector, respectively. Prkch and Ctla4 point mutations were generated using Quikchange II Site-directed Mutagenesis Kit (Stratagene). Ctla4 mutants with cytoplasmic tail deletions (amino acid 183-223 and 192-223) were generated via PCR amplification.
Mice and primary cell cultures
C57BL/6 (B6; CD45.2+), B6.SJL (CD45.1+) and Prkch−/− (CD45.2+)22 mice were housed and maintained under specific pathogen-free conditions, and manipulated according to guidelines approved by the LIAI Animal Care Committee and the Animal Care and Use Committee of The Scripps Research Institute. The Prkch−/−mice are now available from the Jackson Laboratories (B6.Cg-Prkch<tm1.1Gasc>/J). Foxp3-IRES-eGFP (FIG) mice were obtained from the Jackson Lab. Prkch−/− × Foxp3-IRES-eGFP (Prkch−/−-FIG) mice16 were generated by crossing FIG and Prkch−/− mice22. CD4+ T cells were isolated by anti-CD4 (BD Biosciences) positive selection, and were cultured in RPMI-1640 medium (Mediatech, Inc.) supplemented with 10% heat-inactivated fetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, 1 mM MEM nonessential amino acids, and 100 U/ml each of penicillin G and streptomycin (Life Technologies).
Immunoprecipitation and immunoblotting
Simian virus 40 large T antigen-transfected human leukemic Jurkat T cells (JTAg) and MCC-specific hybridoma T cells were described previously9; these cell lines have not been recently STR profiled but tested negative for mycoplasma contamination. JTAg cells in logarithmic growth phase were transfected with plasmid DNAs by electroporation and incubated for 48 h. Transfected cells were stimulated with an anti-human CD3 (OKT3) mAb and recombinant CD86-Fc in the presence of a cross-linking Ab for 5 min. Cell lysis in 1% NP-40 lysis buffer (50 mM Tris-HCl, 50 mM NaCl, 5 mM EDTA), immunoprecipitation, and immunoblotting were carried out as previously described9.
Enzyme-linked immunoabsorbent assay (ELISA)
Serum IgE was quantified using capture and biotinylated mAbs from CALTAG Laboratories, as previously described47. Autoantibodies specific for double-stranded DNA (dsDNA) and histone were determined using plates coated with salmon sperm DNA (Life Technologies) or calf thymus histone (Roche), respectively. Detection was carried out using biotinylated anti-mouse IgG, streptavidin conjugated HRP, and ABTS substrates (Bio-Rad). Relative Ig serum levels were calculated by dividing the absorbance values of experimental samples by the negative control values.
Isolation of mRNA, cDNA synthesis and real-time PCR
Total RNA was extracted from sorted CD4+GFP- and CD4+GFP+ cells of FIG mice using the RNeasy kit (Qiagen). RNA was used to synthesize cDNA by the SuperScript III FirstStrand cDNA synthesis kit (Life Technologies). Gene expression was determined using real-time PCR with iTaq SYBR Green (Bio-Rad) in the presence of the following primer sets for mouse Ctla4 (Forward: 5’ ACTCATGTACCCACCGCCATA 3’; Reverse: 5’ GGGCATGGTTCTGGATCAAT 3’), Prkch (Forward: 5’ CAAGCATTTTACCAGGAAGCG 3’; Reverse: 5’ TGTTTCCCAAATACTCCCCAG 3’) and the housekeeping gene β-actin (ACTB). Relative gene expression levels were determined in triplicates and calculated using the 2-ΔΔCt and normalized to the level of ACTB.
Microscopy
CD4+ T cells were purified from AND-Tg Rag2−/− mice and stimulated by immobilized anti-CD3ε (145-2C11; 10 μg/ml) and anti-CD28 (PV-1; 1 μg/ml) in the presence of mouse recombinant IL-2 (10 ng/ml) and human recombinant TGFβ (5 ng/ml) for 3 d. The cells were retrovirally transduced with eGFP-tagged mouse PKC-θ or PKC-η for 24 h one day after the initial stimulation. On d 4 or later, the cells were sorted on a FACSAria (BD) to obtain homogenous population of highly GFP+ cells, which were maintained in culture. B cells purified from B10. BR mice and stimulated by LPS (Difco; 10 μg/ml) plus MCC (1 μM) were added to the cultured T cells for 1 d. Dead cells were removed by Lympholite-M (Cedarlane), and the CD4+ Treg cells were prestained by DyLight650-labeled anti-TCRβ (H57) Fab, conjugated with MCC-pulsed LPS-stimulated B cells for 5-10 min, fixed with 2% PFA, and imaged by confocal microscopy (Leica SP5) with ProLong gold antifade reagent with DAPI (Molecular Probes).
In vitro suppression assay
FACS-sorted naïve CD4+CD62L+ Teff cells were labeled with 5 μM of CellTrace Violet according to the manufacturer’s protocol (Life Technologies). CD11c+ splenic DCs were purified according to the manufacturer’s instructions (Miltenyi Biotech). Labeled Teff cells (2 × 104) and splenic DCs (5 × 103) were cocultured for 3 days with CD4+GFP+ Treg cells (0.125-4 × 104) sorted from wild-type or Prkch−/− FIG mice in the presence of an anti-mouse CD3 mAb.
Homeostatic expansion model
Naïve CD4+CD62L+ cells from congenic B6. SJL and CD4+GFP+Treg cells from wild-type and Prkch−/− FIG mice were sorted using ARIA Cell Sorter (BD Biosciences). Two × 106 naïve cells were transferred i.v. alone or together with 0.5 × 106 CD4+GFP+ Treg cells into Rag1−/− mice; this was done blind and assigned randomized with no pre-established inclusion/exclusion criteria. Mice were euthanized 7-10 days post-transfer. Spleens, pLN and mLN were harvested and each population was independently analyzed by flow cytometry. To achieve reasonable power, at least 5 mice/group (15 mice/experiment) were used. Additional mice were added to experiments as appropriate.
B16 melanoma model
Splenocytes from wild-type B6 mice were depleted of CD25+ cells using biotinylated anti-CD25 mAb (clone PC61, eBiosciences) and streptavidin-conjugated beads (BD Biosciences). CD4+GFP+ Treg cells were FACS-sorted from wild-type or Prkch−/− FIG mice. 15 × 106 CD25-depleted splenocytes were adoptively transferred alone or together with 0.5 × 106 wild-type or Prkch−/− CD4+GFP+ Treg cells into recipient Rag1−/− mice; this was done blind and assigned randomized with no pre-established inclusion/exclusion criteria. Two × 105 B16-F10 melanoma cells were inoculated intradermally on the right shaved flanks the next day. Tumor size was measured using an electronic dial caliper 2-3 times/week14. To achieve reasonable power, at least 5 mice/group (15 mice/experiment) were used. Additional mice were added to experiments as appropriate.
BM chimeras
Full-length human wild-type or S28A, S32A mutated Prkch were subcloned into a modified pMIG retroviral vector containing IRES and non-signaling rat CD2 gene (lacking the cytoplasmic tail). BM chimeras were produced in irradiated B6 mice as previously described9. Briefly, BM cells were flushed from the femurs and tibias of Prkch−/−-FIG mice that have been pretreated with 5-fluorouracil to enrich for stem cells. BM cells were cultured in DMEM media (Mediatech, Inc.) containing 10% FBS, IL-3 (20 ng/ml), IL-6 (25 ng/ml) and stem cell factor (SCF;100 ng/ml). Retroviral infections were carried out for 2 consecutive days. 1 × 106 infected BM cells were intravenously injected into irradiated B6 mice. Analyses were performed 10-12 weeks post-reconstitution to determine cells coexpressing GFP (for Foxp3 expression) and rat CD2 (for transgene expression) using anti-rCD2 mAb (clone OX-34, Biolegend). Cells were pooled from spleens and peripheral LN of 4-5 chimeric mice, and enriched for CD4+ cells. GFP+rCD2+double positive Treg cells were sorted on an ARIA Cell Sorter, and their in vivo suppressive function was analyzed in a homeostatic expansion assay as described above.
Treg cell-APC coculture
GFP+ Treg cells (5 × 104) from wild-type or Prkch-/- FIG mice were cultured with CellTrace Violet-labeled splenic CD11c+ APCs (5 × 104) for the indicated times. Cells were carefully collected with cut tips and immediately assayed on an LSRII flow cytometer to determine double-positive (GFP+Violet+) conjugates. For CD86 depletion experiment, Treg cells (5 × 104) were cultured with CD45.2+CD11c+ splenic APCs (2.5 × 104) for 9 h and, after which CD45.1+CD11c+ splenic APC (2.5 × 104) from B6.SJL mice were added for another 9 h. Cells were collected and stained with fluorophore-conjugated Abs specific for CD11c, Annexin V, CD4, CD86, I-Ab, CD45.1, or CD45.2.
SILAC and phosphoproteomic analysis
wild-type and Prkch−/− FIG naïve CD4+ T cells were differentiated into iTreg cells as described above in regular RPMI-1640 medium or medium supplemented with 13C, 15N labeled lysine and arginine for SILAC labeling. FACS-sorted GFP+ Treg cells were left unstimulated or stimulated with anti-CD3 plus -CTLA-4 mAbs for 5 min, wild-type and Prkch−/− FIG cell lysates were mixed at a 1:1 ratio, and 300 μg of the protein mixture was precipitated with 5x volume of cold acetone. Following centrifugation at 14,000 × g (10 min at 4°C), protein pellets were solubilized and reduced with 100 mM Tris-HCl/8 M urea/5 mM tris(2-carboxyethyl)phosphine. Cysteine was alkylated with 10 mM iodoacetamide. The solution was diluted 1:4 and digested with 5 μg of trypsin at 37°C overnight. Digestion was terminated by adding 10% acetonitrile and 2% formic acid, and the resulting peptides were subjected to TiO2 phosphopeptide enrichment as described48. Briefly, phosphopeptides were bound to the TiO2 resin, and eluted with 250 mM NH4HCO3, pH 9. Enriched phosphopeptides were analyzed by the MudPIT LC-MS/MS method49. MS analysis was performed using an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher). A cycle of one full-scan mass spectrum (300-1800 m/z) at a resolution of 60,000 followed by 20 data dependent MS/MS spectra at a 35% normalized collision energy was repeated continuously throughout each step of the multidimensional separation.
MS data were analyzed by the Integrated Proteomics Pipeline - IP2 (Integrated Proteomics Applications; http://www.integratedproteomics.com/). The tandem mass spectra were searched against EBI IPI mouse target/decoy protein database. Protein false discovery rates were controlled below 1% for each sample. In ProLuCID database search, the cysteine carboxyamidomethylation was set as a stable modification, and phosphorylation on serine, threonine, or tyrosine was configured as differential modification. Peptide quantification was performed by Census software, in which the isotopic distributions for both the unlabeled and labeled peptides were calculated and this information was then used to determine the appropriate m/z range from which to extract ion intensities. Phosphopeptides were further evaluated with IP2 phospho analysis module, which computes Ascore50 and Debunker score51.
Statistical analysis
Statistical analyses were performed using, unless otherwise stated, ANOVA with post-hoc Bonfferoni’s corrections. Unless otherwise indicated, data represent the mean ± s.e.m, with P < 0.05 considered statistically significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors would like to acknowledge H. Cheroutre and Y.-C. Liu for helpful discussions. The authors would also like to acknowledge the excellent services provided by the Flow Cytometry Core Unit at LIAI, as well as the Animal Husbandry Units at LIAI and TSRI. This is publication number 1552 from the La Jolla Institute for Allergy and Immunology and 21923 from The Scripps Research Institute. This work was supported by NIH grants CA35299 (AA), GM065230 (NRJG), and P01AI089624 (MK). KFK was supported by LIAI-T1D-CRF 2012 Fellowship and Young Investigator Award #270056 from the Melanoma Research Alliance.
Footnotes
Author Contributions
K.F.K. and G.F. designed experiments, collected data, performed analyses and wrote the paper; J.C., T.Y. and T.S. performed microscopy experiment; A.J.C. was involved in experiments and data collection; S.B. performed and assisted in melanoma studies; Y.Z. and J.Y. did the phosphoproteomic experiment and analyses; G.K. and M.K. provided critical reagents and were involved in study design; N.R. J.G. and A.A. designed the study, analyzed data and wrote the paper.
The authors declare no competing financial interests.
References
- 1.Bennett CL, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 2.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 3.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 4.Zanin-Zhorov A, et al. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science. 2010;328:372–376. doi: 10.1126/science.1186068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins AV, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–210. doi: 10.1016/s1074-7613(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 6.Yokosuka T, et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. 2010;33:326–339. doi: 10.1016/j.immuni.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annu Rev Immunol. 2006;24:65–97. doi: 10.1146/annurev.immunol.24.021605.090535. [DOI] [PubMed] [Google Scholar]
- 8.Kong KF, Altman A. In and out of the bull’s eye: protein kinase Cs in the immunological synapse. Trends Immunol. 2013;34:234–242. doi: 10.1016/j.it.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong KF, et al. A motif in the V3 domain of the kinase PKC-theta determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat Immunol. 2011;12:1105–1112. doi: 10.1038/ni.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JK, et al. Impact of the TCR signal on regulatory T cell homeostasis, function, and trafficking. PLoS One. 2009;4:e6580. doi: 10.1371/journal.pone.0006580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park SG, et al. T regulatory cells maintain intestinal homeostasis by suppressing gammadelta T cells. Immunity. 2010;33:791–803. doi: 10.1016/j.immuni.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chuck MI, Zhu M, Shen S, Zhang W. The role of the LAT-PLC-gamma1 interaction in T regulatory cell function. J Immunol. 2010;184:2476–2486. doi: 10.4049/jimmunol.0902876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spitaler M, Emslie E, Wood CD, Cantrell D. Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity. 2006;24:535–546. doi: 10.1016/j.immuni.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 15.Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haribhai D, et al. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol. 2007;178:2961–2972. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- 17.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuda JL, et al. Systemic activation and antigen-driven oligoclonal expansion of T cells in a mouse model of colitis. J Immunol. 2000;164:2797–2806. doi: 10.4049/jimmunol.164.5.2797. [DOI] [PubMed] [Google Scholar]
- 20.Collison LW, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 22.Fu G, et al. Protein kinase C eta is required for T cell activation and homeostatic proliferation. Sci Signal. 2011;4:ra84. doi: 10.1126/scisignal.2002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tai X, et al. Basis of CTLA-4 function in regulatory and conventional CD4+ T cells. Blood. 2012;119:5155–5163. doi: 10.1182/blood-2011-11-388918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kataoka H, et al. CD25(+)CD4(+) regulatory T cells exert in vitro suppressive activity independent of CTLA-4. Int Immunol. 2005;17:421–427. doi: 10.1093/intimm/dxh221. [DOI] [PubMed] [Google Scholar]
- 25.Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A. 2008;105:10113–10118. doi: 10.1073/pnas.0711106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran DQ, et al. Analysis of adhesion molecules, target cells, and role of IL-2 in human FOXP3+ regulatory T cell suppressor function. J Immunol. 2009;182:2929–2938. doi: 10.4049/jimmunol.0803827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarris M, Andersen KG, Randow F, Mayr L, Betz AG. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity. 2008;28:402–413. doi: 10.1016/j.immuni.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borsellino G, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 29.Zhao ZS, Manser E, Loo TH, Lim L. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol Cell Biol. 2000;20:6354–6363. doi: 10.1128/mcb.20.17.6354-6363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lucanic M, Cheng HJ. A RAC/CDC-42-independent GIT/PIX/PAK signaling pathway mediates cell migration in C. elegans. PLoS Genet. 2008;4:e1000269. doi: 10.1371/journal.pgen.1000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phee H, Abraham RT, Weiss A. Dynamic recruitment of PAK1 to the immunological synapse is mediated by PIX independently of SLP-76 and Vav1. Nat Immunol. 2005;6:608–617. doi: 10.1038/ni1199. [DOI] [PubMed] [Google Scholar]
- 32.Yablonski D, Kane LP, Qian D, Weiss A. A Nck-Pak1 signaling module is required for T-cell receptor-mediated activation of NFAT, but not of JNK. The EMBO journal. 1998;17:5647–5657. doi: 10.1093/emboj/17.19.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uhlig HH, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J Immunol. 2006;177:5852–5860. doi: 10.4049/jimmunol.177.9.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dustin ML. T-cell activation through immunological synapses and kinapses. Immunol Rev. 2008;221:77–89. doi: 10.1111/j.1600-065X.2008.00589.x. [DOI] [PubMed] [Google Scholar]
- 35.Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11:852–863. doi: 10.1038/nri3108. [DOI] [PubMed] [Google Scholar]
- 36.Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of Cd25+Cd4+ regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tivol EA, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 38.Waterhouse P, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 39.Buhlmann JE, Elkin SK, Sharpe AH. A role for the B7-1/B7-2:CD28/CTLA-4 pathway during negative selection. J Immunol. 2003;170:5421–5428. doi: 10.4049/jimmunol.170.11.5421. [DOI] [PubMed] [Google Scholar]
- 40.Ise W, et al. CTLA-4 suppresses the pathogenicity of self antigen-specific T cells by cell-intrinsic and cell-extrinsic mechanisms. Nat Immunol. 2010;11:129–135. doi: 10.1038/ni.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider H, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972–1975. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 42.Rudd CE. The reverse stop-signal model for CTLA4 function. Nat Rev Immunol. 2008;8:153–160. doi: 10.1038/nri2253. [DOI] [PubMed] [Google Scholar]
- 43.Lu Y, Schneider H, Rudd CE. Murine regulatory T cells differ from conventional T cells in resisting the CTLA-4 reversal of TCR stop-signal. Blood. 2012;120:4560–4570. doi: 10.1182/blood-2012-04-421420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peggs KS, Quezada SA, Allison JP. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists. Clin Exp Immunol. 2009;157:9–19. doi: 10.1111/j.1365-2249.2009.03912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206:1717–1725. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim G, et al. Spontaneous colitis occurrence in transgenic mice with altered B7-mediated costimulation. J Immunol. 2008;181:5278–5288. doi: 10.4049/jimmunol.181.8.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Becart S, et al. SLAT regulates Th1 and Th2 inflammatory responses by controlling Ca2+/NFAT signaling. J Clin Invest. 2007;117:2164–2175. doi: 10.1172/JCI31640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H, et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013;9:e1003277. doi: 10.1371/journal.pgen.1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 50.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 51.Lu B, Ruse C, Xu T, Park SK, Yates J., 3rd Automatic validation of phosphopeptide identifications from tandem mass spectra. Anal Chem. 2007;79:1301–1310. doi: 10.1021/ac061334v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.