

Since the discovery of cisplatin by Rosenberg et al. in the 1960s, platinum anticancer drugs have played an important role in cancer chemotherapy.[1] Cisplatin is used to treat a variety of cancers such as testicular, lung, breast, and ovarian. The success of cisplatin has led to the synthesis and biological evaluation of thousands of platinum complexes; however, only two additional complexes, carboplatin and oxaliplatin, are approved for clinical use by the FDA.[2] The clinical efficacy of platinum-based anticancer drugs is limited by their poor pharmacokinetic properties. High doses of platins are needed, which often leads to severe side effects. Furthermore, many tumors display inherent or acquired resistance to platinum-based therapies. There exists a need to develop alternative strategies to effectively deliver platinum drugs to the tumor, with fewer side effects.
Nanomaterials offer one possible solution to this problem. Nanoparticulate imaging and therapeutic agents have several advantages over traditional small-molecule agents; these advantages include high agent loading, tunable size, tailorable surface properties, controllable drug release kinetics, and improved pharmacokinetics.[3-5] Nanoparticles also tend to have increased accumulation in tumors as a result of the enhanced permeability and retention (EPR) effect that results from the leaky tumor neovasculatures. Additionally, nanoparticles can be specifically targeted to cancer cells by surface conjugation of an appropriate ligand to further enhance the accumulation of nanoparticles in tumors. The clinical success of nanoparticle-based therapeutics such as Doxil illustrates the potential of nanomaterials in anticancer drug delivery. Our research group has previously developed nanoscale coordination polymers (NCP) as a potential delivery vehicle for cisplatin prodrugs.[4b, 6] These materials demonstrated high agent loading and comparable cytotoxicity to cisplatin in vitro. However, these materials readily degrade under physiological conditions, thus limiting their in vivo efficacy. We sought to develop a new nanoparticle platform that would possess minimal drug release and nanoparticle decomposition when circulating in the bloodstream, but would also readily release the drugs upon cellular internalization. Herein we report the development of bridged polysilsesquioxane (PSQ) nanoparticles as a platform for oxaliplatin delivery.
PSQs are a type of hybrid material composed of siloxane networks with organic or metal–organic bridging ligands. These materials are synthesized from bis(trialkoxysilanes) ((R′O)3-Si-R-Si-(OR′)3) by sol–gel reactions.[7] PSQs have been well studied as bulk materials for a number of applications, but have only recently been prepared as nanomaterials.[8] PSQs offer similar biocompatibility to silica-based materials, which have been extensively studied for biomedical applications.[9] However, as a homopolymer of (R′O)3-Si-R-Si-(OR′)3, PSQ materials allow much higher drug loadings than silica-based materials with grafted drugs on only their surfaces. The physicochemical properties of PSQ materials can be more easily tuned by changing the monomer properties than in a silica-based material.
The platinum complex [Pt(dach)Cl2(triethoxysilylpropyl succinate)2] ([dachPtSi]; dach = R,R-diaminocyclohexane) was synthesized in 50–60% yield by reacting [Pt(dach)-Cl2(OH)2] with excess triethoxysilylpropyl succinic anhydride in DMSO. The [dachPtSi] monomer contains a PtIV complex, which is known to be inert under nonreducing conditions, but will be rapidly reduced by endogenous molecules to form the active PtII complex.[10] Additionally, [dachPtSi] contains orthogonal carboxylic acid functionalities, which can be used to postsynthetically modify the nanoparticle.
PSQ nanoparticles 1 were synthesized by base-catalyzed sol–gel polymerization in an anionic reverse microemulsion (Scheme 1a). Nanoparticles of 1 were precipitated by the addition of ethanol, followed by repeated washing with ethanol. The nanoparticles of 1 were characterized by transmission electron microscopy (TEM), dynamic light scattering (DLS), thermogravimetric analysis (TGA), and inductively coupled plasma mass spectrometry (ICP-MS). As shown in Figure 1a, TEM analysis of 1 showed spherical nanoparticles of 50–100 nm in diameter. DLS measurements of 1 gave an average diameter of 78 nm (polydispersity index, PDI = 0.074). The zeta potential of 1 was −20.7 mV in phosphatebuffered saline (PBS; Figure 1b, Table 1). TGA showed a 48% weight loss for the organic components (Figure 1c), which is slightly lower than the 52% expected for the homopolymer of [dachPtSi]. We believe that the lower weight loss can be attributed to the presence of sodium ions in 1 (from the surfactant used in the synthesis), which was confirmed by energy dispersive X-ray spectroscopic (EDS) measurements. ICP-MS measurements of 1 gave a Pt loading of 18–24% Pt by weight (expected 24% for the homopolymer), thus giving an active agent loading ([Pt(dach)Cl2]) of 35–47% by weight. Each particle of 1 contains approximately 1.5 × 105 [Pt(dach)Cl2] molecules (see the Supporting Information), which is several thousand times the drug loading of known nanoparticle platforms that deliver PtIV prodrugs.[11] Dye-doped particles of 1 (rhod-1) were obtained by incorporating a fraction of silyl-derived rhodamine B into the microemulsion during the synthesis. Particles of rhod-1 had a similar size distribution as 1 (Table 1) but contained rhodamine B to facilitate confocal imaging studies.
Scheme 1.

a) Generalized scheme showing the formation of PSQ particles 1 from the PtIV precursor. Upon cellular internalization and reaction with endogenous biomolecules, the PtIV complexes in 1 will be reduced, thereby releasing the active PtII agent. b) Surface functionalization of 1 by 1) the siloxane linkage formed between silanol groups and silyl-derived molecules, and 2) the amide linkage formed between carboxylic acid groups and amine-containing molecules.
Figure 1.
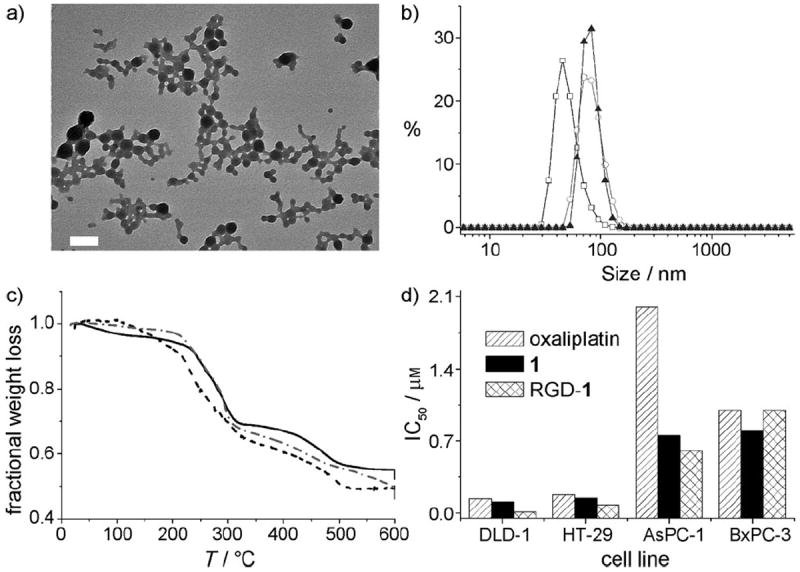
a) TEM micrograph of as-synthesized particles of 1. Scale bar = 200 nm. b) DLS curves for 1 (□), PEG-1 (○), and APEG-1 (▲). c) TGA curves for 1 (——), PEG-1 (---), and APEG-1 (-•-•). d) IC50 values of oxaliplatin, 1, and RGD-1 tested against a panel of four cancer cell lines.
Table 1.
Hydrodynamic diameters and zeta potential of various forms of 1 in PBS.
| Particle | Hydrodynamic diameter [nm] | PDI | Zeta potential [mV] | ||
|---|---|---|---|---|---|
| Intensity average[a] | Number average[b] | Z-average[c] | |||
| 1 | 84.5 ± 0.3 | 62.3 ± 0.2 | 77.8 ± 0.3 | 0.07 ± 0.01 | −20.7 ± 1.4 |
| rhod-1 | 113.9 ± 0.8 | 90.3 ± 3.0 | 105.2 ± 0.5 | 0.09 ± 0.19 | −21. 8 ±1.1 |
| RGD-1 | 102.9 ± 1.8 | 97.5 ± 7.7 | 100.0 ± 2.3 | 0.25 ± 0.05 | −18.2 ± 0.1 |
| PEG-1 | 95.2 ± 6.2 | 82.8 ± 2.9 | 91.3 ± 1.2 | 0.15 ± 0.03 | −5.5 ±1.3 |
| APEG-1 | 94.9 ± 9.8 | 81.6 ± 7.8 | 92.6 ± 0.4 | 0.16 ± 0.05 | −5.0 ± 1.4 |
Intensity average: Intensity-weighted average based on the intensity of light scattered by particles.
Number average: Number-weighted average as derived from the intensity distribution using the Mie theory.
Z-average: Intensity-weighted average obtained by cumulants analysis of DLS data.
We expect that 1 will be stable under normal physiological conditions, but the PtIV prodrug in 1 can be rapidly reduced by endogenous biomolecules, such as glutathione and cysteine, to release [(dach)PtCl2], which can then undergo hydrolysis (assisted by the low intracellular Cl− concentration) and DNA binding (Scheme 1a). Release experiments revealed that 1 is stable in the absence of a reducing agent, with only 10% background release over 24 h (Figure S14). However, with the addition of 10 mm cysteine, approximately 30% of the platin cargo was quickly released, followed by a more gradual release of the cargo. Approximately 80% of the payload was released after two days of incubation with cysteine.
Nanoparticle 1 possesses two surface functional groups—silanol groups and carboxylic acids—which were used for post-synthesis grafting of cyclic arginine–glycine–aspartate (cRGD) peptide and the anisamide ligand (Scheme 1b). Firstly, 1 was functionalized with silyl-derived cRGD by base-catalyzed condensation to afford RGD-1 (Scheme 1b, method 1). The cRGD peptide targets the αvβ3 integrin, which is upregulated in many angiogenic tumor cells.[12] DLS measurements showed an increase in particle size to 100 nm, probably because of mild particle aggregation after the peptide conjugation. The zeta potential increased slightly to −18.2 mV, which is consistent with the grafting of the neutral cRGD peptide.
Nanoparticle 1 was also surface-modified with polyethylene glycol (PEG). PEG is an inert, nontoxic, and nonimmunogenic hydrophilic polymer.[13] Numerous studies of PEG-modified nanoparticles have demonstrated that the PEG coating sterically stabilizes the nanoparticle and imparts stealth properties such as prolonged circulation half-life, reduced serum protein binding, and avoidance of the reticuloendothelial system (RES).[4a,b] PEG was attached to the surface of 1 by an EDC-mediated coupling (EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) between surface carboxylic acids and amino-PEG2000-methyl ether (PEG-1; Scheme 1b, method 2). PEG-1 was characterized by DLS, zeta potential, and TGA. DLS measurements gave a hydrodynamic diameter of 91 nm for PEG-1. The zeta potential of PEG-1 increased to −5.5 mV (Figure 1b, Table 1). This positive shift in the zeta potential to near neutral values is indicative of the formation of a PEG layer that shields the surface charge of the nanoparticles. A slight increase in the particle size was observed for PEG-1, and probably resulted from the extension of the PEG chains from the nanoparticle surface. TGA analysis of PEG-1 gave an increase in organic weight loss of 8%, which corresponds to a coverage of 1 PEG unit per 2.7 nm2 (Figure 1c).
The anisamide (AA) ligand was also attached to the surface of 1 by an amide coupling between the carboxylic acid groups of 1 and amino-PEG2000-AA. The AA ligand is an effective targeting agent for the sigma receptor, an opioid receptor overexpressed on many cancers.[4b, 14] AA-targeted PEG-1 (APEG-1) was prepared by the EDC-mediated coupling of 1 with amino-PEG2000-methyl ether and amino-PEG2000-AA (in a 9:1 wt ratio) under the same conditions as the above-mentioned pegylation reaction. APEG-1 particles exhibited a diameter of 93 nm (determined by DLS) and a zeta potential of −5.0 mV (Figure 1b and Table 1). TGA showed an increase in weight loss of 8% for the organic component, thus suggesting nearly identical PEG surface coverage in APEG-1 to that in PEG-1 (Figure 1c).
The cytotoxicity of 1 was evaluated in vitro against several human colon and pancreatic cancer lines, because oxaliplatin is used clinically to treat colon cancer and has been studied as a potential therapy for pancreatic cancer.[15] Oxaliplatin, 1, and RGD-1 were evaluated against two colon adenocarcinoma cell lines: DLD-1 and HT-29 (Figure 1d). Oxaliplatin was highly effective against the DLD-1 cell line, with an IC50 value of 0.14 μm, while 1 had slightly better efficacy, with an IC50 value of 0.11 μm. RGD-1 showed a tenfold increase in cytotoxicity compared to both 1 and oxaliplatin, and exhibited an extremely low IC50 value of 0.01 μm. Oxaliplatin and 1 showed comparable cytotoxicity (IC50 = 0.175 and 0.145 μm, respectively) against the HT-29 cell line, while RGD-1 was more than twice as cytotoxic (IC50 = 0.081 μm). These results clearly indicate the ability to target particles of 1 to the colon cancer cells with the cRGD ligand.
Two pancreatic ductal adenocarcinoma cell lines, AsPC-1 and BxPC-3, were chosen to evaluate the cytotoxicity of 1, RGD-1, and oxaliplatin (Figure 1d). Nanoparticles 1 and RGD-1 showed IC50 values of 0.75 and 0.6 μm, respectively, against the AsPC-1 cell line. These compounds were about three times as cytotoxic as oxaliplatin (IC50 = 2 μm). Oxaliplatin, 1, and RGD-1 showed comparable cytotoxicity against the BxPC-3 cell line, with IC50 values of 1, 0.8, and 1 μm, respectively. RGD-1 is not expected to show an increase in cytotoxicity compared to 1, as these cell lines do not express high levels of the αvβ3 integrin.[16] Instead, we hypothesized that AA can be used to target AsPC-1 cells, which were shown previously to be sigma-receptor positive.[17] PEG-1 and oxaliplatin showed IC50 values of 0.52 and 0.37 μm, respectively (Figure 2 a), while anisamide targeting reduced the IC50 value of APEG-1 to 0.17 μm, equivalent to a threefold increase in cytotoxicity.
Figure 2.

a) Cell viability curves of oxaliplatin (
 ; IC50 = (0.37 ± 0.02) μm), PEG-1 (▲; IC50 = (0.52 ± 0.08) μm), and APEG-1 (□; IC50 = (0.17 ± 0.01) μm) against AsPC-1 cells. b) Tumor growth inhibition curves for oxaliplatin (◇), PEG-1 (
; IC50 = (0.37 ± 0.02) μm), PEG-1 (▲; IC50 = (0.52 ± 0.08) μm), and APEG-1 (□; IC50 = (0.17 ± 0.01) μm) against AsPC-1 cells. b) Tumor growth inhibition curves for oxaliplatin (◇), PEG-1 (
 ), APEG-1 (○), and PBS (■) administered at 5 mg Pt kg−1 on days 0, 7, and 14 against an AsPC-1 subcutaneous mouse xenograft.
), APEG-1 (○), and PBS (■) administered at 5 mg Pt kg−1 on days 0, 7, and 14 against an AsPC-1 subcutaneous mouse xenograft.
The in vitro cytotoxicity assay results clearly indicated the increased uptake of 1 by DLD-1 and HT-29 human colon cancer cells through integrin receptor targeting and by AsPC-1 human pancreatic cancer cells through sigma receptor targeting. The cellular localization and effect of RGD targeting of 1 was investigated by confocal microscopy. DLD-1 cells showed increased cellular localization and uptake of RGD-rhod-1 particles compared to the untargeted rhod-1 particles (Figure S19). The time dependence of cellular internalization and apoptosis was also investigated by confocal microscopy (Figure 3). AsPC-1 cells were incubated with rhod-1, PEG-rhod-1, and APEG-rhod-1 for either 1 h or 5.5 h before treatment with fluorescein-tagged Annexin V to assess apoptosis. After 1 h of incubation, no fluorescence was evident in the rhodamine B channel for cells incubated with rhod-1, but significant rhodamine B fluorescence was found for the cells incubated with PEG-rhod-1 and APEG-rhod-1 (Figure 3). Consistent with the efficient uptake of PEG-rhod-1 and APEG-rhod-1 particles, the fluorescein channels showed strong green fluorescence in both cases, thus indicating significant cell apoptosis induced by the released Pt drug from the endocytosed particles. In contrast, no apoptosis was observed for the cells incubated with no particles or rhod-1 particles. Confocal images of the cells incubated for 5.5 h with rhod-1 particles indicated a low level of particle uptake and cell apoptosis (much less than the cells that were treated with PEG-rhod-1 and APEG-rhod-1; Figure S20). The confocal microscope imaging studies thus provided additional evidence for the enhanced uptake of PEG-rhod-1 and APEG-rhod-1 particles and triggered release of the platin cargoes in the AsPC-1 cells.
Figure 3.

Laser scanning confocal microscopy images of AsPC-1 cells after incubation with no particle, rhod-1, PEG-rhod-1, and APEG-rhod-1 for 1 h and then treated with Annexin V/FITC. The platin loadings are 5 μm for all the particles. The green fluorescence arises from FITC and the red fluorescence arises from rhodamine B. Scale bars = 50 μm.
Encouraged by the promising in vitro results, we have evaluated the in vivo tumor growth inhibition efficacy of PEG-1 and APEG-1 against a murine model of pancreatic cancer. Female athymic nude mice were injected in both flanks with 1 × 106 AsPC-1 cells and the tumors grew until palpable in both flanks (typically 16–21 days after cell injection). The mice were randomly split into four groups (4–6 mice per group) and received three weekly doses of oxaliplatin (5 mgPtkg−1), PEG-1 (5 mgPtkg−1), APEG-1 (5 mgPtkg−1), or PBS control. Figure 2b shows the effect of the treatments on tumor growth. Mice that received no treatment demonstrated sustained tumor growth, and reached the maximum allowed size 21 days after commencement of treatment. Oxaliplatin appeared to slightly inhibit tumor growth over the study period (75% of the volume of untreated tumors), but the effect was not statistically significant. However, mice that received either PEG-1 or APEG-1 showed a statistically significant reduction in tumor growth (α = 0.06 for PEG-1 and α = 0.02 for APEG-1), thus limiting the average tumor volume to approximately 50%and 40%of the volume for untreated control mice, respectively. The antitumor efficacy of PEG-1 and APEG-1 was confirmed by histological analysis of the resected tumors (Figure 4). Both the untreated and oxaliplatin-treated tumors were very large and composed of large regions of viable tumor cells (Figure 4a, b). The tumors from the oxaliplatin-treated mice showed small necrotic regions around the center of the tumor mass. In contrast, mice that received PSQ nanoparticle treatment (Figure 4c,d) had smaller tumors composed of large regions of necrotic (scarred) tissue. Mice that received APEG-1 showed much larger regions of necrotic tissue. These results suggest that PSQ nanoparticles are potentially useful therapies for the treatment of pancreatic cancer.
Figure 4.

Histology images of resected tumors (with H&E staining) from mice receiving a) saline control, b) oxaliplatin, c) PEG-1, and d) APEG-1. Scale bars = 0.5 mm. The blue-purple dots result from nuclear staining of viable cancer cells whereas pinkish areas indicate the lack of nuclear stains (i.e., necrotic tumor tissue with no viable cancer cells or nuclear material).
In conclusion, we have synthesized PSQ nanoparticles that contain extremely high loadings of oxaliplatin-based chemotherapeutics. These nanoparticles are highly stable under normal physiological conditions, but can be readily reduced to release the platin cargoes in highly reducing tumor microenvironments. The PSQ particles contained both surface silanol and carboxylic acid groups to allow further functionalization with PEG and targeting ligands. The PSQ particles exhibited superior cytotoxicity to oxaliplatin against four cancer cell lines in vitro, and the RGD and anisamide targeting further enhanced the cytotoxicity. In a pancreatic cancer xenograft mouse model, the pegylated and anisamide-targeted PSQ particles showed drastically superior efficacy to oxaliplatin in inhibiting tumor growth. This work highlights the potential of PSQ nanoparticles as excellent delivery vehicles for cancer therapeutics.
Supplementary Material
Acknowledgments
We would like to thank Kathryn deKrafft, Asif Khan, and Honorio Lara for experimental help and acknowledge technical support from the UNC Animal Studies Core.
Footnotes
We acknowledge financial support from NIH (U01-CA151455 and U54-119343).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201104510.
References
- 1.Rosenberg B, VanCamp L, Krigas T. Nature. 1965;205:698. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 2.a) Jung Y, Lippard SJ. Chem Rev. 2007;107:1387. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]; b) Kelland L. Nat Rev Cancer. 2007;7:573. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]; c) Wang D, Lippard SJ. Nat Rev Drug Discovery. 2005;4:307. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 3.a) Yavuz MS, Cheng Y, Chen J, Cobley CM, Zhang Q, Rycenga M, Xie J, Kim C, Song KH, Schwartz AG, Wang LV, Xia Y. Nat Mater. 2009;8:935. doi: 10.1038/nmat2564. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee JE, Lee N, Kim H, Kim J, Choi SH, Kim JH, Kim T, Song IC, Park SP, Moon WK, Hyeon T. J Am Chem Soc. 2010;132:552. doi: 10.1021/ja905793q. [DOI] [PubMed] [Google Scholar]; c) Sajja HK, East MP, Mao H, Wang YA, Nie S, Yang L. Curr Drug Discovery Technol. 2009;6:43. doi: 10.2174/157016309787581066. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cheon J, Lee JH. Acc Chem Res. 2008;41:1630. doi: 10.1021/ar800045c. [DOI] [PubMed] [Google Scholar]; e) Liu R, Zhang Y, Zhao X, Agarwal A, Mueller LJ, Feng P. J Am Chem Soc. 2010;132:1500. doi: 10.1021/ja907838s. [DOI] [PubMed] [Google Scholar]
- 4.a) Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Mol Pharm. 2008;8:505. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rieter WJ, Pott KM, Taylor KML, Lin W. J Am Chem Soc. 2008;130:11584. doi: 10.1021/ja803383k. [DOI] [PubMed] [Google Scholar]; c) Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 5.Li S, Huang L. Mol Pharm. 2008;5:496. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 6.Taylor-Pashow KML, Della Rocca J, Xie Z, Tran S, Lin W. J Am Chem Soc. 2009;131:14261. doi: 10.1021/ja906198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Hu LC, Shea KJ. Chem Soc Rev. 2011;40:688. doi: 10.1039/c0cs00219d. [DOI] [PubMed] [Google Scholar]; b) Shea KJ, Loy DA. Chem Mater. 2001;13:3306. [Google Scholar]
- 8.a) Hu LC, Khiterer M, Huang SJ, Chun Chung Chan J, Davey JR, Shea KJ. Chem Mater. 2010;22:5244. [Google Scholar]; b) Khiterer M, Shea KJ. Nano Lett. 2007;7:2684. doi: 10.1021/nl071087q. [DOI] [PubMed] [Google Scholar]; c) Zhao L, Loy DA, Shea KJ. J Am Chem Soc. 2006;128:14250. doi: 10.1021/ja066047n. [DOI] [PubMed] [Google Scholar]
- 9.a) Taylor-Pashow KML, Della Rocca J, Huxford RC, Lin WB. Chem Commun. 2010;46:5832. doi: 10.1039/c002073g. [DOI] [PubMed] [Google Scholar]; b) Kim T, Momin E, Choi J, Yuan K, Zaidi H, Kim J, Park M, Lee N, McMahon MT, Quinones-Hinojosa A, Bulte JW, Hyeon T, Gilad AA. J Am Chem Soc. 2011;133:2955. doi: 10.1021/ja1084095. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. Nat Mater. 2011;10:389. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ambrogio MW, Thomas CR, Zhao YL, Zink JI, Stoddart JF. Acc Chem Res. 2011 doi: 10.1021/ar200018x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall MD, Mellor HR, Callaghan R, Hambley TW. J Med Chem. 2007;50:3403. doi: 10.1021/jm070280u. [DOI] [PubMed] [Google Scholar]
- 11.a) Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ. J Am Chem Soc. 2007;129:8438. doi: 10.1021/ja073231f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. J Am Chem Soc. 2009;131:14652. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Proc Natl Acad Sci USA. 2008;105:17356. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Min Y, Mao C, Xu D, Liu Y. Chem Commun. 2010;46:8424. doi: 10.1039/c0cc03108a. [DOI] [PubMed] [Google Scholar]
- 12.a) Temming K, Schiffelers RM, Molema G, Kok RJ. Drug Resist Updates. 2005;8:381. doi: 10.1016/j.drup.2005.10.002. [DOI] [PubMed] [Google Scholar]; b) Schottelius M, Laufer B, Kessler H, Wester HJ. Acc Chem Res. 2009;42:969. doi: 10.1021/ar800243b. [DOI] [PubMed] [Google Scholar]
- 13.Zalipsky S. Bioconjugate Chem. 1995;6:150. doi: 10.1021/bc00032a002. [DOI] [PubMed] [Google Scholar]
- 14.a) Aydar E, Palmer CP, Djamgoz MBA. Cancer Res. 2004;64:5029. doi: 10.1158/0008-5472.CAN-03-2329. [DOI] [PubMed] [Google Scholar]; b) Banerjee R, Tyagi P, Li S, Huang L. Int J Cancer. 2004;112:693. doi: 10.1002/ijc.20452. [DOI] [PubMed] [Google Scholar]; c) John CS, Vilner BJ, Geyer BC, Moody T, Bowen WD. Cancer Res. 1999;59:4578. [PubMed] [Google Scholar]; d) Li S, Huang L. Mol Pharm. 2006;3:579. doi: 10.1021/mp060039w. [DOI] [PubMed] [Google Scholar]; e) Nakagawa O, Ming X, Huang L, Juliano RL. J Am Chem Soc. 2010;132:8848. doi: 10.1021/ja102635c. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Vilner BJ, John CS, Bowen WD. Cancer Res. 1995;55:408. [PubMed] [Google Scholar]
- 15.a) Capdevila J, Elez E, Peralta S, Macarulla T, Ramos FJ, Tabernero J. Expert Rev Anticancer Ther. 2008;8:1223. doi: 10.1586/14737140.8.8.1223. [DOI] [PubMed] [Google Scholar]; b) Sanchez SE, Trevino JG. Curr Med Chem. 2008;15:1674. doi: 10.2174/092986708784872348. [DOI] [PubMed] [Google Scholar]; c) Stathopoulos GP, Androulakis N, Souglakos J, Stathopoulos J, Georgoulias V. Anticancer Res. 2008;28:1303. [PubMed] [Google Scholar]
- 16.a) Löhr M, Trautmann B, Gottler M, Peters S, Zauner I, Maier A, Kloppel G, Liebe S, Kreuser ED. Pancreas. 1996;12:248. doi: 10.1097/00006676-199604000-00007. [DOI] [PubMed] [Google Scholar]; b) Mukhopadhyay S, Barnes CM, Haskel A, Short SM, Barnes KR, Lippard SJ. Bioconjugate Chem. 2008;19:39. doi: 10.1021/bc070031k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashiwagi H, McDunn JE, Simon PO, Jr, Goedegebuure PS, Xu J, Jones L, Chang K, Johnston F, Trinkaus K, Hotchkiss RS, Mach RH, Hawkins WG. Mol Cancer. 2007;6:48. doi: 10.1186/1476-4598-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.