

Abstract
Rapid, nongenomic vascular cell and tissue responses to estrogen have been demonstrated for more than a decade. Although the pendulum continues to swing, accumulating evidence, both clinical and pre-clinical, support favorable effects of ovarian steroid hormones in the vascular system. These effects are mediated both by classical steroid hormone receptor-mediated transcriptional modulation, and largely by endothelial plasma membrane-associated estrogen receptor (ER) α, which when engaged triggers a signaling cascade resulting in release of cardioprotective nitric oxide (NO). In addition to full-length ERα (ER66), an N-terminus truncated ERαisoform, ER46, plays a key role in these rapid endothelial responses to 17β-estradiol (E2). We have recently determined that ER46 can be a Type I integral transmembrane molecule. In this review, we discuss ER isoforms, rapid E2-stimulated signaling in the endothelium, the importance of the ER46 transmembrane orientation, and the clinical context of this rapid endothelial signaling.
Keywords: Estrogen, ER46, eNOS, Endothelium
1. Introduction and Clinical Background
Decades of observational clinical studies and epidemiological studies support a protective role for estrogen in the cardiovascular system. Numerous studies have documented clinically favorable effects of estrogen on circulating lipoproteins, endothelial nitric oxide (NO) production, vascular inflammation, and atherosclerotic plaque development. These findings supported the increasing use of hormone replacement in postmenopausal women as preventive therapy for chronic cardiovascular disease, until publication of the Women’s Health Initiative (WHI) results (Rossouw et al., 2002; Anderson, et al., 2004). Having been stopped early due to increased risks of coronary disease, stroke, pulmonary embolism, and breast cancer, the Initiative sparked an extensive critical reevaluation of the observational data supporting hormone replacement therapy (HRT) as a preventive measure.
Attention to the issue of hormone therapy timing emerged in response to the need to reconcile the WHI findings with multiple lines of clinical and basic scientific evidence in support of estrogen’s vascular protective effects. Subgroup analysis of the WHI supports this hypothesis of timing, in which estrogen’s benefits are contingent upon therapy beginning shortly after menopause. Moreover, in the Danish Osteoporosis Prevention Study (DOPS), a multicenter randomized trial, HRT was initiated in healthy perimenopausal or recently postmenopausal women who were followed for more than ten years of treatment. The treatment group had a reduced risk of the combined endpoint of death, heart failure, and myocardial infarction (Schierbeck et al., 2012). The small number of cardiovascular events may limit the degree to which the trial is seen as proof of the timing hypothesis. Nonetheless, the DOPS results, published shortly after the U.S. Preventive Services Task Force recommendation against the use of hormone therapy for the prevention of chronic disease in postmenopausal women, added to the debate concerning the WHI and its influence on clinical practice.
In parallel to the ongoing re-examination of the clinical data, new research utilizing animal models to better dissect the molecular pathways of estrogen receptor signaling is revealing some of the molecular mechanisms whereby estrogen can elicit its widely varying effects. In this regard, the effect of estrogen signaling pathways on chronic inflammation and cytokine signaling, as well its direct effects on the vascular endothelium, have gained particular attention.
Estrogen responses can be cell type-specific, due to variance in estrogen receptor isoform expression and variable recruitment of coregulatory molecules. Moreover, the balance of estrogen isoforms changes with age in some tissues, which has been shown to influence the vascular response to oxidative stress, nitric oxide production, and the process of atherosclerosis. Several new animal models utilizing genetic manipulations of specific receptor isoforms recently have been published, and they offer new insights as to how these isoforms exert different effects within the cardiovascular system (see below).
Selective estrogen receptor modulators (SERMs) represent a major advance in clinical practice by taking advantage of the ability to differentially modulate estrogen effects with varying degrees of tissue selectivity (Riggs and Hartmann, 2003). The selectivity is made possible by the endogenous variation in ER expression in different tissues, as well as tissue-specific variations in expression and action of ER coregulators. Further elucidation of the molecular biology of the cell type-specific signaling events are needed to advance selective estrogen receptor modulation to the point of offering vascular protective benefit while minimizing the known risks of long-term HRT. To this end, a better characterization of expression levels and estrogen receptor isoform signaling is revealing molecular mechanisms as they relate to the clinically-derived HRT timing hypothesis. These studies highlight estrogen’s direct action on the endothelium, which is characterized by rapid, non-nuclear signaling through membrane-associated effector molecules. These non-genomic pathways, specifically in endothelial cells, comprise a key homeostatic switch favoring NO synthesis and opposing inflammation and thrombosis. Quantitative coronary angiography in humans demonstrates that physiological levels of estradiol modulate coronary vascular function in a short-term endothelial-dependent manner (Gilligan et al., 1994).
The health of the endothelium is governed by sum of ligand-dependent and ligand-independent transcriptional regulation together with the rapid non-genomic signaling effects of estrogen receptors. Animal model-based studies of ER isoforms provide the best insight to date as to how clinical observations such as that of the WHI may finally be reconciled with longstanding scientific knowledge of the cardiovascular protective effects of estrogen, with implications for aging and preventive therapy.
2. ER Isoforms
Distinct ERα isoforms are expressed in a cell- and tissue-specific manner. An N-terminus truncated short isoform of ERα, ER46, is abundant in human endothelial cells (ECs) (Russell et al., 2000; Haynes et al., 2003; Li et al., 2003), and expressed in osteoblasts (Denger et al., 2001; Longo et al., 2004), as well as breast and endometrial cell lines (Zivadinovic and Watson, 2005; Márquez and Pietras, 2001). A 50 kDa ERα isoform has been identified in hypothalamic neurons and astrocytes (Gorosito et al., 2008; Bondar et al., 2009; Dominguez et al., 2010). ER36 has been found in breast carcinomas (Wang et al., 2006; Shi et al., 2009).
These ERα isoforms are generated from multiple ERα mRNA transcripts regulated by alternative promoter usage and alternative splicing, as well as from alternative translation initiation (Flouriot et al., 1998; Kos et al., 2001; Hirata et al., 2003). The ERα gene has eight coding exons, eight untranslated 5′-exons in the upstream region of exon 1, and an intronic exon S located between exons 3 and 4 (Hirata et al., 2003). Seven human estrogen promoters (A-F and T) have been defined (Thompson et al., 1997; Kos et al., 2001; Brand et al., 2002). ER46, lacking the first ERα coding exon (exon 1A), is transcribed from two different ERα promoters (E and F) and produced by splicing of exon 1E directly to exon 2 (Flouriot et al., 2000). Multiple E- and F-transcript ERα isoforms have been detected and these transcript variants are tissue-specific. Expression of the E variants is restricted to MCF-7 cells and adult liver. F variants have been found in both reproductive and non-reproductive organs. To date, two ERα E-transcript and six F-transcript isoforms have been identified (Ishii et al., 2013). ERα mRNA 5′-untranslated regions (5′-UTRs) result in multiple upstream open reading frames and affect translational efficiency (Kos et al., 2002). It has recently been shown that the amount of translated ERα protein may be dictated by the inclusion (or exclusion) of non-coding internal exons in E and F variants, without modulating ERα promoter activity (Ishii et al., 2013). These multiple mechanisms of RNA expression, processing and translation provide a level of complexity resulting in numerous receptor isoforms, potentially directing varying cellular responses to hormonal stimuli.
3. Vascular ER Isoforms
The two estrogen receptors (ERs), ERα and β, are encoded by separate genes and share high sequence homology. In their role as transducers of genomic signaling, they bind the same estrogen response elements (ERE), and may signal in either ligand-dependent (as classical nuclear receptors) or ligand-independent manners. They also exert transcriptional regulation by association with other DNA-bound elements (Truss and Beato, 1993). In endothelial cells, estrogen receptor activation drives an anti-atherogenic gene program (Mendelsohn and Karas, 1999). In addition to both classical ligand-activated transcriptional activation and growth factor-induced ligand-independent transcriptional activation, we focus here on plasma membrane-initiated, rapid non-genomic signaling, resulting in the activation of endothelial nitric oxide synthase (eNOS), and enhanced NO production.
Both ERα and ERβ are expressed in ECs and vascular smooth muscle cells (SMCs), although expression level heterogeneity is significant. ERβ expression in ECs and SMCs increases at the site of vascular injury in a carotid denudation model, however, ERβ-KO mice have maintained E2-stimulated repression of the neointimal vascular remodeling response to injury (Mäkelä et al., 1999). Reduced expression of both receptors has been observed in atherosclerotic carotid arteries, in which context estradiol is diminished in its capacity to suppress vascular smooth muscle cell proliferation (Losordo et al., 1994; Nakamura et al., 2004). While such findings, together with clinical observations, suggested an atheroprotective function of estrogen receptor signaling, particularly in the case of ERα, the mechanistic understanding began to take shape with generation of several ERα knockout mice over the last two decades. While complete ERα knockout mice are deficient in the E2-stimulated increase in NO production, ERβ knockout mice do not show such defect. We thus focus here on ERα and its isoforms, and their endothelial functions. Physiologically functional ERα isoforms have been studied through the use of targeted gene manipulation in mice. Over twenty years ago, the first exon of ERα was deleted, and subsequent analysis revealed that these mice retained expression of N-terminally truncated 55 and 49 kDa proteins (Lubahn et al., 1993; Couse et al., 1995). The initially confounding, but ultimately fortuitous, findings from the deletion of ERα exon one led to discovery of physiologically relevant ERα splice isoforms. The retention of these alternatively spliced ERα receptors in the knockout mouse allowed preservation of some of the effects of estradiol, including the induction of endothelial NO production, and re-endothelialization following endovascular injury (Iafrati et at. 1997; Brouchet et al., 2001; Darblade et al., 2002; Pare et al., 2002). The deletion of ERα by targeted gene disruption of exon 2 yields a true null lacking these persistent effects.
17β-estradiol acts on a subpopulation of membrane-associated estrogen receptors to activate multiple signaling cascades, converging on the induction of eNOS (Chambliss et al., 2010). This pathway promotes endothelial migration and proliferation in vitro, and increases carotid artery re-endothelialization in a mouse model of perivascular injury. This non-nuclear pathway does not induce the uterotropic response (Kousteni et al. 2002), raising the therapeutically valuable possibility of selectively promoting the vascular protective signaling pathways, separate from reproductive system effects.
ERα-AF10 mice were generated by deletion of the first exon of ERα, resulting in loss of the N-terminal activation function domain AF-1 (Billon-Galés et al., 2009). These mice have intact endothelial NO production, as well as re-endothelialization in response to vascular injury. Moreover, in contrast to the complete deletion of ERα, loss of AF1 does not affect responsiveness to E2 with respect to fatty streak deposit in the LDLR−/− atherosclerosis model. In this model, endogenous estrogen reduces aortic atherogenesis by half as compared to ovarectomized mice, and the deletion of AF1 does not block this protection. ERα-AF10 mice do not have significantly different plasma cholesterol profiles compared to control, supporting previous observations that the atheroprotective effect of E2 seems to be a direct arterial effect (Elhage et al., 1997). The AF1 domain does not have a recognized role in the localization of ER to the plasma membrane and membrane-initiated steroid signaling; rather, it is the AF2 domain that is most essential to these functions (Kousteni et al., 2001; Razandi et al., 2003).
The dispensability of AF1 domain, as demonstrated by ERα-AF10, implicated AF2 as having an essential function in the atheroprotective effects of ERα. This led the same investigators to create ERα-AF20 mice, lacking the C-terminal activation function 2 domain (Billon-Galés et al., 2011). While this deletion had no effect on E2-induced re-endothelialization following vascular injury, the protective effect of E2 in the aortic atherosclerosis model was partially lost, as evidenced by increased fatty streak deposition, collagen accumulation, and macrophage infiltration. This is likely a consequence of the AF-2 deletion on receptor targeting and function, both membrane- and nuclear-initiated. Expression analysis of several genes important in atherogenesis demonstrates a role for the AF2 domain in E2-regulated transcription as well.
4. Rapid ER Signaling in Endothelium
We utilized a membrane-impermeant form of E2 to demonstrate plasma membrane binding of estradiol in human vascular ECs. This binding is inhibited by a classical ERα antagonist and competed by free E2. Ligand binding at the plasma membrane rapidly triggers a signaling cascade that includes activation of the kinases c-Src, PI-3 kinase and Akt, resulting in eNOS-mediated enhanced production of NO (Haynes et al., 2003; Li et al., 2003). In fact, elevations in endothelial cGMP can be seen as early as 5 minutes after E2 stimulation (Russell et al., 2000). An N-terminus truncated ERα isoform, ER46, has been identified at the EC plasma membrane (Figtree et al., 2003; Li et al., 2003). Murine aorta studies demonstrate a requirement for Akt, ERK, PI-3 kinase, and c-Src in E2-induced eNOS activation, NO production, and endothelium-dependent vasodilation (Florian et al., 2004; Guo et al., 2005; Li et al., 2007).
Posttranslational modifications of ER and c-Src are important for eNOS activation. The posttranslational palmitoylation at Cys447 directs membrane localization of ER46, protein-protein interactions and eNOS activation. We have defined a functional signaling complex containing c-Src, PI3-K and ER46 in ECs (Haynes et al., 2003; Li et al., 2003; Kim and Bender, 2005; Li et al., 2007). Membrane-impermeant E2 triggers phosphorylation of the c-Src Tyr419 active site, followed by eNOS phosphorylation at Ser1177. c-Src also must be lipid modified for plasma membrane targeting. Overexpression of the myristoylation c-Src Gly2Ala mutant in ER46-cotransfected COS-7 cells significantly abolishes both c-Src and ER46 plasma membrane localization, demonstrating their interdependence for protein localization at the plasma membrane (Li et al., 2007). The colocalization and association of ER46 with caveolin-1 has been shown in human ECs, cancer cells and myometrial cells (Li et al., 2003; Acconcia et al., 2005; Kiss et al., 2005). Based on these findings, we (and others) developed a signaling model in which the ER46-centered molecular complex, including c-Src, PI3-K, Akt, Hsp90 and eNOS, resides in caveolae at the plasma membrane (Kim and Bender, 2005). Based on more recent findings, this model has been modified (see below).
ERα palmitoylation site mutant mice (C451A-ERα) have been used to demonstrate the importance of the aforementioned plasma membrane targeting in vivo. In contrast to WT mice, the mesenteric arteries from these mice fail to rapidly vasodilate in response to E2. Furthermore, endothelial repair and eNOS activation are diminished in C451A-ERα mice (Adlanmerini et al., 2014). ERα-AF20 mice (deletion of C-terminal residues 543-549), with an intact palmitoylation site, maintain their E2-induced re-endothelialization after carotid injury (Adlanmerini et al., 2014), further supporting the importance of posttranslational lipid modification and membrane targeting of the receptor.
In cell reconstitution studies, ER46 triggers eNOS activation more efficiently than full-length ERα (ER66) at the plasma membrane (Li et al., 2003). The binding affinity of E2 to ER46 appears to be slightly higher than to ER66 in a eukaryotic cell-free expression system. In an intact cell system, palmitoylation inhibition reduces E2-binding affinity, with a greater effect on ligand binding to ER46 (5-fold) than on ER66 (2.7-fold) (Lin et al., 2013). This is further evidence that plasma membrane targeting, especially of ER46, is important for maximal ligand binding and consequent signaling responses.
5. ER46 as an Integral Transmembrane Protein
Our membrane-impermeant E2 studies, as well as anti-ERα antibody binding to non-permeabilized cells, raised the possibility that ectodomain-expressing transmembrane ERs might exist. Ecliptic pHuorin is a pH-sensitive GFP mutant with a range of fluorescence, lower to higher, between pH 7.0 and 8.0, but which is not fluorescent at pH <6.0 (Miesenböck et al., 1998). When expressing recombinant, pHluorin-fused ER46 in immortalized human ECs (EA.hy926 cells), live plasma membrane confocal imaging by total internal reflection fluorescence microscopy (TIRFM) demonstrates much greater fluorescence with pHluorin fused to the N-, rather than the C-, terminus. This is consistent with an N-terminal ectodomain (medium pH set at 7.8), and a C-terminal cytoplasmic domain (intracellular pH measured at 7.2). The power of this mutant GFP includes the ability to rapidly change fluorescence intensity when pHluorin is exposed to pH changes in the extracellular medium. Indeed, the fluorescence of pHluorin-ER46 (N-terminus-fused) is rapidly and completely quenched with a 7.8 to 6.5 extracellular pH change, with minimal change in the fluorescence intensity of ER46-pHluorin (C-terminus-fused) (Figure 1A, Kim et al., 2011). Overall expression levels of the N- and C- terminal recombinant proteins were similar. These data are consistent with the ability of ER46 to conform to a Type I integral transmembrane protein. This orientation was biochemically confirmed by tryptic proteolysis at exposed ectodomain-containing Arg287 and Lys302 (or 303) sites (Kim et al., 2011). Isoleucine-386 is at the center of ER46’s projected transmembrane hydrophobic core domain (Val376-Val392, Figure 1B). Mutation of this Ile386 is predicted to abolish the molecule’s transmembrane orientation. An N-terminal pHluorin-fused ER46 Ile386Cys mutant, when expressed in human ECs, is minimally fluorescent, compared to the WT receptor, or to ER46 with a conservative Ile386Val mutation, and loses the dynamic fluorescence alteration with a pH change (Figure 1A, C). Three other isoleucine-to-cysteine point mutations, at the ectodomain (Ile358), the transmembrane domain (Ile389), and the cytoplasmic domain (Ile424), have no effect on N-terminal-fused ER46 fluorescence intensity (Figure 1C) (Kim et al., 2011).
Figure 1.

pHluorin-fused recombinant ER46 and its mutant and topological model of ER46 in human ECs. (A) TIRFM live images of extracellular pH change effect on EA.hy926 (EA) cells expressing pHluorin-fused recombinant proteins at extracellular pH 7.8, 6.5 and then 7.4 with NH4Cl. pH values in parentheses indicate the intracellular pH. (B) Model of ER46 with an N-terminal ectodomain from Met174 to Gln375, based on the PSORT II transmembrane prediction program. (C) Comparative TIRFM fluorescence levels of EA cells transfected with pHluorin-ER46, ER46-pHluorin, pHluorin-ER46-Ile386Cys, pHluorin-ER46-Ile386Val, pHluorin-ER46-Ile358Cys, pHluorin-ER46-Ile389Cys and pHluorin-ER46-Ile424Cys at extracellular pH 7.8, with mean pixel determination of n=10, ± S.D. *p<0.001. Figures reproduced in part from Kim et al., (2011).
These results support an N-terminus out, C-terminus in, orientation, with a critical residue(s) conferring transmembrane capability. To address whether this change in orientation affects rapid estrogen-induced signaling, NO detection by 4-amino-5-methylamino-2′,7′-difluorescein (DAF-FM) confocal imaging was performed in live ECs. E2-stimulated, ER46 WT-expressing ECs efficiently produce NO, as expected, whereas the E2-induced signaling response is greatly impaired in the transmembrane mutant ER46-Ile386Cys-expressing cells (Figure 2). This provides strong evidence that the transmembrane orientation of ER46 is important for its rapid signal transducing properties in response to estrogen.
Figure 2.
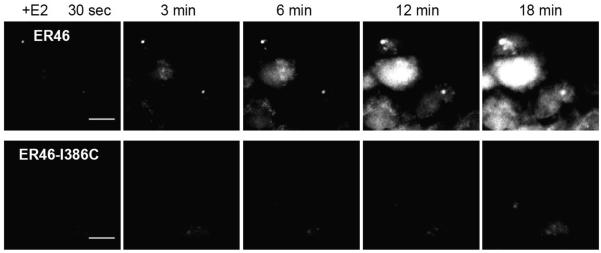
NO detection in live COS-7 cells. COS-7 cells were transfected with plasmids encoding eNOS and either ER46 or ER46-Ile386Cys. Sequential imaging was performed at 37 °C after loading cells with DAF-FM diacetate. 30 nM E2 was added at 30 sec of imaging, with imaging time points indicated. Scale bar = 20 μm. Reproduced from Kim et al., (2011).
E2-ERα ligand binding domain (LBD) co-crystallization defined the phenolic hydroxyl of E2’s A ring within a cavity formed by ERα helix 3 (Met342 to Leu354) and helix 6 (Trp383 to Arg394), with direct E2 binding to Glu353 (Brzozowski et al., 1997). In our transmembrane orientation model, Glu353 and helix3 are available within the ectodomain, whereas helix 6 is not, making the formation of the polar cavity, described above, for E2 binding unlikely. Thus, it will be critical to perform structural analyses, in the future, to fully define the basis of ectodomain binding by ligand.
Given this new information regarding ER46’s ability to achieve a transmembrane orientation, and to efficiently signal when in this conformation, it is important to emphasize that this likely comprises one subset of cellular ERs, in addition to nuclear, cytosolic and caveolae-associated subsets. The dominance of any one of these localized subsets undoubtedly relates to the cell/tissue type, state of cell activation, and available protein-protein interaction binding partners. Defining the hierarchy of these influences, as it relates to receptor trafficking and localization, will create some new targeting opportunities.
6. Summary and Future Directions
Estrogen receptor isoforms are abundant, widely expressed and highly variable in form and function. The field has only begun to understand the relevance and importance of various isoforms in cell and organ physiology and pathology. In the case of the endothelium, mouse genetics have informed us that smaller splice isoforms of ER66 can be fully functional in the vascular system, enabling vascular protective effects of estrogen. We have focused on ER46, abundant in ECs isolated from a wide variety of vessels, and shown that, remarkably, it can conform to a transmembrane orientation which, in vitro, enhances its E2-induced rapid signaling responses. There remains much to learn, including how the ligand-receptor orientation participates in crosstalk with nuclear (genomic) responses, whether this transmembrane molecule shuttles to other intracellular compartments, and many other unanswered cell biological questions. Most importantly, we see the presence of an isoform ectodomain as a novel molecular therapeutic targeting opportunity, one that may have selective effects on, for example, the cardiovascular system.
Acknowledgements
This work was supported by the NIH R01 HL61782, T32HL007950 and by the Raymond and Beverly Sackler Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acconcia F, Ascenzi P, Bocedi A, Spisni E, Tomasi V, Trentalance A, Visca P, Marino M. Palmitoylation-dependent estrogen receptor α membrane localization: regulation by 17β-estradiol. Mol. Biol. Cell. 2005;16:231–237. doi: 10.1091/mbc.E04-07-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond-Letron I, Guihot AL, Boudou F, Sautier L, Vessières E, Kim SH, Lière P, Fontaine C, Krust A, Chambon P, Katzenellenbogen JA, Gourdy P, Shaul PW, Henrion D, Arnal JF, Lenfant F. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E283–290. doi: 10.1073/pnas.1322057111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N, Lewis CE, Manson J, Margolis K, Ockene J, O'Sullivan MJ, Phillips L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G, Stefanick ML, Van Horn L, Wactawski-Wende J, Wallace R, Wassertheil-Smoller S, Women's Health Initiative Steering Committee Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- Billon-Galés A, Fontaine C, Filipe C, Douin-Echinard V, Fouque MJ, Flouriot G, Gourdy P, Lenfant F, Laurell H, Krust A, Chambon P, Arnal JF. The transactivating function 1 of estrogen receptor α is dispensable for the vasculoprotective actions of 17β-estradiol. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2053–2058. doi: 10.1073/pnas.0808742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billon-Galés A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, Berges H, Gadeau AP, Lenfant F, Gourdy P, Chambon P, Arnal JF. Activation function 2 (AF2) of estrogen receptor-alpha is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc. Natl. Acad. Sci. U.S.A. 2011;108:13311–13316. doi: 10.1073/pnas.1105632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar G, Kuo J, Hamid N, Micevych P. Estradiol-induced estrogen receptor-α trafficking. J. Neurosci. 2009;29:15323–15330. doi: 10.1523/JNEUROSCI.2107-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand H, Kos M, Denger S, Flouriot G, Gromoll J, Gannon F, Reid G. A novel promoter is involved in the expression of estrogen receptor α in human testis and epididymis. Endocrinology. 2002;143:3397–3404. doi: 10.1210/en.2001-210832. [DOI] [PubMed] [Google Scholar]
- Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-α but not estrogen receptor-β. Circulation. 2001;103:423–428. doi: 10.1161/01.cir.103.3.423. [DOI] [PubMed] [Google Scholar]
- Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, Madak-Erdogan Z, Maggi A, Dineen SP, Roland CL, Hui DY, Brekken RA, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW. Non-nuclear estrogen receptor α signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J. Clin. Invest. 2010;120:2319–2330. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB, Smithies O, Korach KS. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol. Endocrinol. 1995;9:1441–1454. doi: 10.1210/mend.9.11.8584021. [DOI] [PubMed] [Google Scholar]
- Darblade B, Pendaries C, Krust A, Dupont S, Fouque MJ, Rami J, Chambon P, Bayard F, Arnal JF. Estradiol alters nitric oxide production in the mouse aorta through the α-, but not β-, estrogen receptor. Circ. Res. 2002;90:413–419. doi: 10.1161/hh0402.105096. [DOI] [PubMed] [Google Scholar]
- Denger S, Reid G, Kos M, Flouriot G, Parsch D, Brand H, Korach KS, Sonntag-Buck V, Gannon F. ERα gene expression in human primary osteoblasts: evidence for the expression of two receptor proteins. Mol. Endocrinol. 2001;15:2064–2077. doi: 10.1210/mend.15.12.0741. [DOI] [PubMed] [Google Scholar]
- Dominguez R, Micevych P. Estradiol rapidly regulates membrane estrogen receptor alpha levels in hypothalamic neurons. J. Neurosci. 2010;30:12589–12596. doi: 10.1523/JNEUROSCI.1038-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhage R, Arnal JF, Pieraggi MT, Duverger N, Fiévet C, Faye JC, Bayard F. 17β-estradiol prevents fatty streak formation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1997;17:2679–2684. doi: 10.1161/01.atv.17.11.2679. [DOI] [PubMed] [Google Scholar]
- Figtree GA, McDonald D, Watkins H, Channon KM. Truncated estrogen receptor α46-kDa isoform in human endothelial cells. Circulation. 2003;107:120–126. doi: 10.1161/01.cir.0000043805.11780.f5. [DOI] [PubMed] [Google Scholar]
- Florian M, Lu Y, Angle M, Magder S. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids. 2004;69:637–645. doi: 10.1016/j.steroids.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Flouriot G, Griffin C, Kenealy M, Sonntag-Buck V, Gannon F. Differentially expressed messenger RNA isoforms of the human estrogen receptor-α gene are generated by alternative splicing and promoter usage. Mol. Endocrinol. 1998;12:1939–1954. doi: 10.1210/mend.12.12.0209. [DOI] [PubMed] [Google Scholar]
- Flouriot G, Brand H, Denger S, Metivier R, Kos M, Reid G, Sonntag-Buck V, Gannon F. Identification of a new isoform of the human estrogen receptor-alpha (hER-α) that is encoded by distinct transcripts and that is able to repress hER-α activation function 1. EMBO J. 2000;19:4688–4700. doi: 10.1093/emboj/19.17.4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilligan DM, Quyyumi AA, Cannon RO., 3rd Effects of physiological levels of estrogen on coronary vasomotor function in postmenopausal women. Circulation. 1994;89:2545–2551. doi: 10.1161/01.cir.89.6.2545. [DOI] [PubMed] [Google Scholar]
- Gorosito SV, Lorenzo AG, Cambiasso MJ. Estrogen receptor alpha is expressed on the cell-surface of embryonic hypothalamic neurons. Neuroscience. 2008;154:1173–1177. doi: 10.1016/j.neuroscience.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors α and β. J. Biol. Chem. 2005;280:19704–19710. doi: 10.1074/jbc.M501244200. [DOI] [PubMed] [Google Scholar]
- Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, Collinge M, Sessa WC, Bender JR. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J. Biol. Chem. 2003;278:2118–2123. doi: 10.1074/jbc.M210828200. [DOI] [PubMed] [Google Scholar]
- Hirata S, Shoda T, Kato J, Hoshi K. Isoform/variant mRNAs for sex steroid hormone receptors in humans. Trends Endocrinol. Metab. 2003;14:124–129. doi: 10.1016/s1043-2760(03)00028-6. [DOI] [PubMed] [Google Scholar]
- Iafrati MD, Karas RH, Aronovitz M, Kim S, Sullivan TR, Jr., Lubahn DB, O’Donnell TF, Jr., Korach KS, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor-α-deficient mice. Nat. Med. 1997;3:545–548. doi: 10.1038/nm0597-545. [DOI] [PubMed] [Google Scholar]
- Ishii H, Kobayashi M, Munetomo A, Miyamoto T, Sakuma Y. Novel splicing events and post-transcriptional regulation of human estrogen receptor α E isoforms. J. Steroid Biochem. Mol. Biol. 2013 2013 Jan;133:120–128. doi: 10.1016/j.jsbmb.2012.09.027. [DOI] [PubMed] [Google Scholar]
- Kim KH, Bender JR. Rapid, estrogen receptor-mediated signaling: why is the endothelium so special? Sci. STKE. 2005;288:pe28. doi: 10.1126/stke.2882005pe28. [DOI] [PubMed] [Google Scholar]
- Kim KH, Toomre D, Bender JR. Splice isoform estrogen receptors as integral transmembrane proteins. Mol. Biol. Cell. 2011;22:4415–4423. doi: 10.1091/mbc.E11-05-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss AL, Turi A, Müllner N, Kovács E, Botos E, Greger A. Oestrogen-mediated tyrosine phosphorylation of caveolin-1 and its effect on the oestrogen receptor localization: An in vivo study. Mol. Cell. Endocrinol. 2005;245:128–137. doi: 10.1016/j.mce.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Kos M, Reid G, Denger S, Gannon F. Minireview: genomic organization of the human ERα gene promoter region. Mol. Endocrinol. 2001;15:2057–2063. doi: 10.1210/mend.15.12.0731. [DOI] [PubMed] [Google Scholar]
- Kos M, Denger S, Reid G, Gannon F. Upstream open reading frames regulate the translation of the multiple mRNA variants of the estrogen receptor α. J. Biol. Chem. 2002;277:37131–37138. doi: 10.1074/jbc.M206325200. [DOI] [PubMed] [Google Scholar]
- Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen B, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O'Brien CA, Plotkin L, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–846. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor α variant (ER46) in human endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4807–4812. doi: 10.1073/pnas.0831079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Hisamoto K, Kim KH, Haynes MP, Bauer PM, Sanjay A, Collinge M, Baron R, Sessa WC, Bender JR. Variant estrogen receptor-c-Src molecular interdependence and c-Src structural requirements for endothelial NO synthase activation. Proc. Natl. Acad. Sci. U.S.A. 2007;104:16468–16473. doi: 10.1073/pnas.0704315104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AH, Li RW, Ho EY, Leung GP, Leung SW, Vanhoutte PM, Man RY. Differential ligand binding affinities of human estrogen receptor-α isoforms. PLoS One. 2013;8:e63199. doi: 10.1371/journal.pone.0063199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo M, Brama M, Marino M, Bernardini S, Korach KS, Wetsel WC, Scandurra R, Faraggiana T, Spera G, Baron R, Teti A, Migliaccio S. Interaction of estrogen receptor α with protein kinase C alpha and c-Src in osteoblasts during differentiation. Bone. 2004;34:100–111. doi: 10.1016/j.bone.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Losordo DW, Kearney M, Kim EA, Jekanowski J, Isner JM. Variable expression of the estrogen receptor in normal and atherosclerotic coronary arteries of premenopausal women. Circulation. 1994;89:1501–1510. doi: 10.1161/01.cir.89.4.1501. [DOI] [PubMed] [Google Scholar]
- Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11162–11166. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä S, Savolainen H, Aavik E, Myllärniemi M, Strauss L, Taskinen E, Gustafsson JA, Häyry P. Differentiation between vasculoprotective and uterotrophic effects of ligands with different binding affinities to estrogen receptors α and β. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7077–7082. doi: 10.1073/pnas.96.12.7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez DC, Pietras RJ. Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cancer cells. Oncogene. 2001;20:5420–5430. doi: 10.1038/sj.onc.1204729. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Suzuki T, Miki Y, Tazawa C, Senzaki K, Moriya T, Saito H, Ishibashi T, Takahashi S, Yamada S, Sasano H. Estrogen receptors in atherosclerotic human aorta: inhibition of human vascular smooth muscle cell proliferation by estrogens. Mol. Cell. Endocrinol. 2004;219:17–26. doi: 10.1016/j.mce.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, Mendelsohn ME. Estrogen receptor-α mediates the protective effects of estrogen against vascular injury. Cir. Res. 2002;90:1087–1092. doi: 10.1161/01.res.0000021114.92282.fa. [DOI] [PubMed] [Google Scholar]
- Pendaries C, Darblade B, Rochaix P, Krust A, Chambon P, Korach KS, Bayard F, Arnal JF. The AF-1 activation-function of ERα may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc. Natl. Acad. Sci. U.S.A. 2002;99:2205–2210. doi: 10.1073/pnas.042688499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, Levin ER. Identification of a structural determinant necessary for the localization and function of estrogen receptor α at the plasma membrane. Mol. Cell. Biol. 2003;23:1633–1646. doi: 10.1128/MCB.23.5.1633-1646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs BL, Hartmann LC. Selective estrogen-receptor modulators - mechanisms of action and application to clinical practice. N. Engl. J. Med. 2003;348:618–629. doi: 10.1056/NEJMra022219. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J, Writing Group for the Women’s Health Initiative Investigators Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Russell KS, Haynes MP, Sinha D, Clerisme E, Bender JR. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5930–5935. doi: 10.1073/pnas.97.11.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierbeck LL, Rejnmark L, Tofteng CL, Stilgren L, Eiken P, Mosekilde L, Køber L, Jensen JE. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ. 2012;345:e6409. doi: 10.1136/bmj.e6409. [DOI] [PubMed] [Google Scholar]
- Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, Wang Z, Xie Y. Expression of ER-α36, a novel variant of estrogen receptor α, and resistance to tamoxifen treatment in breast cancer. J. Clin. Oncol. 2009;27:3423–3429. doi: 10.1200/JCO.2008.17.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, McPherson LA, Carmeci C, deConinck EC, Weigel RJ. Identification of two estrogen receptor transcripts with novel 5' exons isolated from a MCF7 cDNA library. J. Steroid Biochem. Mol. Biol. 1997;62:143–153. doi: 10.1016/s0960-0760(97)00029-0. [DOI] [PubMed] [Google Scholar]
- Truss M, Beato M. Steroid hormone receptors: interaction with deoxyribonucleic acid and transcription factors. Endocr. Rev. 1993;14:459–479. doi: 10.1210/edrv-14-4-459. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-α, hER-α36: Transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9063–9068. doi: 10.1073/pnas.0603339103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivadinovic D, Watson CS. Membrane estrogen receptor-α levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res. 2005;7:R130–144. doi: 10.1186/bcr959. [DOI] [PMC free article] [PubMed] [Google Scholar]