

Abstract
Mutations in genes encoding widely expressed nuclear envelope proteins often lead to diseases that manifest in specific tissues. Lamina-associated polypeptide 1 (LAP1) is an integral protein of the inner nuclear membrane that is expressed in most cells and tissues. Within the nuclear envelope, LAP1 interacts physically with lamins, torsinA and emerin, suggesting it may serve as a key node for transducing signals across the inner nuclear membrane. Indeed, recent in vivo studies in genetically modified mice strongly support functional links between LAP1 and both torsinA (in neurons) and emerin (in muscle). These studies suggest that tissue-selective diseases caused by mutations in genes encoding nuclear envelope proteins may result, at least in part, from the selective disruption of discrete nuclear envelope protein complexes.
Keywords: nuclear envelope, lamin, nuclear membrane, muscular dystrophy, dystonia
1. Introduction
1.1. Nuclear envelope membranes
The nuclear envelope is composed of the nuclear membranes, the nuclear pore complexes and the nuclear lamina (Figure 1). The nuclear membranes are composed of three continuous but morphologically distinct domains in interphase cells: inner, outer and pore. The outer nuclear membrane is directly continuous with the endoplasmic reticulum membrane, with which it shares ribosomes. It is separated from the inner nuclear membrane by the perinuclear space, which is continuous with the lumen of the rough endoplasmic reticulum. The pore membranes connect the inner and outer nuclear membranes at the nuclear pore complexes. The lamina is a meshwork of intermediate filament proteins called lamins, and is localized primarily at the inner aspect of the inner nuclear membrane.
Figure 1.
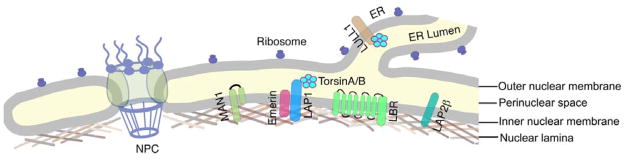
Schematic diagram of the nuclear envelope showing the nuclear membranes, nuclear lamina and a nuclear pore complex (NPC). The outer nuclear membrane contains ribosomes on its outer surface just like the directly contiguous endoplasmic reticulum (ER). The ER lumen is directly continuous with the perinuclear space. Selected proteins concentrated in the inner nuclear membrane and LULL1 in ER membrane are shown along with torsinA and torsin B (TorsinA/B) in the perinuclear space. LAP1 is shown interacting with lamins, emerin and torsin A/B.
As the nuclear membranes are actually a single interconnected membrane system, one could imagine that integral proteins synthesized on the rough endoplasmic reticulum would be randomly distributed among them. However, certain integral proteins are concentrated in each of these membranes in interphase cells, either as a result of binding to resident structures in specific domains or by active transport mechanisms [1–4]. Approximately 80 transmembrane proteins are concentrated in the inner nuclear membrane [5]. Many of the integral proteins of the inner nuclear membrane bind to nuclear lamins, which likely contributes to their retention within the nuclear envelope. While the nuclear envelope proteome may vary to some extend among different cell types, integral proteins of the inner nuclear membrane typically exhibit a near-ubiquitous pattern of expression throughout different tissues [5–6]. This pattern implies that such proteins have fundamental roles in maintaining nuclear structure or supporting critical nuclear functions, indicating that alterations in the genes encoding these proteins is likely to be lethal or to cause widespread pathology. Surprisingly, however, the opposite is the case; a range of discoveries link mutations in these genes to human diseases that exhibit striking tissue specificity, typically of muscle, adipose or neural tissue.
1.2. Laminopathies/nuclear envelopathies
In 1994, Toniolo and colleagues reported that mutations in the gene encoding a widely expressed, previously uncharacterized integral membrane protein they named emerin were responsible for X-linked Emery-Dreifuss muscular dystrophy [7]. Subsequent research showed that emerin was localized to the inner nuclear membrane [8,9]. This was the first demonstration that mutations in a gene encoding an integral protein of the inner nuclear membrane widely expressed in many cells and tissues could cause tissue-selective disease. This tissue specificity is even more surprising considering that most pathogenic mutations cause loss of emerin expression, not subtle structural alterations [7–9,10].
Subsequent discoveries further strengthened the theme of tissue selective disease arising from mutations in genes encoding widely expressed nuclear envelope proteins [11,12]; these diseases are now referred to as “laminopathies” or “nuclear envelopathies” [11,12]. Perhaps the most dramatic example involves mutations in the gene encoding lamins A and C, which encodes extrinsic proteins of the inner nuclear membrane that are building blocks of the nuclear lamina. Distinct mutations in this gene cause over a dozen different diseases, which predominantly affect striated muscle, adipose tissue or peripheral nerve, whereas some mutations disrupt multiple tissues and produce a phenotype of accelerated aging (“progeria”) [11,12]. Among these diseases is autosomal dominant Emery-Dreifuss muscular dystrophy, which phenocopies X-linked Emery-Dreifuss muscular dystrophy caused by mutation in the gene encoding emerin [13]. In addition to emerin and lamins A and C, mutations in genes encoding other widely expressed proteins of the inner nuclear membrane cause tissue-selective diseases. Heterozygous mutations in the gene encoding LBR, an integral protein of the inner nuclear membrane [14], cause Pelger-Huët anomaly, which affects only blood neutrophils [15]. Mutations in the gene encoding MAN1, another integral protein of the inner nuclear membrane [16], cause sclerosing bone dysplasias with or without skin involvement, but without any other major organ pathology [17].
The mechanisms responsible for the apparent tissue-selective functions of nuclear envelope proteins remain for the most part unknown. To obtain insights into this question, we explored the biology of lamina-associated polypeptide 1 (LAP1), an integral protein of the inner nuclear membrane. Our work suggests that the function of protein complexes in the nuclear envelope, rather than of individual proteins per se, may underlie the tissue-selective pathology of the laminopathies/nuclear envelopathies.
2. LAP1
2.1. Discovery
LAP1 was first identified as three polypeptide antigens recognized by a monoclonal antibody generated against rat liver nuclear envelope protein extracts [18]. This antibody recognized antigens with apparent molecular masses of approximately 75 kDa, 68 kDa and 55 kDa in rat liver nuclear envelopes. Biochemical extractions showed the antigens to be integral membrane proteins and immunofluorescence and immuno-electron microscopy localized them to the inner nuclear membrane. Biochemical extraction experiments showed them to be associated with the nuclear lamina and subsequent experiments demonstrated that they bind assembled lamins in vitro [19]. The proteins with apparent molecular masses of approximately 75kDa, 68 kDa and 55 kDa were named lamina-associated polypeptides 1A–C (LAP1A, LAP1B and LAP1C).
Analysis of the cloned rat LAP1C cDNA demonstrated that it encoded a type II integral membrane protein composed of 506 amino acids [20]. Further sequence analysis showed the human LAP1B cDNA encoded a protein of 584 amino acids with similar topology [21]. Human and mouse LAP1B are 66% identical in amino acid sequence. All of the isoforms arise from a single gene by alternative RNA splicing, have a single transmembrane segment, and lack amino-terminal signal sequences. This results in a topology with the amino-terminus facing the nucleoplasm and the carboxyl-terminus within the perinuclear space (“type II” transmembrane proteins; Figure 2). This topology predicts that the amino-terminal domain binds lamins within the nucleus, an interaction likely responsible for LAP1 concentration in the inner nuclear membrane. The amino-terminal domain of LAP1 is also in a position to bind nucleoplasmic proteins such as PP1 [22], as well as the nucleoplasmic domains of other integral inner nuclear membrane proteins. The carboxyl-terminal domain within the perinuclear space has the potential to bind to proteins translocated into the endoplasmic reticulum and the luminal domains of other integral proteins of the inner nuclear membrane.
Figure 2.
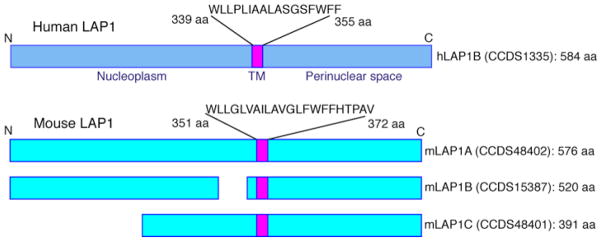
Schematic diagrams showing the deduced structures and membrane topologies of LAP1 isoforms from human and mice. The diagrams have been constructed based on data in the Consensus CDS (CCDS) database (http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi) and the CCDS ID is given for each protein shown. For human LAP1, the completed sequence of LAP1B (hLAP1B) is in the database. The other isoforms, which are not shown in the figure, have incomplete cDNA sequences in the database. The regions of hLAP1B located in nucleoplasm and perinuclear space are indicated in blue and the transmembrane segment within the inner nuclear membrane is indicated in violet with its amino acid sequence (single letter codes) shown. For mouse LAP1, completed sequences for all three isoforms (mLAP1A, mLAP1B and mLAP1C) are in the database. Their regions located in the nucleoplasm and perinuclear space are shown in light blue and transmembrane segment in violet. A number with aa following it indicates the amino acid residue in the protein isoform and the total number of amino acids in each isoform is indicated at the far left.
Analysis of LAP1 mRNA and protein in mouse embryo and adult tissues demonstrates LAP1 isoforms to have an essentially ubiquitous expression pattern [18,23–25]. Similar to the monoclonal antibody originally raised against LAP1 [18], a rabbit polyclonal antibody generated against 16 peptides of the carboxyl-terminus recognizes three LAP1 isoforms in various mouse tissues but also recognizes a non-specific band of approximately 65 kDa [23, 24]. This rabbit polyclonal antibody also recognizes three isoforms of human LAP1 [26]. The expression of different isoforms is differentially regulated in different tissues and during developmental stages. The small LAP1C isoform is abundant in cell lines and mouse embryos but the longer LAP1A and LAP1B isoforms are abundant in differentiated adult tissues [18,24–26].
2.2. Interacting proteins in addition to lamins
2.2.1. Torsins
After its identification as a lamina-associated protein, LAP1 was shown to interact with torsinA [23]. A mutation in the gene TOR1A encoding torsinA that removes a single glutamic acid causes DYT1 dystonia, a dominantly inherited central nervous system-selective disease characterized by prolonged involuntary twisting movements [27]. TorsinA is a member of a family of AAA+ (ATPase associated with different cellular activities) proteins localized to the lumen of the endoplasmic reticulum. However, the pathogenic torsinA variant preferentially concentrates in the perinuclear space [28,29], which is continuous with the endoplasmic reticulum lumen. Using a cell-based screen, Goodchild and Dauer [23] identified LAP1 as a torsinA-interacting protein in the nuclear envelope. This screen also identified an endoplasmic reticulum-localized torsinA-interacting protein with a luminal domain highly homologous to that of LAP1 and termed luminal domain like LAP1 (LULL1). Differences in the amino-terminal regions of LAP1 and LULL1 appear to determine whether they concentrate in the inner nuclear membrane or main endoplasmic reticulum membrane system [30]. TorsinA variants with decreased ATPase activity bind more efficiently to LAP1 [23,30]. In addition to torsinA, the mammalian torsin family members torsinB, torsin2 and torsin3 also bind to LAP1 [24]. Kim et al. [24] demonstrated differences in the tissue distribution of different torsin family members that appear responsible for the neural-selective nuclear membrane abnormalities characteristic in torsinA null mice. TorsinA is expressed at greater levels in neural tissues compared to non-neural tissues, whereas a strikingly opposite pattern is observed for torsinB. The expression pattern of torsin3a is similar to torsinB, whereas torsin2a is expressed at similar levels in all tissues examined.
2.2.2. Emerin
Using an unbiased screen to identify proteins in cellular extracts that bind to LAP1, Shin et al. [26] identified emerin. The nucleoplasmic domains of these transmembrane proteins of the inner nuclear membrane likely mediate their interaction. In a subset of LAP1 null fibroblasts examined by immunofluorescence microscopy, emerin mislocalized to distinct foci along the nuclear envelope, rather than in a smooth rim-like pattern. A-type lamins co-localized with emerin in these foci. In contrast, the sub-cellular distribution of the integral inner nuclear membrane proteins LBR and LAP2beta, a nuclear pore complex protein Nup98 and lamin B1 were normal in the LAP1 null cells. These results show that LAP1 is required for normal localization of a population of emerin and A-type lamin complexes within the inner nuclear membrane and highlight the specificity of these effects.
2.2.3. The LAP1 complex in the nuclear envelope
The protein interaction data for LAP1 suggest that it can exist as a complex in the nuclear envelope with A-type lamins, torsins and emerin (see Figure 1). The existence of such a complex suggests that LAP1 may cooperate functionally with these proteins in tissues where they play a critical role. The differential expression of the components of this complex in different tissues may lead to changes in the complex’s composition and function. For example, torsinA is expressed at relatively higher levels in neuronal tissues whereas torsinB and torsin3 are expressed at higher levels in non-neuronal tissues [24,31].
2.3 Functional interactions of LAP1
2.3.1. TorsinA
Deletion of torsinA from mice or mice homozygous for the DYT1 dystonia-causing mutation exhibit striking morphological abnormalities of neuronal nuclear membranes (termed nuclear envelope “blebs”), whereas nuclear envelope morphology is normal in all non-neuronal cell types [25]. To test whether LAP1 participates in this torsinA-related pathology and examine its potential role in the neuronal specific nuclear envelope defects, Kim et al. [24] generated mice with a germline deletion of the gene encoding LAP1 (named Tor1aip1). The nuclear envelopes of neural tissue or cultured neurons from these mice exhibited morphological abnormalities that appear similar to those in torsinA mutant mice. Three-dimensional electron microscopic tomographic reconstructions of the neuronal nuclear envelopes demonstrated that the nuclear membrane alterations are identical in torsinA and LAP1 null mice. The fact that LAP1 null mice phenocopy the perinatal lethality and neuronal nuclear envelope abnormalities seen in torsinA knockout mutant mice suggests strongly that they functionally interact in neurons.
In contrast to the neural specific abnormalities of torsinA mutant mice, LAP1 null mice exhibited nuclear envelope blebs in all cell and tissue types examined. Moreover, while the nuclear envelopes of torsinA null fibroblasts appears normal, reducing LAP1 levels by 50% on a torsinA null background leads to nuclear envelope blebs in fibroblasts [24]. These findings further support a close functional relationship between torsinA and LAP1. Knockdown of torsinB from fibroblasts lacking torsinA also leads to abnormal nuclear envelope morphology, suggesting that torsinA and torsinB share redundant functions in conjunction with LAP1.
2.3.2. Emerin
Loss of function of emerin in humans leads to X-linked Emery-Dreifuss muscular dystrophy and phenotypically similar myopathies [7,32,33]. These diseases are characterized by dilated cardiomyopathy and skeletal muscular dystrophy. The interaction of LAP1 with emerin prompted Shin et al. [26] to conditionally delete LAP1 from striated muscle of mice. These mice developed profound muscular dystrophy and cardiomyopathy leading to early lethality. In contrast, deletion of LAP1 from hepatocytes caused some alterations in nuclear size, but did not affect lifespan or overtly alter liver function. These findings suggested that LAP1 plays an essential role in striated muscle maintenance, perhaps in conjunction with emerin.
Whereas emerin loss in humans causes myopathy and cardiomyopathy, mice lacking emerin are for the most part normal with only very subtle muscle abnormalities [34,35]. Analysis of LAP1 and emerin in skeletal muscle revealed discordant expression between these species that may explain this phenomenon. Compared to mouse tissue, human skeletal muscle has significantly more emerin and relatively little LAP1 [26]. Strikingly, depletion of LAP1 from muscle of emerin null mice leads to myopathy that is much more severe than that resulting from loss of LAP1 alone, with the double knockout mice demonstrating significantly more pronounced pathology and shorter lifespans [26]. Additionally, in emerin null mice, reducing LAP1 levels by approximately 50% (similar to levels in human tissue), causes evidence of muscle damage. LAP1 and emerin therefore appear to function together in skeletal muscle (a similarly analysis has yet to be done in heart). In mice, the relative overabundance of LAP1 may compensate for emerin loss of function, and explain the lack of phenotype in emerin null mice. The LAP1-emerin complex may therefore be analogous to the utrophin-dystrophin complex in striated muscle [36,37]. In humans, loss of dystrophin expression leads to Duchenne muscular dystrophy, a severe and lethal myopathy [38, 39]. In contrast, mice lacking dystrophin develop minimal muscle pathology. However, depletion of the dystrophin-associated protein utrophin causes a profound myopathy [36,37]. These findings have led to efforts to ameliorate Duchenne muscular dystrophy by increasing levels of utrophin [40] and the related strategy of increasing LAP1 levels may benefit patients with X-linked Emery-Dreifuss muscular dystrophy.
3. LAP1 and its potential role in tissue-selective human disease
The discoveries that LAP1 functionally interacts with torsinA and emerin suggest that it may play a role in tissue-selective human diseases caused by mutations in the gene encoding these proteins. One possibility is that LAP1 could be a modifier protein in DYT1 dystonia or muscular dystrophy/cardiomyopathy caused by loss of emerin. While this is currently an untested hypothesis, the existence of modifier genes is suggested by the clinical data. DYT1 dystonia caused by a unique mutation in the gene encoding torsinA has only penetrance of approximately 30% and the symptoms are of variable clinical severity in different patients [41]. Genetic background also affects the phenotype of mice carrying the disease-causing torsinA variant [42]. In the case of emerin loss of function, the disease severity is also highly variable [43,44] and, while the usual phenotype is that of Emery-Dreifuss muscular dystrophy, some patients present with a limb-girdle involvement or minimal skeletal muscle disease and early-onset cardiomyopathy [32,33,45–47]. Based on the interactions of LAP1 with torsinA and emerin, it is logical to hypothesize that polymorphisms in its human gene (TOR1AIP1) alter its level of expression or function and act as a modifier in these patients.
Mutation in the gene encoding LAP1 may also cause disease. In a presentation at the 18th International World Muscle Society Congress in October 2013, three affected members of a Turkish family with an autosomal recessive limb-girdle muscular dystrophy with joint contractures were reported to have a mutation in the gene encoding LAP1 that generates a premature stop codon [48]. Screening for mutations in the gene encoding LAP1 in patients with muscular dystrophy, cardiomyopathy and/or dystonia may therefore identify new laminopathies/nuclear envelopathies.
4. Conclusions
LAP1 was identified in 1988 as an integral protein of the inner nuclear membrane associated with the nuclear lamina but its function remained elusive. Subsequent protein interaction studies begun about 20 years after its discovery revealed that LAP1 binds to torsinA and emerin. These findings led to experiments in model mice and cellular systems that showed LAP1 functions with torsinA in neurons and with emerin in muscle. As such, the gene encoding LAP1 is now a logical candidate as a modifier in dystonia and muscular dystrophy and may be a disease-causing gene in its own right, linking additional human diseases to nuclear envelope dysfunction.
Highlights.
LAP1 is a ubiquitously-expressed inner nuclear membrane protein that interacts with lamins, torsins and emerin in the nuclear envelope
LAP1 functions in striated muscle maintenance
Polymorphisms in the LAP1 gene may modulate the phenotypes of DYT1 dystonia and Emery-Dreifuss muscular dystrophy and mutations in the gene may cause muscular dystrophy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Soullam B, Worman HJ. Signals and structural features involved in integral membrane protein targeting to the inner nuclear membrane. J Cell Biol. 1995;130:15–27. doi: 10.1083/jcb.130.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmer L, Worman HJ. Inner nuclear membrane proteins: functions and targeting. Cell Mol Life Sci. 2001;58:1741–7. doi: 10.1007/PL00000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King MC, Lusk CP, Blobel G. Karyopherin-mediated import of integral inner nuclear membrane proteins. Nature. 2006;442:1003–7. doi: 10.1038/nature05075. [DOI] [PubMed] [Google Scholar]
- 4.Lusk CP, Blobel G, King MC. Highway to the inner nuclear membrane: rules for the road. Nat Rev Mol Cell Biol. 2007;8:414–20. doi: 10.1038/nrm2165. [DOI] [PubMed] [Google Scholar]
- 5.Schirmer EC, Florens L, Guan T, Yates JR, 3rd, Gerace L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 2003;301:1380–2. doi: 10.1126/science.1088176. [DOI] [PubMed] [Google Scholar]
- 6.de Las Heras JI, Meinke P, Batrakou DG, Srsen V, Zuleger N, Kerr AR, Schirmer EC. Tissue specificity in the nuclear envelope supports its functional complexity. Nucleus. 2013 doi: 10.4161/nucl.26872. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–7. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 8.Nagano A, Koga R, Ogawa M, Kurano Y, Kawada J, Okada R, Hayashi YK, Tsukahara T, Arahata K. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat Genet. 1996;12:254–9. doi: 10.1038/ng0396-254. [DOI] [PubMed] [Google Scholar]
- 9.Manilal S, Nguyen TM, Sewry CA, Morris GE. The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet. 1996;5:801–8. doi: 10.1093/hmg/5.6.801. [DOI] [PubMed] [Google Scholar]
- 10.Yates JR, Wehnert M. The Emery-Dreifuss muscular dystrophy mutation database. Neuromuscul Disord. 1999;9:199. [PubMed] [Google Scholar]
- 11.Worman HJ, Bonne G. “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. 2007;313:2121–33. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell. 2009;17:626–38. doi: 10.1016/j.devcel.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 13.Bonne G, Di Barletta MR, Varnous S, Bécane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–8. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 14.Worman HJ, Evans CD, Blobel G. The lamin B receptor of the nuclear envelope inner membrane: a polytopic protein with eight potential transmembrane domains. J Cell Biol. 1990;111:1535–42. doi: 10.1083/jcb.111.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Müller D, Vayá A, Aznar J, Ware RE, Sotelo Cruz N, Lindner TH, Herrmann H, Reis A, Sperling K. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huët anomaly) Nat Genet. 2002;31:410–4. doi: 10.1038/ng925. [DOI] [PubMed] [Google Scholar]
- 16.Lin F, Blake DL, Callebaut I, Skerjanc IS, Holmer L, McBurney MW, Paulin-Levasseur M, Worman HJ. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J Biol Chem. 2000;275:4840–7. doi: 10.1074/jbc.275.7.4840. [DOI] [PubMed] [Google Scholar]
- 17.Hellemans J, Preobrazhenska O, Willaert A, Debeer P, Verdonk PC, Costa T, Janssens K, Menten B, Van Roy N, Vermeulen SJ, Savarirayan R, Van Hul W, Vanhoenacker F, Huylebroeck D, De Paepe A, Naeyaert JM, Vandesompele J, Speleman F, Verschueren K, Coucke PJ, Mortier GR. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet. 2004;36:1213–8. doi: 10.1038/ng1453. [DOI] [PubMed] [Google Scholar]
- 18.Senior A, Gerace L. Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J Cell Biol. 1988;107:2029–36. doi: 10.1083/jcb.107.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foisner R, Gerace L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell. 1993;73:1267–79. doi: 10.1016/0092-8674(93)90355-t. [DOI] [PubMed] [Google Scholar]
- 20.Martin L, Crimaudo C, Gerace L. cDNA cloning and characterization of lamina-associated polypeptide 1C (LAP1C), an integral protein of the inner nuclear membrane. J Biol Chem. 1995;270:8822–8. doi: 10.1074/jbc.270.15.8822. [DOI] [PubMed] [Google Scholar]
- 21.Kondo Y, Kondoh J, Hayashi D, Ban T, Takagi M, Kamei Y, Tsuji L, Kim J, Yoneda Y. Molecular cloning of one isotype of human lamina-associated polypeptide 1s and a topological analysis using its deletion mutants. Biochem Biophys Res Commun. 2002;294:770–8. doi: 10.1016/S0006-291X(02)00563-6. [DOI] [PubMed] [Google Scholar]
- 22.Santos M, Rebelo S, Van Kleeff PJ, Kim CE, Dauer WT, Fardilha M, da Cruz E Silva OA, da Cruz E Silva EF. The nuclear envelope protein, lap1b, is a novel protein phosphatase 1 substrate. PLoS One. 2013;8:e76788. doi: 10.1371/journal.pone.0076788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodchild RE, Dauer WT. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol. 2005;168:855–62. doi: 10.1083/jcb.200411026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim CE, Perez A, Perkins G, Ellisman MH, Dauer WT. A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc Natl Acad Sci USA. 2010;107:9861–6. doi: 10.1073/pnas.0912877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–32. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Shin JY, Méndez-López I, Wang Y, Hays AP, Tanji K, Lefkowitch JH, Schulze PC, Worman HJ, Dauer WT. Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev Cell. 2013;26:591–603. doi: 10.1016/j.devcel.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–8. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 28.Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA. 2004;101:847–52. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Alegre P, Paulson HL. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci. 2004;24:2593–601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naismith TV, Dalal S, Hanson PI. Interaction of torsinA with its major binding partners is impaired by the dystonia-associated deltaGAG deletion. J Biol Chem. 2009;284:27866–74. doi: 10.1074/jbc.M109.020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jungwirth M, Dear ML, Brown P, Holbrook K, Goodchild R. Relative tissue expression of homologous torsinB correlates with the neuronal specific importance of DYT1 dystonia-associated torsinA. Hum Mol Genet. 2010;19:888–900. doi: 10.1093/hmg/ddp557. [DOI] [PubMed] [Google Scholar]
- 32.Talkop UA, Talvik I, Sõnajalg M, Sibul H, Kolk A, Piirsoo A, Warzok R, Wulff K, Wehnert MS, Talvik T. Early onset of cardiomyopathy in two brothers with X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 2002;12:878–81. doi: 10.1016/s0960-8966(02)00134-7. [DOI] [PubMed] [Google Scholar]
- 33.Astejada MN, Goto K, Nagano A, Ura S, Noguchi S, Nonaka I, Nishino I, Hayashi YK. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol. 2007;26:159–64. [PMC free article] [PubMed] [Google Scholar]
- 34.Melcon G, Kozlov S, Cutler DA, Sullivan T, Hernandez L, Zhao P, Mitchell S, Nader G, Bakay M, Rottman JN, Hoffman EP, Stewart CL. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet. 2006;15:637–51. doi: 10.1093/hmg/ddi479. [DOI] [PubMed] [Google Scholar]
- 35.Ozawa R, Hayashi YK, Ogawa M, Kurokawa R, Matsumoto H, Noguchi S, Nonaka I, Nishino I. Emerin-lacking mice show minimal motor and cardiac dysfunctions with nuclear-associated vacuoles. Am J Pathol. 2006;168:907–17. doi: 10.2353/ajpath.2006.050564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–27. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 37.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–38. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 38.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 39.Hoffman EP, Knudson CM, Campbell KP, Kunkel LM. Subcellular fractionation of dystrophin to the triads of skeletal muscle. Nature. 1987;330:754–8. doi: 10.1038/330754a0. [DOI] [PubMed] [Google Scholar]
- 40.Ruegg UT. Pharmacological prospects in the treatment of Duchenne muscular dystrophy. Curr Opin Neurol. 2013;26:577–84. doi: 10.1097/WCO.0b013e328364fbaf. [DOI] [PubMed] [Google Scholar]
- 41.Fahn S. The genetics of idiopathic torsion dystonia. Int J Neurol. 1991–1992;25–26:70–80. [PubMed] [Google Scholar]
- 42.Tanabe LM, Martin C, Dauer WT. Genetic background modulates the phenotype of a mouse model of DYT1 dystonia. PLoS One. 2012;7:e32245. doi: 10.1371/journal.pone.0032245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ben Yaou R, Toutain A, Arimura T, Demay L, Massart C, Peccate C, Muchir A, Llense S, Deburgrave N, Leturcq F, Litim KE, Rahmoun-Chiali N, Richard P, Babuty D, Recan-Budiartha D, Bonne G. Multitissular involvement in a family with LMNA and EMD mutations: Role of digenic mechanism? Neurology. 2007;68:1883–94. doi: 10.1212/01.wnl.0000263138.57257.6a. [DOI] [PubMed] [Google Scholar]
- 44.Muntoni F, Bonne G, Goldfarb LG, Mercuri E, Piercy RJ, Burke M, Yaou RB, Richard P, Récan D, Shatunov A, Sewry CA, Brown SC. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain. 2006;129:1260–1268. doi: 10.1093/brain/awl062. [DOI] [PubMed] [Google Scholar]
- 45.Sakata K, Shimizu M, Ino H, Yamaguchi M, Terai H, Fujino N, Hayashi K, Kaneda T, Inoue M, Oda Y, Fujita T, Kaku B, Kanaya H, Mabuchi H. High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation. 2005;111:3353–3358. doi: 10.1161/CIRCULATIONAHA.104.527184. [DOI] [PubMed] [Google Scholar]
- 46.Ura S, Hayashi YK, Goto K, Astejada MN, Murakami T, Nagato M, Ohta S, Daimon Y, Takekawa H, Hirata K, Nonaka I, Noguchi S, Nishino I. Limb-girdle muscular dystrophy due to emerin gene mutations. Arch Neurol. 2007;64:1038–1041. doi: 10.1001/archneur.64.7.1038. [DOI] [PubMed] [Google Scholar]
- 47.Karst ML, Herron KJ, Olson TM. X-linked nonsyndromic sinus node dysfunction and atrial fibrillation caused by emerin mutation. J Cardiovasc Electrophysiol. 2008;19:510–515. doi: 10.1111/j.1540-8167.2007.01081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kayman-Kurekci G, Talim B, Korkusuz P, Sayar N, Sarioglu T, Oncel I, Sharafi P, Gundesli H, Balci-Hayta B, Purali N, Serdaroglu P, Topaloglu H, Dincer P. TorsinA-interacting protein1 /lamina-associated polypeptide 1B in a form of limb girdle muscular dystrophy: a novel gene related to nuclear envelopathies. Late Breaking News Abstracts 18th International Congress of the World Muscle Society; 1–5 October 2013; Asilomar Conference Grounds, California, USA. [Google Scholar]