

Abstract
The pregnancy disorders associated with placental ischemia share many similar pathological and pathophysiological features and are associated with the failure to deliver adequate nutrients and oxygen to the placenta. The origins of this deficiency are a subject of intense study. In this presentation I review the genesis and consequences of this pathology addressing similarities and difference with the different disorders and addressing current gaps in our knowledge.
Introduction
The failure to deliver adequate oxygen and nutrients to the placenta not only compromises fetal well being but also activates a number of pathophysiological abnormalities that affect maternal health and compounds the fetal problems. In this presentation I will discuss the genesis and consequences of abnormal placental perfusion and attempt to identify questions, which could guide productive future research.
The genesis of placental ischemia
The establishment of an intervillus circulation
When the embryo implants into the decidua it is originally present in a relatively avascular setting.1 There is no direct vascular contact with the intervillus space which is fluid filled. Nutrition is provided primarily by the endometrial glands. During this time, cytotrophoblast differentiation, which appears to depend upon an increased pO2, does not occur but rather the cytotrophoblast proliferates extensively.2 This increased proliferation is proposed to result in villus plugging of the terminal spiral arteries preventing intervillus perfusion. At about 8-10 weeks of gestation the maternal vessels begin to remodel eliminating the cytotrophoblast plugging and allowing perfusion of the intervillus space. In addition to increasing nutrient and oxygen delivery to the embryo the resultant increased perfusion has two particularly relevant consequences. The advent of increased oxygen delivery results in oxidative stress, which must be buffered, and the increase in pO2 converts cytotrophoblast to an invasive phenotype.1, 2
Physiological modification of the spiral arteries perfusing the intervillus space
The remodeling of the spiral arteries that leads to perfusion of the intervillus space results from dramatic modifications of the maternal spiral arteries that supply the placental site. Spiral arteries in non-pregnant women are the terminal branches of the uterine arteries that supply blood to the endometrium. They are fairly characteristic, small, muscular arteries, richly innervated and sensitive to humoral and neural signals.3 During normal pregnancy, between 10 and 20 weeks of gestation, these vessels undergo physiological changes.4 The most obvious modification (Figure 1) is a dramatic increase in terminal luminal diameter, which increases 5-10 fold.5 The vessel wall structure also changes (Figure 2). The muscular and elastic components of the vessel are eliminated resulting in a vessel that has become a flaccid dilated tube.6 This modification extends to the inner third of the myometrium. This depth of invasion is important. Near the junction of the mucosal lining of the uterus (endometrium in non-pregnant woman and decidua in pregnancy) there is a segment of vascular smooth muscle that is proposed to serve as a functional sphincter.7 During menses this portion of the vessel constricts to terminate bleeding that occurs when the spiral artery is disrupted by endometrial shedding.3 As a result of the physiological remodeling this “sphincter” is lost. Combined with the loss of smooth muscle in the spiral artery in the decidual and the inner third of the myometrium this portion of the vessel loses its capacity to contract in response to humoral or neural signals. Another change of unknown significance is increased formation of vascular shunts between arteries and veins below the placenta.
Figure 1. Failed spiral artery remodeling.
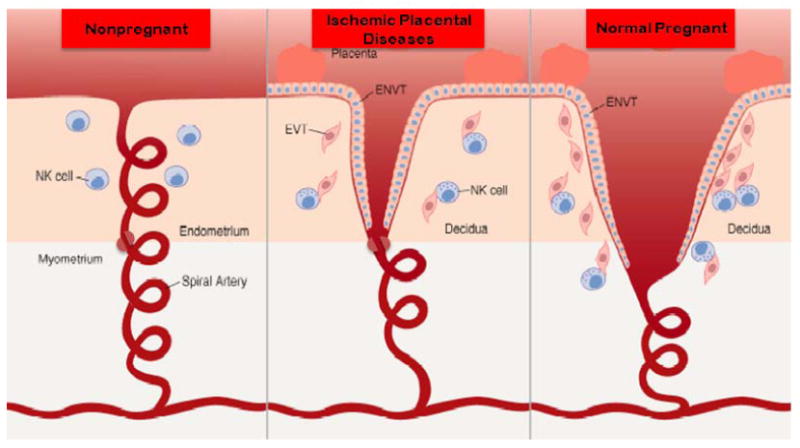
During pregnancy the spiral arteries that underlie the placenta, which in non-pregnant women are typical, small muscular arteries containing smooth muscle and an inner elastic lamina, undergo physiological remodeling. This includes terminal dilatation, the loss of the internal elastic lamina and the loss of smooth muscle. This change extends into the inner third of the myometrium resulting in the loss of a condensation of vascular smooth muscle near the myometrial decidual junction. This “sphincter” is proposed to be responsible for terminating blood flow at the time of menses. In preeclampsia this process is not complete. The terminal dilatation is not as extensive and the removal of smooth muscle is not complete and does not extend beyond the decidua leaving the functional vascular sphincter intact (Reprinted from Parham P. NK cells and trophoblasts: Partners in pregnancy. J Exp Med. 2004; 200(8): 951-5).51
Figure 2. Spiral arterial changes in normal pregnancy.
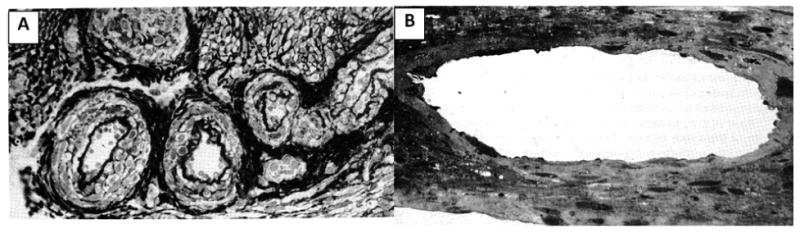
(A) Section of spiral arterioles at the junction of endometrium and myometrium in a nonpregnant individual. Note inner elastic lamina and smooth muscle. (B) Section of spiral arteriole in the same scale and from the same location during pregnancy. Note markedly increased diameter and absence of inner elastic lamina and smooth muscle (Reprinted from Sheppard B, Bonnar J. Uteroplacental arteries and hypertensive pregnancy. In: Bonnar J, MacGillivray I, Symonds G, editors. Pregnancy Hypertension. Baltimore: University Park; 1980. p. 205).52
Abnormal modification of the spiral arteries in settings of “placental ischemia”
In a subset of pregnant women these physiological modifications do not occur (Fig 1). The terminal portions of the vessels do not dilate normally, smooth muscle remains in the vessel wall and the modification of the vessels does not extend beyond the decidual layer resulting in the maintenance of the “functional sphincter”. These findings are very similar in pregnancies with preeclampsia, intrauterine growth restriction,8 recurrent pregnancy loss and in one third of cases of preterm birth9 and likely in a percentage of “normal pregnancies”.10
Functional consequences of remodeling and failed remodeling
For many years it was concluded that the major impact of the remodeling of the spiral arteries was to increase perfusion of the intervillus space. To a certain extent this is true since it is the remodeling which likely “unplugs” the spiral arteries occluded by cytotrophoblast.3 However, the concept that the striking increase in the terminal diameter of the spiral artery results in a dramatic increase of perfusion (Poiseuille's law, flow increases with the fourth power of the radius) is not appropriate, since the dilatation is limited to the terminal portion of the vessel. The major impact of the terminal dilatation as elegantly described by Graham Burton is to reduce the velocity of the blood delivered to the intervillus space.3 This velocity, which can be modeled to be 1-2 meters/second as a result of the striking increase of the uterine blood flow accompanying pregnancy, is reduced to 10 centimeters/second by the terminal dilatation. This reduced velocity is essential to protect the delicate placental villi, which in the hemochorial placenta are in direct contact with the spiral artery blood flow. Moreover, the reduced velocity of perfusion is necessary to allow time for extraction of oxygen. Thus, the potential consequence of the failed terminal dilatation is villus damage with an increased shedding of villus fragments into maternal blood and reduced extraction of oxygen from in the intervillus space. The villus damage is also proposed to lead to the release of procoagulants into the intervillus space with activation of the coagulation cascade, vascular occlusion and placental infarction. Atherosis (Fig 3) is another vascular abnormality compromising placental perfusion that is more common in the setting of placental ischemia.11 This phenomenon results in the occlusion of uterine vessels, including the spiral arteries and vessels proximal to them, with foam cells similar to those seen with atherosclerotic changes in systemic vessels.12 These cells also contain lipids similar to those found in atherosclerotic plaques.13 The atherotic change is especially interesting in view of the findings of the metabolic syndrome in nonpregnant women with a history of preeclampsia14 or who were delivered preterm.15 Burton3 also proposes that hormonally induced changes in uterine vessels proximal to the spiral arteries may be important to result in adequate placental perfusion and that these modifications are blunted in settings of “placental ischemia”.
Figure 3. Atherosis.
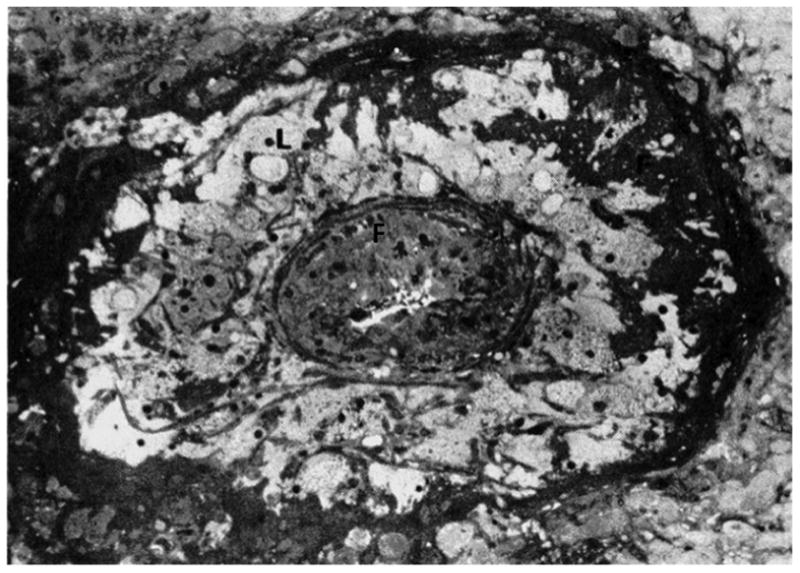
Numerous lipid-laden cells (L) and fibrin deposition (F) are present in the media of this occluded decidual vessel(Reprinted from Sheppard B, Bonnar J. Uteroplacental arteries and hypertensive pregnancy. In: Bonnar J, MacGillivray I, Symonds G, editors. Pregnancy Hypertension. Baltimore: University Park; 1980. p. 205).52
Based upon reduced extraction of oxygen with the failed vascular remodeling it is very possible that with an ischemic placenta the blood in the intervillus space may actually be hyperoxic. The abnormalities in response to failed remodeling also result in oxidative stress secondary to a hypoxic reperfusion scenario. The spiral arteries with residual vascular smooth muscle maintain the potential to respond to neural or humoral signals. In this regard the failure of vascular remodeling to extend beyond the decidua resulting in the maintenance of the functional sphincter present at the myometrial mucosal junction further facilitates oxidative stress.
The genesis of failed vascular remodeling
What are the controls of normal spiral artery remodeling and what goes awry in disorders associated with placental ischemia? These remain open questions in implantation research. There are important clues largely from the study of disorders with failed remodeling but there is no clear story and minimal consensus.
Clue number one is the association of abnormal remodeling with reduced trophoblast invasion. This has led to the conclusion that material produced by trophoblast may be necessary for the remodeling process to occur normally.6 However, even this conclusion is not universal. 3 Clue number two relates to the unique immunological interaction of the mother and her genetically dissimilar fetus. With hemochorial placentation the genetically dissimilar mother and fetus are in intimate vascular contact with trophoblast cells invading into the maternal spiral arteries.6 This interaction is also present in the decidua. Decidua contains abundant maternal immune cells in close contact with invasive fetal trophoblast. This would seem to be a set up for immunological conflict. An immunological interaction is supported by the well recognized protective effect of maternal exposure to paternal antigen prior to pregnancy. Thus, for example, first pregnancies without prior exposure to fetal (paternal) antigens as part of the normal pregnancy associated intermingling of maternal and fetal (paternal) tissues, pregnancies occurring after only a brief exposure to paternal antigen in semen, and the use of barrier contraception are associated with increased rates of preeclampsia.16
The immunological theory has concentrated largely on the relationship of immunological mediation of trophoblast invasion. Ashley Moffett17 has studied the relationship with fetal trophoblast of a unique form of natural killer cells (uNK) that constitute 70% of inflammatory cells in the decidua. She compared the expression for the most polymorphic of the major histocompatibility antigens on trophoblast, HLA-C, and receptors on uNK. The role of uNK in both implantation and vascular remodeling was suggested by studies demonstrating that if uNK are knocked out in genetically modified mice normal spiral artery remodeling does not occur.18 In studies of the relationship of HLA-C to uNK it appears that this interaction can result in either stimulatory or inhibitor effects on trophoblast function depending on which of a particular maternal uNK KIR receptor and HLA-C isotypes are present. The combination of a mother who is homozygous for KIR-A and a fetus with HLA-C2 is associated with an increased risk of preeclampsia, growth restriction and recurrent abortion, disorders associated with placental ischemia.17 Attempts to determine mechanisms by which this might occur demonstrate that when HLA-C2 interacts with KIR-B but not with KIR-A production of cytokines that stimulate trophoblast migration is increased.19
Other activators have also been suggested as relevant. It is proposed that the angiogenic factors PlGF and VEGF act to stimulate remodeling of the spiral arteries. Although this is interesting in light of the demonstrated relationship of low PlGF and high concentrations of the decoy receptor for VEGF and PlGF, s-Flt, in preeclampsia, the time sequence does not fit well. The higher concentration of s-Flt is not present until after the remodeling of the spiral arteries is proposed to take place,20 while the concentrations of PlGF are minimally different at this time.21 The cardiac protease, corin, which functions to activate atrial naturetic factor (ANP) is expressed in the pregnant mouse uterus and when knocked out to results in impaired invasion and spiral artery remodeling and a preeclampsia like syndrome.22 The same effect is present in the ANP knockout mouse, while in in vitro studies indicated increased matrigel invasion by human trophoblast in response to ANP. The concentration of corin is also higher in uterine tissue from pregnant than non-pregnant women while pro-ANP, which would be converted by corin to active ANP, is increased in uterine tissues from women with preeclampsia. This interesting data remains to be confirmed. How it may relate to immunological differences has not been explored.
Other suggested mediators of invasion of vascular remodeling and trophoblastic invasion based largely on associations and in vitro experiments include uric acid and several cytokines and cytokine antagonists.23-28 In general (but not universally)23 inflammatory cytokines are proposed to inhibit invasion. Although the subject of intense study the control of spiral artery remodeling is far from resolved with several pathways suggested which likely interact to mediate this vitally important process.
Pathophysiological consequences of “placental ischemia”
Local effects
Oxidative Stress
The alterations of the placental blood supply associated with placental ischemia lead to local and systemic pathophysiological changes. The changes in the spiral arteries and in uterine vessels proximate to these vessels reduce to some extent the delivery of nutrients and oxygen to the intervillus space. As described, this is not nearly as striking as has been posited in the past.3 It appears that the increased velocity of blood passing through the intervillus space is more pertinent to the reduced oxygen and nutrient delivery. Because of reduced time in the intervillus space there is reduced extraction of gasses and nutrients. Oxygen and nutrient delivery are further reduced by decreased placental exchange secondary to the occlusion of placental site vessels by atherotic changes and placental damage due to the increased velocity of blood.3 This leads to the interesting possibility that because of reduced extraction, the blood in the intervillus space is hyperoxic exposing the fetal surface of the placenta to increased oxygen concentration while at the same time reduced extraction renders the majority of the placenta hypoxic. It is proposed that this combination and the residual responsiveness of the incompletely remodeled spiral arteries results in the generation of oxidative stress.29
Oxidative stress occurs when the concentration of reactive oxygen species exceeds the buffering capacity of available antioxidants.30 It can obviously occur with excessive generation of reactive oxygen species or reduced availability of antioxidant activity either due to reduced concentrations of labile antioxidants or enzymes with antioxidant activity. All of these factors appear be present in placentas with placental ischemic syndromes. 29, 31 Free radicals associated with reactive oxygen species are extraordinarily toxic and damage protein, lipids and nucleic acids.30, 32 Once the delicate balance of pro and antioxidants is disturbed an almost explosive feed forward generation of reactive oxygen species ensues.
In settings of hypoxia reperfusion, such as is present with failed spiral artery remodeling, hypoxia leads to the inability to utilize ATP. Increased ATP breakdown and the ultimate degradation of these products by the xanthine oxidase/dehydrogenase (XOD/XO) system to form uric acid, generates the reactive oxygen product superoxide and H2O2 that can generate reactive oxygen species.33 In a setting of hypoxia, tissues, including trophoblast31 increase the amount and activity of XOD/XO further increasing the generation of reactive oxygen species.34 Further free radical generation occurs secondary to the activation of the NADPH oxidase system by either preexisting inflammatory mediators or those released in response to the tissue injury by free radicals.34 Interestingly, NADPH oxidase can also be activated by angiotensin,35 to which sensitivity is increased in preeclampsia36 and by angiotensin AT1 receptor activating autoantibodies that are increased in women with preeclampsia.37 NADPH oxidase generates superoxide, which in addition to its other activities acts to uncouple the enzyme responsible for nitric oxide (NO) generation, nitric oxide synthase (NOS) with the result that NOS also begins to generate free radicals.35 The resulting reduction in NO production reduces NO as does the consumption of NO by the neutralization of free radicals. To compound the problem, hypoxia down regulates one of the primary enzymes responsible for the degradation of reactive oxygen species, superoxide dismutase.38 Labile antioxidants are consumed by the excess free radicals and the situation is reinitiated with reperfusion but now in a setting of reduced antioxidant protection.
Endoplasmic Reticulum Stress
Endoplasmic reticulum stress (ER stress) is a pathophysiological phenomenon closely linked with oxidative stress.39 Reduced oxygen delivery can result in both oxidative and endoplasmic reticulum stress.32 Both can be induced by inflammatory mediators and both can produce these signaling molecules. ER stress is a cellular mechanism to reduce protein synthesis in settings in which nutrient and oxygen delivery are not sufficient to fully process proteins resulting a characteristic response, the “unfolded protein response” (UFR). As a homeostatic mechanism it is proposed that it occurs with a less severe insult than oxidative stress. Interestingly ER stress can also generate oxidative stress. ER stress can also be induced by a myriad of other signals including cytokines, elevated lipids, and high blood sugar. With UFR, protein synthesis stops, which Graham Burton has proposed is the genesis of small placentas with fetal growth restriction. With severe stress conditions, UFR can result in cellular apoptotic death. The release of inflammatory mediators, generation of reactive oxygen species, reduced protein synthesis and induction of apoptotic cell death have local as well as systemic impact.
Systemic Effects
Preeclampsia
Failed spiral artery remodeling with subsequent placental hypoxia, oxidative and endoplasmic reticulum stress may have systemic as well as local effects. The two-stage model of preeclampsia posits that the placental alterations associated with failed spiral artery remodeling result the in the release of placental products into the maternal circulation that induce the pathophysiological features of preeclampsia.14, 40 There are several candidate signals for the preeclampsia syndrome secondary to the pathophysiological consequences of failed remodeling. Oxidized lipids formed as a result of oxidative stress are incorporated into the membrane of syncitiotrophoblast. The associated apoptotic effect of endoplasmic reticulum stress with the necrotic effects of oxidative stress and hypoxia augment structural changes secondary to oxidation to facilitate shedding of syncitiotrophoblast microparticles (STBM) into the maternal circulation. There is some shedding of STBM in normal pregnancy that might serve an important role in information transfer.41 However, the particles released in response to failed spiral artery remodeling are quantitatively and likely qualitatively different.42 The number of particles are much higher in preeclampsia.43 Qualitatively the STBM from the placenta associated with failed spiral artery remodeling likely contain oxidized lipids that could transfer oxidative stress systemically. Further with preeclampsia an increased number of the particles that are released are necrotic rather than apoptotic and have toxic cellular effects not present with apoptotic particles.44, 45 Thus, excess number of STBM induce a more robust inflammatory response further augmented by the qualitative changes of the particles that may also have additional toxic properties.
Angiogenic and antiangiogenic factors produced by placenta are also influenced by hypoxia and oxidative stress.46 The antiangiogenic factor soluble VEGF receptor 1 (s-Flt) is increased with hypoxia.46 This decoy receptor sequesters the angiogenic factors, VEGF and PlGF preventing interaction with their receptors.47 It is also likely the PlGF production is also reduced since the circulating concentration of PlGF is lower in women destined to be preeclamptic prior to the increase in s-Flt.48
Why are there no systemic findings in other disorders with placental ischemia?
Despite what are thought to be similar placental and placental bed pathological findings in the disorders of placental ischemia, substantial maternal systemic manifestations are limited to preeclampsia.8 There are several suggested explanations but no definitive answer. First, the pathological findings may not be similar and the problem may be with poorly classified diseases.8 It may also be a quantitative rather than qualitative difference. For example, Graham Burton49 proposes that in IUGR the placental pathophysiology is not sufficient to generate oxidative stress, which does occur in preeclampsia resulting in systemic findings. Further, in many studies of IUGR measurement of relevant analytes indicate results intermediate between normal and preeclampsia.50 It is also possible that there are qualitative differences with different materials released from the stressed placenta. It is proposed that only in preeclampsia is the STBM from necrotic trophoblast.44, 45 In other disorders and in normal pregnancy these particles are from apoptotic trophoblast. We have favored the concept that the difference resides with the mother with only some women having the “maternal constitutional factors” that result in the response to placental products that leads to maternal pathophysiology.14 With these many possibilities it is apparent that the true answer is not established and perhaps may be a “little of each”.
Insights and future directions
The disorders associated with placental ischemia are the major problems of pregnancy. Understanding the detailed pathophysiological features of implantation and vascular remodeling would have a major impact to predict and prevent these problems. In my review it is evident that although there is extensive knowledge about these important topics, large and vital knowledge gaps need to be filled. The mechanisms of normal and abnormal trophoblast invasion and how they relate to spiral artery vascular remodeling are important topics for resolution. The control of spiral artery remodeling which is likely due to more than normal trophoblast invasion must be resolved. Despite the value of “lumping” the placental ischemic disorders we must determine what differentiates IUGR, preterm delivery, recurrent pregnancy loss and preeclampsia. Especially pertinent is the elucidation of the components responsible for maternal systemic pathophysiology being related largely to preeclampsia.
Approaches to the resolution require the use of state of the art techniques including microscopic analyses that utilize quantitative methodologies. Where possible examinations of markers and pathological changes should be done both before and during full disease and with rigid accurate diagnoses. Further, we should begin to look to “big science” to use collaborative studies to provide sufficient sample size and different populations to answer these questions accurately and with attention to potential diversity.
Acknowledgments
NIH PO1 HD30367 supported this work
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burton GJ, Jauniaux E, Charnock-Jones DS. The influence of the intrauterine environment on human placental development. Int J Dev Biol. 2010;54(2-3):303–12. doi: 10.1387/ijdb.082764gb. [DOI] [PubMed] [Google Scholar]
- 2.Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science. 1997;277:1669–72. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 3.Burton GJ, Woods AW, Jauniaux E, Kingdom JCP. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. 2009;30(6):473–82. doi: 10.1016/j.placenta.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pijnenborg R, Vercruysse L, Hanssens A. The uterine spiral arteries in human pregnancy: Facts and controversies. Placenta. 2006;27(9-10):939–58. doi: 10.1016/j.placenta.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Brosens I. A study of the spiral arteries of the decidua basalis in normotensive and hypertensive pregnancies. J Obstet Gynaecol Br Commonw. 1964;71:222–30. doi: 10.1111/j.1471-0528.1964.tb04270.x. [DOI] [PubMed] [Google Scholar]
- 6.Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27(9-10):939–58. doi: 10.1016/j.placenta.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Brosens JJ, Pijnenborg R, Brosens IA. The myometrial junctional zone spiral arteries in normal and abnormal pregnancies: a review of the literature. American Journal of Obstetrics & Gynecology. 2002;187(5):1416–23. doi: 10.1067/mob.2002.127305. [DOI] [PubMed] [Google Scholar]
- 8.Roberts J, Escudero C. The placenta in preeclampsia. Pregnacy Hypertension. 2012;1(2(2)):72–83. doi: 10.1016/j.preghy.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arias F, Rodriquez L, Rayne SC, Kraus FT. Maternal placental vasculopathy and infection: two distinct subgroups among patients with preterm labor and preterm ruptured membranes. Am J Obstet Gynecol. 1993;168(2):585–91. doi: 10.1016/0002-9378(93)90499-9. [DOI] [PubMed] [Google Scholar]
- 10.Aardema MW, Oosterhof H, Timmer A, van Rooy I, Aarnoudse JG. Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta. 2001;22(5):405–11. doi: 10.1053/plac.2001.0676. [DOI] [PubMed] [Google Scholar]
- 11.Khong TY, Pearce JM, Robertson WB. Acute atherosis in preeclampsia: Maternal determinants and fetal outcome in the presence of the lesion. Am J Obstet Gynecol. 1987;157:360–3. doi: 10.1016/s0002-9378(87)80172-2. [DOI] [PubMed] [Google Scholar]
- 12.Staff AC, Dechend R, Pijnenborg R. Learning From the Placenta Acute Atherosis and Vascular Remodeling in Preeclampsia-Novel Aspects for Atherosclerosis and Future Cardiovascular Health. Hypertension. 2010;56(6):1026–34. doi: 10.1161/HYPERTENSIONAHA.110.157743. [DOI] [PubMed] [Google Scholar]
- 13.Staff AC, Ranheim T, Khoury J, Henriksen T. Increased contents of phospholipids, cholesterol, and lipid peroxides in decidua basalis in women with preeclampsia. Am J Obstet Gynecol. 1999;180(3 Part 1):587–92. doi: 10.1016/s0002-9378(99)70259-0. [DOI] [PubMed] [Google Scholar]
- 14.Roberts JM, Hubel CA. The Two Stage Model of Preeclampsia: Variations on the Theme. Placenta. 2009;30:S32–S7. doi: 10.1016/j.placenta.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Catov JM, Bodnar LM, Kip KE, Hubel C, Ness RB, Harger G, et al. Early pregnancy lipid concentrations and spontaneous preterm birth. Am J Obstet Gynecol. 2007;197(6):610.e1–7. doi: 10.1016/j.ajog.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 16.Roberts J. Pregnancy related hypertension. In: Creasy R, Resnik R, Iams JD, editors. Maternal-Fetal Medicine: Principles and Practice. 6th. Philadelphia: Saunders Elsevier; 2009. pp. 650–88. [Google Scholar]
- 17.Colucci F, Boulenouar S, Kieckbusch J, Moffett A. How does variability of immune system genes affect placentation? Placenta. 2011;32(8):539–45. doi: 10.1016/j.placenta.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guimond MJ, Wang BP, Croy BA. Engraftment of Bone Marrow From Severe Combined Immunodeficient (Scid) Mice Reverses the Reproductive Deficits in Natural Killer Cell-Deficient Tg-Epsilon-26 Mice. J Exp Med. 1998;187(2):217–23. doi: 10.1084/jem.187.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiong S, Sharkey AM, Kennedy PR, Gardner L, Farrell LE, Chazara O, et al. Maternal uterine NK cell-activating receptor KIR2DS1 enhances placentation. J Clin Invest. 2013;123(10):4264–72. doi: 10.1172/JCI68991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powers RW, Roberts JM, Cooper KM, Gallaher MJ, Frank MP, Harger GF, et al. Maternal serum soluble fms-like tyrosine kinase 1 concentrations are not increased in early pregnancy and decrease more slowly postpartum in women who develop preeclampsia. Am J Obstet Gynecol. 2005;193(1):185–91. doi: 10.1016/j.ajog.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 21.Powers RW, Roberts JM, Plymire DA, Pucci D, Datwyler SA, Laird DM, et al. Low Placental Growth Factor Across Pregnancy Identifies a Subset of Women With Preterm Preeclampsia Type 1 Versus Type 2 Preeclampsia? Hypertension. 2012;60(1):239–46. doi: 10.1161/HYPERTENSIONAHA.112.191213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Y, Wang W, Dong N, Lou J, Srinivasan DK, Cheng W, et al. Role of corin in trophoblast invasion and uterine spiral artery remodelling in pregnancy. Nature. 2012;484(7393):246–50. doi: 10.1038/nature10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Fan DX, Duan J, Li MQ, Zhu XY, Jin LP. Thymic stromal lymphopoietin downregulates NME1 expression and promotes invasion in human trophoblasts via the activation of STAT3 signaling pathway. Clin Immunol. 2012;143(1):88–95. doi: 10.1016/j.clim.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 24.Bell MJ, Roberts JM, Founds SA, Jeyabalan A, Terhorst L, Conley YP. Variation in endoglin pathway genes is associated with preeclampsia: a case-control candidate gene association study. Bmc Pregnancy and Childbirth. 2013;13 doi: 10.1186/1471-2393-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mano Y, Kotani T, Shibata K, Matsumura H, Tsuda H, Sumigama S, et al. The Loss of Endoglin Promotes the Invasion of Extravillous Trophoblasts. Endocrinology. 2011;152(11):4386–94. doi: 10.1210/en.2011-1088. [DOI] [PubMed] [Google Scholar]
- 26.Champion H, Innes BA, Robson SC, Lash GE, Bulmer JN. Effects of interleukin-6 on extravillous trophoblast invasion in early human pregnancy. Mol Hum Reprod. 2012;18(8):391–400. doi: 10.1093/molehr/gas010. [DOI] [PubMed] [Google Scholar]
- 27.Anton L, Brown AG, Parry S, Elovitz MA. Lipopolysaccharide induces cytokine production and decreases extravillous trophoblast invasion through a mitogen-activated protein kinase-mediated pathway: possible mechanisms of first trimester placental dysfunction. Hum Reprod. 2012;27(1):61–72. doi: 10.1093/humrep/der362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitley GSJ, Cartwright JE. Cellular and Molecular Regulation of Spiral Artery Remodelling: Lessons from the Cardiovascular Field. Placenta. 2010;31(6):465–74. doi: 10.1016/j.placenta.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poston L, Raijmakers MT. Trophoblast oxidative stress, antioxidants and pregnancy outcome--a review. Placenta. 2004;25(Suppl A):S72–8. doi: 10.1016/j.placenta.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Schulz E, Gori T, Munzel T. Oxidative stress and endothelial dysfunction in hypertension. Hypertension Research - Clinical & Experimental. 2011;34(6):665–73. doi: 10.1038/hr.2011.39. [DOI] [PubMed] [Google Scholar]
- 31.Many A, Hubel CA, Fisher SJ, Roberts JM, Zhou Y. Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am J Pathol. 2000;156(1):321–31. doi: 10.1016/S0002-9440(10)64733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burton GJ, Jauniaux E. Oxidative stress. Best Practice & Research in Clinical Obstetrics & Gynaecology. 2011;25(3):287–99. doi: 10.1016/j.bpobgyn.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelley EE, Khoo NKH, Hundley NJ, Malik UZ, Freeman BA, Tarpey MM. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic Biol Med. 2010;48(4):493–8. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al Ghouleh I, Khoo NKH, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, et al. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med. 2011;51(7):1271–88. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–4. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 36.Gant NF, Daley GL, Chand S, Whalley PJ, MacDonald PC. A study of angiotensin II pressor response throughout primigravid pregnancy. J Clin Invest. 1973;52(11):2682–9. doi: 10.1172/JCI107462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103(7):945–52. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackson RM, Parish G, Ho YS. Effects of hypoxia on expression of superoxide dismutases in cultured ATII cells and lung fibroblasts. Am J Physiol. 1996;271(6 Pt 1):L955–62. doi: 10.1152/ajplung.1996.271.6.L955. [DOI] [PubMed] [Google Scholar]
- 39.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197(7):857–67. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Redman CWG. Current topic: pre-eclampsia and the placenta. Placenta. 1991;12:301–08. doi: 10.1016/0143-4004(91)90339-h. [DOI] [PubMed] [Google Scholar]
- 41.Redman CWG, Sargent IL. Microparticles and immunomodulation in pregnancy and preeclampsia. J Reprod Immunol. 2007;76(1-2):61–7. doi: 10.1016/j.jri.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 42.Gardiner C, Tannetta DS, Simms CA, Harrison P, Redman CWG, Sargent IL. Syncytiotrophoblast microvesicles released from pre-eclampsia placentae exhibit increased tissue factor activity. PLoS ONE. 2011;6(10):e26313. doi: 10.1371/journal.pone.0026313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goswami D, Tannetta DS, Magee LA, Fuchisawa A, Redman CWG, Sargent IL, et al. Excess syncytiotrophoblast microparticle shedding is a feature of early-onset pre-eclampsia, but not normotensive intrauterine growth restriction. Placenta. 2006;27(1):56–61. doi: 10.1016/j.placenta.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Burton GJ, Jones CJP. Syncytial Knots, Sprouts, Apoptosis, and Trophoblast Deportation from the Human Placenta. Taiwanese Journal of Obstetrics & Gynecology. 2009;48(1):28–37. doi: 10.1016/S1028-4559(09)60032-2. [DOI] [PubMed] [Google Scholar]
- 45.Huppertz B, Kingdom J, Caniggia I, Desoye G, Black S, Korr H, et al. Hypoxia favours necrotic versus apoptotic shedding of placental syncytiotrophoblast into the maternal circulation. Placenta. 2003;24(2-3):181–90. doi: 10.1053/plac.2002.0903. [DOI] [PubMed] [Google Scholar]
- 46.Mutter WP, Karumanchi SA. Molecular mechanisms of preeclampsia. Microvasc Res. 2008;75(1):1–8. doi: 10.1016/j.mvr.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andraweera PH, Dekker GA, Roberts CT. The vascular endothelial growth factor family in adverse pregnancy outcomes. Hum Reprod Update. 2012;18(4):436–57. doi: 10.1093/humupd/dms011. [DOI] [PubMed] [Google Scholar]
- 48.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350(7):672–83. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 49.Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS. Placental Endoplasmic Reticulum Stress and Oxidative Stress in the Pathophysiology of Unexplained Intrauterine Growth Restriction and Early Onset Preeclampsia. Placenta. 2009;30:S43–S8. doi: 10.1016/j.placenta.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roberts DJ, Post MD. The placenta in pre-eclampsia and intrauterine growth restriction. J Clin Pathol. 2008;61(12):1254–60. doi: 10.1136/jcp.2008.055236. [DOI] [PubMed] [Google Scholar]
- 51.Parham P. NK cells and trophoblasts: Partners in pregnancy. J Exp Med. 2004;200(8):951–5. doi: 10.1084/jem.20041783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheppard B, Bonnar J. Uteroplacental arteries and hypertensive pregnancy. In: Bonnar J, MacGillivray I, Symonds G, editors. Pregnancy Hypertension. Baltimore; University Park: 1980. p. 205. [Google Scholar]