

Abstract
Biomolecular systems have been modeled at a variety of scales, ranging from explicit treatment of electrons and nuclei to continuum description of bulk deformation or velocity. Many challenges of interfacing between scales have been overcome. Multiple models at different scales have been used to study the same system or calculate the same property (e.g., channel conductance). Accurate modeling of biochemical processes under in vivo conditions and the bridging of molecular and subcellular scales will likely soon become reality.
Introduction
The biological functions carried out by proteins and nucleic acids involve motions that occur on disparate spatial and temporal scales (Table 1). In enzyme-catalyzed reactions, bond breaking and formation proceed through the rearrangement of electrons and nuclei. The activities of the enzymes may be regulated by the binding of other proteins. The enzymes and regulators may all be components of higher complexes. These components and their transitory complexes constitute the crowded, heterogeneous macromolecular milieus in cellular compartments, which could in turn influence the behaviors of the constituents. In some cases protein molecules may directly bind to a 1-dimensional (e.g., genomic DNA or actin filament) or 2-dimensional (cell membrane in particular) surface. Here even stronger mutual influence of the protein molecules and the surface can be expected. It is apparent that a model based on a single type of physics and using a uniform spatial scale would not be capable of describing this multitude of biological processes and providing fundamental understanding. Consequently multiscale modeling of biomolecular systems has flourished in recent years.
Table 1.
Motions involved in a few representative biological processes
| Biological process | Motions involved |
|---|---|
| enzyme catalysis | rearrangement of electrons and nuclei in active site; conformational change of protein matrix; diffusion of substrate and product |
| replication, transcription, and translation | assembly and disassembly of multi-component machines; binding and unbinding of protein factors; complementary base-pairing; priming or initiation and polymer chain elongation; local and large-scale conformational transitions of components; nucleoside triphosphate hydrolysis; translocation along a nucleotide sequence |
| ion conductance | ion diffusion, translocation, binding and unbinding; rearrangement of pore-lining regions in response to an arriving or leaving permeant ion; stimulus-triggered sensor motion; propagation of motion from sensor to channel gate; reorganization of annular lipids upon channel gating |
| membrane remodeling | membrane attachment and insertion of membrane-shaping proteins; oligomerization of these proteins; bending, undulation, fission and fusion of surrounding membranes |
The importance of multiscale modeling is fittingly recognized by the award of this year’s Nobel Prize in Chemistry to Martin Karplus, Michael Levitt, and Arieh Warshel for “Development of Multiscale Models for Complex Chemical Systems.” These Nobel Laureates laid some of the foundations for ongoing research. In particular, the original concept and implementation of combined quantum mechanics/molecular mechanics (QM/MM) simulations [1,2] still serve as a guide in the study of enzyme activities [3,4] and as an inspiration for modeling at other scales. The idea of coarse-graining [5] is at the core of much current research.
Other foundational developments include the projection-operator formalism of Zwanzig [6] and Mori [7], the umbrella sampling method of Torrie and Valleau [8] for calculating the potential of mean force, and the particle insertion method of Widom [9] for calculating the excess chemical potential. Via the projection-operator formalism, one can project out the “irrelevant” degrees of freedom and focus on the motion of the “relevant” degrees of freedom. The umbrella sampling method provides a practical way to find the potential of mean force governing these relevant degrees of freedom. The particle insertion method, originally developed for simple fluids, has been extended to model the effects of the crowded macromolecular milieus on the thermodynamics and kinetics of “test” proteins [10,11].
Space will not allow for an exhaustive coverage of the recent progress in multiscale modeling and simulations of biomolecular systems. The following survey will focus on the strategies for interfacing different scales and some illustrative applications. The interested reader is referred to other recent reviews on related topics [4,12–25].
Modeling at different scales
The essence of multiscale modeling is captured by a quote attributed to Einstein: “Everything should be made as simple as possible, but not simpler.” If one wants to study bond breaking and formation, one must work with a quantum mechanical model that governs the rearrangement of the electrons and nuclei involved (Fig. 1a). On the other hand, when studying the conformational transitions of a protein molecule, it suffices to use Newton’s equation for the motion of the atoms (Fig. 1b). One may further coarse-grain the model, representing groups of atoms (e.g., amino-acid residues) by single beads, enabling simulations of more extensive conformational changes and over longer timescales [12,14–25] (Fig. 1c).
Fig. 1.

Models at different scales. (a) A quantum mechanical model. (b) A molecular mechanics all-atom model. (c) A coarse-grained model. (d) A rigid-body model for a concentrated protein mixture. (e) In a continuum model for a lipid bilayer, inward movement of two protein monomers (dashed circles) in the upper leaflet induces a velocity field (arrows) in the lower leaflet. The last panel is reproduced from Ref. [35] with permission from The Royal Society of Chemistry.
Effectively, by coarse-graining one freezes the internal motions within the groups modeled by the beads. In an extreme form of coarse-graining, internal motions of a whole protein domain, a whole protein molecule, or a whole protein complex are frozen. Then each such unit is treated as a rigid body and only the overall translation and rotation are modeled explicitly (Fig. 1d). With the rigid-body treatment, the rate constant for the site-specific binding of an enzyme to the whole ribosome has been calculated [26] and simulations of highly concentrated protein mixtures mimicking the bacterial cytoplasm have been carried out [27–29].
When a protein is bound to a DNA molecule or a lipid bilayer, the mutual influence can extend a long range. In these cases it can be fruitful to model the extended surfaces as 1-dimensional or 2-dimensional continuum. For example, DNA has been modeled as an elastic rod [30,31], and lipid bilayers have been modeled as an elastic membrane [32–34] or a structureless fluid sheet [35] (Fig. 1e). The system is no longer described by discrete particles, but by continuous “fields,” which can be the bulk deformation or velocity at an arbitrary position on the surface. In addition, flexible peptide linkers connecting protein domains have been modeled as a continuous polymer chain [36].
Schemes of interfacing between scales
In a multiscale model, one effectively is dealing with variables that evolve over (supposedly) different timescales. Essential to any multiscale modeling is the separate treatment of the fast evolving and slowly evolving variables, perhaps assuming different equations of motion. A general scheme for the separation of variables can be illustrated by a system specified by fast variables r and slow variables R. Let the state of the system be described by the time-dependent probability density p(r,R,t), governed by the following equation of evolution:
| (1) |
where
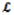 (r,R) is an operator serving to transform p(r,R,t) in the (r,R) space. In treating r, one makes use of the fact that R evolves slowly on the timescale of r and solves for the time-dependent probability density of r at a fixed R: p1(r,t|R), according to
(r,R) is an operator serving to transform p(r,R,t) in the (r,R) space. In treating r, one makes use of the fact that R evolves slowly on the timescale of r and solves for the time-dependent probability density of r at a fixed R: p1(r,t|R), according to
| (2) |
where
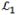 (r|R) is the part of
(r,R) containing only transformation in the r subspace.
(r|R) is the part of
(r,R) containing only transformation in the r subspace.
In treating R, one assumes that on its timescale the evolution of r is fast so that the latter always relaxes to the equilibrium distribution: p1eq(r|R). That is, one approximates the full probability density as
| (3) |
which can be formally derived via the projection-operator formalism [6,7]. The evolution in R is then governed by
| (4) |
where
| (5) |
Different flavors of this general scheme for the separation of variables will be found below.
Because the fast and slow variables are coupled, one must deal with the interfacing of the models at different scales. Interfacing strategies can be placed into two broad classes [12,14,20]. In one, known as sequential (or hierarchical or message-passing; Fig. 2), one first carries out simulations for the full model with explicit treatment for both the fast and slow variables. Information from these simulations is used to parameterize a reduced model for only the slow variables [see eq (5)]. The latter then becomes the subject of study. All-atom molecular mechanics models, coarse-grained models, and rigid-body models can all be viewed as reduced versions of fuller models (the full quantum mechanics model in the first case and all-atom molecular mechanics models in the second and third cases; Fig. 2a–c), although parameterization is often supplemented by experimental data and empirical choices [12,14–25,28,29]. In principle, the equation of motion for a coarse-grained model can be derived by the projection-operator formalism from the Newtonian dynamics of an all-atom model [37]. Implicit solvent models can similarly be viewed as reduced versions of explicit solvent models. The one case where simulations of a full model can provide all the information for parameterization is a reduced model for one or a few reaction coordinates (Fig. 2d). Here, from the simulations of the full model, one can calculate the potential of mean force for the reaction coordinate (e.g., an interatomic distance) via umbrella sampling [8] and parameters for its dynamics (e.g., the effective friction coefficient) by fitting time-correlation functions.
Fig. 2.

Information passing from high- to low-resolution models. (a) Calculations on a quantum mechanical model can help determine the energy function of a molecular mechanics all-atom model. (b) A similar passage from an all-atom model to a coarse-grained model. (c) All-atom simulations of a protein in explicit solvent yield diffusion constants for overall translation and rotation. (d) From all-atom simulations, the potential of mean force U(r) and effective friction coefficient γ for a reaction coordinate r can be obtained (adapted from Schaad et al. [91]).
The second broad class of interfacing strategies is known as hybrid (or concurrent or mixed-resolution), where different parts of a system are modeled at different scales (perhaps following different equations of motion). QM/MM models are classical examples [1,2] (Fig. 3a). More recent hybrid models include those combining an atomistic representation for a protein molecule (or an “active” region thereof) and a coarse-grained representation for the solvent (plus membrane) environment (or the rest of the protein molecule) [38–45] (Fig. 3b); those combining a rigid-body model for protein domains and a continuous-polymer or coarse-grained model for a loop or linker [36,46] (Fig. 3c); and those combining a rigid-body model for membrane proteins and a continuum model for the surrounding membrane [35] (Fig. 3d). When different regions of the same molecule are modeled at different resolutions, as in QM/MM simulations of enzyme catalysis [4], the boundary layer, consisting of covalently bonded atoms, requires great care to ensure proper coupling between the regions. Electrostatic interactions between regions modeled at different resolutions, atomistic and coarse-grained in particular [45], still pose significant challenges.
Fig. 3.

Hybrid multiscale models that mix (a) quantum mechanical and molecular mechanical; (b) all-atom (for protein) and coarse-grained (for lipid and water); (c) coarse-grained (for linker) and rigid-body (for protein domains); (d) rigid-body (for protein monomers) and continuum (for lipid bilayer) representations. The last panel is reproduced from Ref. [35] with permission from The Royal Society of Chemistry.
By coarse-graining one eliminates energy barriers associated with degrees of freedom internal to the groups of atoms represented by single beads, therefore the energy landscape is flattened and becomes easier to traverse. Raising the temperature has a similar effect, which forms the basis of temperature replica exchange [47], where simulations at high temperatures are used to facilitate conformational sampling at a desired low temperature, through on-the-fly swap of replicas simulated at a range of temperatures. Analogous consideration led to the development of resolution replica exchange [48–51], where simulations of coarse-grained models drive the simulation of an atomistic model (Fig. 4a). This method has so far been applied only to simple systems like short peptides and its potential remains to be exploited. The same premise is behind a serial combination of coarse-grained and atomistic simulations, where extensive coarse-grained simulations are used to produce seed conformations to initiate subsequent atomistic simulations. The serial combination has been used to study much larger systems including membrane proteins [52–54]. In effect, the coarse-grained simulations evolve slow variables [eq (4)] whereas the atomistic simulations evolve fast variables [eq (2)].
Fig. 4.

Computational gains from the use of separated simulations (a) at different resolutions, (b) in different regions, or (c) of different components (taken from Zhou and Qin [11]).
An interesting alternative to resolution replica exchange was recently developed [55]. In this “multiscale enhanced sampling” scheme, an energy term that couples the atomistic model and the coarse-grained model was introduced. The Hamiltonian replica exchange method [56] was then adopted, in which replicas were assigned various coupling strengths, with zero coupling resulting in the pure atomistic model. This scheme was applied to study the folding of a β-hairpin [55] and the disorder-to-order transition of a loop in a protein [57], and has been generalized to path sampling [58].
Instead of fixed partitioning into high- and low-resolution parts, sometimes switching between alternative partitions during the course of a simulation can result in a significant gain in computational speed without sacrificing accuracy. For example, when simulating the binding of a ligand to a protein, one can treat the whole protein as rigid when the ligand is far away but treat the loops around the binding site as flexible when the ligand is near (Fig. 4b upper panels). Interestingly, for calculating the binding rate constant one can even completely separate the simulations in the outer and inner regions, according to a method called BDflex [59]. Through simulations in which the ligand is confined to the outer region while the whole protein is treated as rigid and the boundary between the outer and inner regions as absorbing, one obtains the rate constant for absorption on the boundary (Fig. 4b lower left panel). Subsequently the rate constant for ligand binding is obtained from simulations in which the ligand is confined to the inner region (Fig. 4b lower right panel). This time the loops are treated as flexible and the boundary as partially absorbing, with the extent of absorption determined by the rate constant for absorption from the outer simulations.
The postprocessing approach for modeling the effects of macromolecular “crowders” on the thermodynamics and kinetics of a test protein [10,60] is another example of separating simulations at different scales. While such effects can in principle be calculated through simulations where the test protein and the crowders are mixed (Fig. 4c left panel), there are distinct advantages (in particular, enabling all-atom representations) when one first carries out separate simulations of the test protein and of the crowders and then postprocesses the simulations (Fig. 4c right panel). Postprocessing entails weighting each conformation in the protein simulation according to the Boltzmann factor of the excess chemical potential arising from the interactions of the test protein with the crowders. The latter quantity can be calculated according to Widom’s particle insertion method [9], but such a calculation is very costly [28]. A practical method has now been developed, by expressing the protein-crowder interactions as correlation functions and evaluating them via fast Fourier transform [61].
Illustrative applications
In many cases, multiple models are applied to study the same system at different scales, resulting in a more comprehensive understanding. One example is the M2 proton channel of Influenza A virus. This tetrameric protein, with 97 residues in each subunit, is essential for viral replication and is an established drug target. Quantum mechanical calculations were used to model the pH-dependent conformations of the His37-Trp41 tetrameric cluster [62], which embodies the pH sensor, proton selectivity filter, and primary gate. QM/MM molecular dynamics simulations were used to explore the local stability of alternative conformations of the His37-Trp41 cluster [63]. Through a number of all-atom molecular dynamics studies, the motion of the permeant proton along the channel pore was characterized [64]; the role of Val27 as a secondary gate was proposed [65]; helix bending around Gly34 was revealed and suggested to be coupled to channel gating [66,67]; and inhibitors that target drug-resistant M2 mutants were designed [68]. The rate of proton transport was calculated by modeling the gated binding to and unbinding from the His34 tetrad as diffusion-limited reactions, leading to quantitative rationalization of experimentally observed current-voltage and current-pH relations as well as solvent isotope effect [69,70].
In addition to the binding/unbinding reaction based approach [69], ion conductance across transmembrane channels has been calculated from models at a variety of scales [71]. The most detailed are all-atom molecular dynamics simulations, from which channel conductance can be calculated by counting the number of complete ion-crossing events [72–74]. One step down the resolution hierarchy are models that treat the conducting ions explicitly by Brownian dynamics simulations, but the channel protein, membrane, and solvent as static dielectric continuum [75,76]. A further approximation is to replace the discrete ions of each species by a continuous charge density and treat ion-ion interactions in a mean-field way; the resulting Poisson-Nernst-Planck model continues to find use [77–79]. Lastly one can model ion permeation as the diffusion of one or a few ions in a 1- or 3-dimensional potential of mean force [80–83]; this potential of mean force can be obtained from all-atom molecular dynamics simulations.
Concluding remarks
Clearly, models at different scales can all contribute to the fundamental understanding of complex biomolecular systems. Different spatial scales may evolve over different timescales according to different equations of motion. From a technical standpoint, artificial coupling to a low-resolution model can facilitate conformational sampling of a high-resolution model [55,57,58]. In other cases, separation of the simulations in different regions [59] or of different components [61] can be designed for efficient calculation of biophysical properties. While the partitioning into high- and low-resolution parts is fixed in most current studies, switchable or adaptive partitioning is being developed [84–86]. Iterative information exchange between high and low-resolution models has also proven useful [24]. All these activities produce an equalization of the resolution hierarchy.
This year’s Nobel Prize in Chemistry recognizes what is already achieved through multiscale modeling, and much more can be expected to come. By focusing high-resolution modeling on key components of complex systems, such as “test” proteins in crowded cellular milieus, results with increasing accuracy will be attainable, including those for biochemical processes under in vivo conditions. On the other hand, with further coarse-graining, it will be realistic to bridge the molecular and subcellular scales [87–90].
Highlights.
Many challenges of interfacing between scales have been overcome.
Coupling between scales can be introduced or restricted, both to gain computational efficiency.
Multiple models at different scales allow comprehensive understanding of a single system.
Accurate modeling of in vivo conditions is becoming realistic.
Bridging molecular and subcellular scales will be possible via coarse-graining.
Acknowledgments
This work was supported in part by Grants GM58187 and GM88187 from the National Institutes of Health. I thank Xiaodong Pang for help with preparing the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warshel A, Levitt M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 2.Field MJ, Bash PA, Karplus M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J Comput Chem. 1990;11:700–733. [Google Scholar]
- 3.Gao JL. Hybrid quantum and molecular mechanical simulations: An alternative avenue to solvent effects in organic chemistry. Acc Chem Res. 1996;29:298–305. [Google Scholar]
- 4.Senn HM, Thiel W. QM/MM methods for biomolecular systems. Angew Chem Int Ed Engl. 2009;48:1198–1229. doi: 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- 5.Levitt M, Warshel A. Computer simulation of protein folding. Nature. 1975;253:694–698. doi: 10.1038/253694a0. [DOI] [PubMed] [Google Scholar]
- 6.Zwanzig R. Memory effects in irreversible thermodynamics. Phys Rev. 1961;124:983–992. [Google Scholar]
- 7.Mori H. Transport, collective motion, and Brownian motion. Prog Theor Phys. 1965;33:423–455. [Google Scholar]
- 8.Torrie GM, Valleau JP. Non-physical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J Comput Phys. 1977;23:187–199. [Google Scholar]
- 9.Widom B. Some topics in theory of fluids. J Chem Phys. 1963;39:2808–2812. [Google Scholar]
- 10.Qin S, Zhou HX. Atomistic modeling of macromolecular crowding predicts modest increases in protein folding and binding stability. Biophys J. 2009;97:12–19. doi: 10.1016/j.bpj.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou HX, Qin S. Simulation and modeling of crowding effects on the thermodynamic and kinetic properties of proteins with atomic details. Biophys Rev. 2013;5:207–215. doi: 10.1007/s12551-013-0101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayton GS, Noid WG, Voth GA. Multiscale modeling of biomolecular systems: In serial and in parallel. Curr Opin Struct Biol. 2007;17:192–198. doi: 10.1016/j.sbi.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Hu H, Yang W. Free energies of chemical reactions in solution and in enzymes with ab initio quantum mechanics/molecular mechanics methods. Annu Rev Phys Chem. 2008;59:573–601. doi: 10.1146/annurev.physchem.59.032607.093618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherwood P, Brooks BR, Sansom MS. Multiscale methods for macromolecular simulations. Curr Opin Struct Biol. 2008;18:630–640. doi: 10.1016/j.sbi.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayton GS, Voth GA. Systematic multiscale simulation of membrane protein systems. Curr Opin Struct Biol. 2009;19:138–144. doi: 10.1016/j.sbi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tozzini V. Multiscale modeling of proteins. Acc Chem Res. 2010;43:220–230. doi: 10.1021/ar9001476. [DOI] [PubMed] [Google Scholar]
- 17.Kamerlin SC, Vicatos S, Dryga A, Warshel A. Coarse-grained (multiscale) simulations in studies of biophysical and chemical systems. Annu Rev Phys Chem. 2011;62:41–64. doi: 10.1146/annurev-physchem-032210-103335. [DOI] [PubMed] [Google Scholar]
- 18.de Pablo JJ. Coarse-grained simulations of macromolecules: From DNA to nanocomposites. Annu Rev Phys Chem. 2011;62:555–574. doi: 10.1146/annurev-physchem-032210-103458. [DOI] [PubMed] [Google Scholar]
- 19.Takada S. Coarse-grained molecular simulations of large biomolecules. Curr Opin Struct Biol. 2012;22:130–137. doi: 10.1016/j.sbi.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Saunders MG, Voth GA. Coarse-graining of multiprotein assemblies. Curr Opin Struct Biol. 2012;22:144–150. doi: 10.1016/j.sbi.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Riniker S, Allison JR, van Gunsteren WF. On developing coarse-grained models for biomolecular simulation: a review. Phys Chem Chem Phys. 2012;14:12423–12430. doi: 10.1039/c2cp40934h. [DOI] [PubMed] [Google Scholar]
- 22.Noid WG. Perspective: Coarse-grained models for biomolecular systems. J Chem Phys. 2013;139:090901. doi: 10.1063/1.4818908. [DOI] [PubMed] [Google Scholar]
- 23.Cui Q, Zhang L, Wu Z, Yethiraj A. Generation and sensing of membrane curvature: Where materials science and biophysics meet. Curr Opin Solid State Mater Sci. 2013;17:164–174. [Google Scholar]
- 24.Saunders MG, Voth GA. Coarse-graining methods for computational biology. Annu Rev Biophys. 2013;42:73–93. doi: 10.1146/annurev-biophys-083012-130348. [DOI] [PubMed] [Google Scholar]
- 25.Meier K, Choutko A, Dolenc J, Eichenberger AP, Riniker S, van Gunsteren WF. Multi-resolution simulation of biomolecular systems: A review of methodological issues. Angew Chem Int Ed Engl. 2013;52:2820–2834. doi: 10.1002/anie.201205408. [DOI] [PubMed] [Google Scholar]
- 26.Qin S, Zhou HX. Dissection of the high rate constant for the binding of a ribotoxin to the ribosome. Proc Natl Acad Sci U S A. 2009;106:6974–6979. doi: 10.1073/pnas.0900291106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ando T, Skolnick J. Crowding and hydrodynamic interactions likely dominate in vivo macromolecular motion. Proc Natl Acad Sci U S A. 2010;107:18457–18462. doi: 10.1073/pnas.1011354107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.McGuffee SR, Elcock AH. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput Biol. 2010;6:e1000694. doi: 10.1371/journal.pcbi.1000694. an atomically detailed model of the bacterial cytoplasm, generated from rigid-body Brownian dynamics simulations of the 50 most abundant macromolecules. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mereghetti P, Gabdoulline RR, Wade RC. Brownian dynamics simulation of protein solutions: Structural and dynamical properties. Biophys J. 2010;99:3782–3791. doi: 10.1016/j.bpj.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villa E, Balaeff A, Schulten K. Structural dynamics of the lac repressor-DNA complex revealed by a multiscale simulation. Proc Natl Acad Sci U S A. 2005;102:6783–6788. doi: 10.1073/pnas.0409387102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31•.Lillian TD, Taranova M, Wereszczynski J, Andricioaei I, Perkins NC. A multiscale dynamic model of DNA supercoil relaxation by topoisomerase IB. Biophys J. 2011;100:2016–2023. doi: 10.1016/j.bpj.2011.03.003. an elastic rod model for supercoiled DNA, with boundary conditions set by the potential of mean force calculated from all-atom molecular dynamics simulations of a topoisomerase-DNA complex, to study supercoil relaxation by the topoisomerase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayton GS, Blood PD, Voth GA. Membrane remodeling from N-BAR domain interactions: Insights from multi-scale simulation. Biophys J. 2007;92:3595–3602. doi: 10.1529/biophysj.106.101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arkhipov A, Yin Y, Schulten K. Four-scale description of membrane sculpting by BAR domains. Biophys J. 2008;95:2806–2821. doi: 10.1529/biophysj.108.132563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watson MC, Penev ES, Welch PM, Brown FL. Thermal fluctuations in shape, thickness, and molecular orientation in lipid bilayers. J Chem Phys. 2011;135:244701. doi: 10.1063/1.3660673. [DOI] [PubMed] [Google Scholar]
- 35•.Camley BA, Brown FLH. Diffusion of complex objects embedded in free and supported lipid bilayer membranes: Role of shape anisotropy and leaflet structure. Soft Matter. 2013;9:4767–4779. diffusion constants of membrane-bound proteins are calculated by modeling them as rigid objects coupled to the surrounding membrane and solvent fluids via no-slip boundary conditions. [Google Scholar]
- 36.Zhou HX. The affinity-enhancing roles of flexible linkers in two-domain DNA-binding proteins. Biochemistry. 2001;40:15069–15073. doi: 10.1021/bi015795g. [DOI] [PubMed] [Google Scholar]
- 37.Izvekov S. Microscopic derivation of particle-based coarse-grained dynamics. J Chem Phys. 2013;138:134106. doi: 10.1063/1.4795091. [DOI] [PubMed] [Google Scholar]
- 38.Neri M, Anselmi C, Cascella M, Maritan A, Carloni P. Coarse-grained model of proteins incorporating atomistic detail of the active site. Phys Rev Lett. 2005;95:218102. doi: 10.1103/PhysRevLett.95.218102. [DOI] [PubMed] [Google Scholar]
- 39.Shi Q, Izvekov S, Voth GA. Mixed atomistic and coarse-grained molecular dynamics: Simulation of a membrane-bound ion channel. J Phys Chem B. 2006;110:15045–15048. doi: 10.1021/jp062700h. [DOI] [PubMed] [Google Scholar]
- 40.Gohlke H, Thorpey MF. A natural coarse graining for simulating large biomolecular motion. Biophys J. 2006;91:2115–2120. doi: 10.1529/biophysj.106.083568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orsi M, Noro MG, Essex JW. Dual-resolution molecular dynamics simulation of antimicrobials in biomembranes. J R Soc Interface. 2011;8:826–841. doi: 10.1098/rsif.2010.0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han W, Schulten K. Further optimization of a hybrid united-atom and coarse-grained force field for folding simulations: Improved backbone hydration and interactions between charged side chains. J Chem Theory Comput. 2012;8:4413–4424. doi: 10.1021/ct300696c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Predeus AV, Gul S, Gopal SM, Feig M. Conformational sampling of peptides in the presence of protein crowders from AA/CG-multiscale simulations. J Phys Chem B. 2012;116:8610–8620. doi: 10.1021/jp300129u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riniker S, Eichenberger AP, van Gunsteren WF. Structural effects of an atomic-level layer of water molecules around proteins solvated in supra-molecular coarse-grained water. J Phys Chem B. 2012;116:8873–8879. doi: 10.1021/jp304188z. [DOI] [PubMed] [Google Scholar]
- 45.Wassenaar TA, Ingolfsson HI, Priess M, Marrink SJ, Schafer LV. Mixing MARTINI: Electrostatic coupling in hybrid atomistic-coarse-grained biomolecular simulations. J Phys Chem B. 2013;117:3516–3530. doi: 10.1021/jp311533p. [DOI] [PubMed] [Google Scholar]
- 46•.Wade RC, Luty BA, Demchuk E, Madura JD, Davis ME, Briggs JM, McCammon JA. Simulation of enzyme-substrate encounter with gated active sites. Nat Struct Biol. 1994;1:65–69. doi: 10.1038/nsb0194-65. pioneering study of protein-ligand binding kinetics, mixing coarse-grained model for loops around the binding site and rigid-body model for the rest of the protein. [DOI] [PubMed] [Google Scholar]
- 47.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 48.Lwin TZ, Luo R. Overcoming entropic barrier with coupled sampling at dual resolutions. J Chem Phys. 2005;123:194904. doi: 10.1063/1.2102871. [DOI] [PubMed] [Google Scholar]
- 49.Lyman E, Ytreberg FM, Zuckerman DM. Resolution exchange simulation. Phys Rev Lett. 2006;96:028105. doi: 10.1103/PhysRevLett.96.028105. [DOI] [PubMed] [Google Scholar]
- 50.Christen M, van Gunsteren WF. Multigraining: An algorithm for simultaneous fine-grained and coarse-grained simulation of molecular systems. J Chem Phys. 2006;124:154106. doi: 10.1063/1.2187488. [DOI] [PubMed] [Google Scholar]
- 51.Liu P, Shi Q, Lyman E, Voth GA. Reconstructing atomistic detail for coarse-grained models with resolution exchange. J Chem Phys. 2008;129:114103. doi: 10.1063/1.2976663. [DOI] [PubMed] [Google Scholar]
- 52.Thogersen L, Schiott B, Vosegaard T, Nielsen NC, Tajkhorshid E. Peptide aggregation and pore formation in a lipid bilayer: A combined coarse-grained and all atom molecular dynamics study. Biophys J. 2008;95:4337–4347. doi: 10.1529/biophysj.108.133330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stansfeld PJ, Hopkinson R, Ashcroft FM, Sansom MS. PIP2-binding site in Kir channels: Definition by multiscale biomolecular simulations. Biochemistry. 2009;48:10926–10933. doi: 10.1021/bi9013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bucher D, Hsu YH, Mouchlis VD, Dennis EA, McCammon JA. Insertion of the Ca2+-independent phospholipase A2 into a phospholipid bilayer via coarse-grained and atomistic molecular dynamics simulations. PLoS Comput Biol. 2013;9:e1003156. doi: 10.1371/journal.pcbi.1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moritsugu K, Terada T, Kidera A. Scalable free energy calculation of proteins via multiscale essential sampling. J Chem Phys. 2010;133:224105. doi: 10.1063/1.3510519. [DOI] [PubMed] [Google Scholar]
- 56.Fukunishi H, Watanabe O, Takada S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: Application to protein structure prediction. J Chem Phys. 2002;116:9058–9067. [Google Scholar]
- 57••.Moritsugu K, Terada T, Kidera A. Disorder-to-order transition of an intrinsically disordered region of sortase revealed by multiscale enhanced sampling. J Am Chem Soc. 2012;134:7094–7101. doi: 10.1021/ja3008402. coupling to low-resolution model is introduced to facilitate conformational sampling of high-resolution model. [DOI] [PubMed] [Google Scholar]
- 58.Fujisaki H, Shiga M, Moritsugu K, Kidera A. Multiscale enhanced path sampling based on the Onsager-Machlup action: Application to a model polymer. J Chem Phys. 2013;139:054117. doi: 10.1063/1.4817209. [DOI] [PubMed] [Google Scholar]
- 59•.Greives N, Zhou HX. BDflex: A method for efficient treatment of molecular flexibility in calculating protein-ligand binding rate constants from Brownian dynamics simulations. J Chem Phys. 2012;137:135105. doi: 10.1063/1.4756913. separate simulations of protein-ligand binding in outer and inner regions are designed, to ensure adequate treatment of loop flexibility when the ligand is near. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qin S, Minh DD, McCammon JA, Zhou HX. Method to predict crowding effects by postprocessing molecular dynamics trajectories: Application to the flap dynamics of HIV-1 protease. J Phys Chem Lett. 2010;1:107–110. doi: 10.1021/jz900023w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61••.Qin S, Zhou HX. FFT-based method for modeling protein folding and binding under crowding: Benchmarking on ellipsoidal and all-atom crowders. J Chem Theory Comput. 2013;9:4633–4643. doi: 10.1021/ct4005195. protein-crowder interaction energies are expressed as correlation functions and evaluated via FFT, enabling accurate modeling of crowding effects under in vivo conditions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong H, Yi M, Cross TA, Zhou HX. Calculations and validation of the pH-dependent structures of the His37-Trp41 quartet, the heart of acid activation and proton conductance in the M2 protein of Influenza A virus. Chem Sci. 2013;4:2776–2787. doi: 10.1039/C3SC50293G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63•.Dong H, Fiorin G, Degrado WF, Klein ML. Exploring histidine conformations in the M2 channel lumen of the Influenza A virus at neutral pH via molecular simulations. J Phys Chem Lett. 2013;4:3067–3071. doi: 10.1021/jz401672h. conformational space of M2 protein is explored by both QM/MM and all-atom molecular dynamics simulations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen H, Wu Y, Voth GA. Proton transport behavior through the influenza A M2 channel: Insights from molecular simulation. Biophys J. 2007;93:3470–3479. doi: 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yi M, Cross TA, Zhou HX. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J Phys Chem B. 2008;112:7977–7979. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yi M, Cross TA, Zhou HX. Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proc Natl Acad Sci U S A. 2009;106:13311–13316. doi: 10.1073/pnas.0906553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khurana E, Dal Peraro M, DeVane R, Vemparala S, DeGrado WF, Klein ML. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc Natl Acad Sci U S A. 2009;106:1069–1074. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Klein ML, et al. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J Am Chem Soc. 2011;133:12834–12841. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69•.Zhou HX. A theory for the proton transport of the influenza virus M2 protein: Extensive test against conductance data. Biophys J. 2011;100:912–921. doi: 10.1016/j.bpj.2011.01.002. a theory for rate of proton transport, based on functional mechanism suggested by quantum mechanical calculations and with parameters supplied by all-atom molecular dynamics simulations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou HX. Mechanistic insight into the H2O/D2O isotope effect in the proton transport of the influenza virus M2 protein. J Membr Biol. 2011;244:93–96. doi: 10.1007/s00232-011-9402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roux B, Allen T, Berneche S, Im W. Theoretical and computational models of biological ion channels. Q Rev Biophys. 2004;37:15–103. doi: 10.1017/s0033583504003968. [DOI] [PubMed] [Google Scholar]
- 72.Chakrabarti N, Ing C, Payandeh J, Zheng N, Catterall WA, Pomes R. Catalysis of Na+ permeation in the bacterial sodium channel NaVAb. Proc Natl Acad Sci U S A. 2013;110:11331–11336. doi: 10.1073/pnas.1309452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu J, Modi N, Kleinekathoefer U. Simulation of ion transport through an N-acetylneuraminic acid-inducible membrane channel: From understanding to engineering. J Phys Chem B. 2013;117:15966–15975. doi: 10.1021/jp408495v. [DOI] [PubMed] [Google Scholar]
- 74.Ulmschneider MB, Bagneris C, McCusker EC, Decaen PG, Delling M, Clapham DE, Ulmschneider JP, Wallace BA. Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc Natl Acad Sci U S A. 2013;110:6364–6369. doi: 10.1073/pnas.1214667110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee KI, Jo S, Rui H, Egwolf B, Roux B, Pastor RW, Im W. Web interface for Brownian dynamics simulation of ion transport and its applications to beta-barrel pores. J Comput Chem. 2012;33:331–339. doi: 10.1002/jcc.21952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen R, Chung SH. Molecular dynamics simulations of scorpion toxin recognition by the Ca2+-activated potassium channel KCa3. 1. Biophys J. 2013;105:1829–1837. doi: 10.1016/j.bpj.2013.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choudhary OP, Ujwal R, Kowallis W, Coalson R, Abramson J, Grabe M. The electrostatics of VDAC: Implications for selectivity and gating. J Mol Biol. 2010;396:580–592. doi: 10.1016/j.jmb.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng Q, Chen D, Wei GW. Second-order Poisson Nernst-Planck solver for ion channel transport. J Comput Phys. 2011;230:5239–5262. doi: 10.1016/j.jcp.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dyrka W, Bartuzel MM, Kotulska M. Optimization of 3D Poisson-Nernst-Planck model for fast evaluation of diverse protein channels. Proteins. 2013;81:1802–1822. doi: 10.1002/prot.24326. [DOI] [PubMed] [Google Scholar]
- 80.Berneche S, Roux B. A microscopic view of ion conduction through the K+ channel. Proc Natl Acad Sci U S A. 2003;100:8644–8648. doi: 10.1073/pnas.1431750100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou HX. Equivalence of two approaches for modeling ion permeation through a transmembrane channel with an internal binding site. J Chem Phys. 2011;134:135101. doi: 10.1063/1.3575585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu F, Hummer G. Drying transition in the hydrophobic gate of the GLIC channel blocks ion conduction. Biophys J. 2012;103:219–227. doi: 10.1016/j.bpj.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Teo I, Schulten K. A computational kinetic model of diffusion for molecular systems. J Chem Phys. 2013;139:121929. doi: 10.1063/1.4820876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heyden A, Lin H, Truhlar DG. Adaptive partitioning in combined quantum mechanical and molecular mechanical calculations of potential energy functions for multiscale simulations. J Phys Chem B. 2007;111:2231–2241. doi: 10.1021/jp0673617. [DOI] [PubMed] [Google Scholar]
- 85.Heyden A, Truhlar DG. Conservative algorithm for an adaptive change of resolution in mixed atomistic/coarse-grained multiscale simulations. J Chem Theory Comput. 2008;4:217–221. doi: 10.1021/ct700269m. [DOI] [PubMed] [Google Scholar]
- 86.Nielsen SO, Bulo RE, Moore PB, Ensing B. Recent progress in adaptive multiscale molecular dynamics simulations of soft matter. Phys Chem Chem Phys. 2010;12:12401–12414. doi: 10.1039/c004111d. [DOI] [PubMed] [Google Scholar]
- 87.Arkhipov A, Freddolino PL, Schulten K. Stability and dynamics of virus capsids described by coarse-grained modeling. Structure. 2006;14:1767–1777. doi: 10.1016/j.str.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 88.Grime JM, Voth GA. Early stages of the HIV-1 capsid protein lattice formation. Biophys J. 2012;103:1774–1783. doi: 10.1016/j.bpj.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.May ER, Brooks CL., 3rd On the morphology of viral capsids: Elastic properties and buckling transitions. J Phys Chem B. 2012;116:8604–8609. doi: 10.1021/jp300005g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kekenes-Huskey PM, Liao T, Gillette AK, Hake JE, Zhang Y, Michailova AP, McCulloch AD, McCammon JA. Molecular and subcellular-scale modeling of nucleotide diffusion in the cardiac myofilament lattice. Biophys J. 2013;105:2130–2140. doi: 10.1016/j.bpj.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schaad O, Zhou HX, Szabo A, Eaton WA, Henry ER. Simulation of the kinetics of ligand binding to a protein by molecular dynamics: geminate rebinding of nitric oxide to myoglobin. Proc Natl Acad Sci U S A. 1993;90:9547–9551. doi: 10.1073/pnas.90.20.9547. [DOI] [PMC free article] [PubMed] [Google Scholar]