

Abstract
Heterozygous germline mutations in PHOX2B, a transcriptional regulator of sympathetic neuronal differentiation, predispose to diseases of the sympathetic nervous system, including neuroblastoma and congenital central hypoventilation syndrome (CCHS). Whereas the PHOX2B variants in CCHS largely involve expansions of the second polyalanine repeat within the C-terminus of the protein, those associated with neuroblastic tumors are nearly always frameshift and truncation mutations. To test the hypothesis that the neuroblastoma-associated variants exert their effects through loss or gain of protein-protein interactions, we performed a large-scale yeast two-hybrid screen using both wild-type (WT) and six different mutant PHOX2B proteins against over 10,000 human genes. The neuronal calcium sensor protein HPCAL1 (VILIP-3) exhibited strong binding to WT PHOX2B and a CCHS-associated polyalanine expansion mutant, but only weakly or not at all to neuroblastoma-associated frameshift and truncation variants. We demonstrate that both WT PHOX2B, the neuroblastoma-associated R100L missense and the CCHS-associated alanine expansion variants induce nuclear translocation of HPCAL1 in a Ca2+-independent manner, while the neuroblastoma-associated 676delG frameshift and K155X truncation mutants impair subcellular localization of HPCAL1, causing it to remain in the cytoplasm. HPCAL1 did not appreciably influence the ability of WT PHOX2B to transactivate the DBH promoter, nor did it alter the decreased transactivation potential of PHOX2B variants in 293T cells. Abrogation of the PHOX2B-HPCAL1 interaction by shRNA knockdown of HPCAL1 in neuroblastoma cells expressing PHOX2B led to impaired neurite outgrowth with transcriptional profiles indicative of inhibited sympathetic neuronal differentiation. Our results suggest that certain PHOX2B variants associated with neuroblastoma pathogenesis, because of their inability to bind to key interacting proteins such as HPCAL1, may predispose to this malignancy by impeding the differentiation of immature sympathetic neurons.
Keywords: PHOX2B, HPCAL1, neuroblastoma, neuronal calcium sensor, differentiation, network perturbation
INTRODUCTION
Neuroblastomas are embryonal tumors of the peripheral sympathetic nervous system that typically arise in the sympathetic ganglia and adrenal glands. Although these tumors are primarily sporadic, a genetic predisposition is seen in about 1% of cases (1). Germline mutations in the anaplastic lymphoma kinase receptor ALK, and in the homeodomain transcription factor PHOX2B, account for the majority of familial neuroblastoma cases (1). Their clinical presentation varies widely, from a symptomless mass to disseminated disease that often becomes resistant to all available therapies. The tumor cells are characterized by a spectrum of differentiation, ranging from undifferentiated, immature cells to partially differentiated cells that have begun the transition to sympathetic neurons. This feature has led to the hypothesis that these tumors develop because of perturbations in genes affecting neuronal differentiation (2).
PHOX2B is thought to function as a master regulator of sympathetic neuron development (3). Indeed, loss of PHOX2B in mice leads to failure of formation of the sympathetic ganglia and the absence of cells expressing tyrosine hydroxylase (TH) and dopamine beta-hydroxylase (DBH), key enzymes in catecholamine biosynthesis (4) and markers of terminal differentiation of sympathetic neurons (5). Similarly, overexpression of PHOX2B promotes the differentiation of avian neural crest and human neuroblastoma cells, the latter in the presence of retinoic acid (6, 7). Heterozygous germline mutations in PHOX2B predispose to neuroblastoma and other neurocristopathies, such as congenital central hypoventilation syndrome (CCHS) and Hirschprung’s disease (HSCR) (8–11), characterized by absent or reduced autonomic innervation in the brain and intestine, respectively.
The PHOX2B gene encodes a 314-amino-acid protein that includes a homeodomain DNA-binding region and two polyalanine repeats of 9 and 20 alanines within the C-terminal end. Mutations in the majority of CCHS patients are in-frame expansions (from +5 to +13 alanine residues) of the second polyalanine repeat (polyalanine repeat expansion mutations, PARM) (8), whereas those associated with tumors of neural crest origin tend to be (i) missense alterations in the homeodomain, (ii) insertions or deletions within the third exon (C-terminus) that alter the reading frame of the gene or (iii) nonsense mutations leading to a truncated protein that lacks the C-terminus (collectively called non-PARMs) (8–13). A detailed understanding of how mutant alleles of the same gene predispose to different diseases has been difficult to acquire. Several studies have shown that the neuroblastoma-associated PHOX2B variants can increase the proliferation of immature sympathetic neurons and inhibit their differentiation both in vitro and in vivo (7, 14–16). However, even among the spectrum of neuroblastoma-associated PHOX2B variants, it is not clear how missense mutations within the homeodomain versus frame-shift and truncation mutations that alter the C-terminus of the protein can predispose to the development of neuroblastoma.
It is well established that proteins do not function in isolation, but are typically associated with macromolecular complexes and signaling pathways that are part of highly interconnected interaction networks (17). Thus, disease-causing mutations in protein-coding regions can lead to either total loss of protein expression or to the expression of mutant proteins that alter one or a few specific interactions (18–20). Aberrant protein-protein interactions represent a largely unexplored mechanism by which diverse PHOX2B mutations could give rise to different disease phenotypes. Indeed, perturbations of the underlying, wild-type (WT) PHOX2B protein-protein interaction network may explain disease phenotypes that cannot be attributed to mutations that directly impact the transactivation of key target genes. Although three WT PHOX2B-interacting proteins, all transcription factors or coactivators, have been identified to date (14, 21, 22), such studies have not uncovered altered protein-protein interactions involving PHOX2B variants.
We therefore considered that PHOX2B mutations may exert their tumorigenic effects by preventing or modifying the binding of interacting proteins or perhaps by recruiting novel interactors. To test this hypothesis, we performed a high-throughput yeast two-hybrid screen using both WT PHOX2B and representative examples of both neuroblastoma- and CCHS-associated variants. This search identified a neuronal calcium sensor protein, HPCAL1 (alternatively known as VILIP-3) that interacts with WT and CCHS-associated variant PHOX2B proteins, but only weakly or not at all with the neuroblastoma-associated frameshift and truncation variants. We demonstrate that PHOX2B binding with HPCAL1 is important for the nuclear transport of the calcium sensor and for mediating the effects of PHOX2B on sympathetic neuronal differentiation. These findings implicate mutational disruption of the PHOX2B-HPCAL1 interaction as a contributing factor to neuroblastoma predisposition.
RESULTS
The neuronal calcium sensor HPCAL1 is a binding partner of PHOX2B
To identify proteins that interact with PHOX2B, we performed a large-scale high-throughput ORFeome-based yeast two-hybrid (Y2H) screen, using the full-length WT PHOX2B and 6 different PHOX2B variants (Figure 1a). These included neuroblastoma-associated variants with missense mutations within the homeodomain region (R100L and R141G) (11), truncation of the entire third exon (K155X) (8), and frameshift mutations within the third exon (721del20 and 676delG) (10, 23), as well as a +7 alanine repeat expansion mutation associated with CCHS (AlaExp) (24). The WT and mutant PHOX2B cDNAs were cloned into Gateway-compatible Y2H destination vectors to generate GAL4 DNA-binding domain (DB) and activation domain (AD) fusion proteins (25). These clones were screened against the human ORFeome v.3.1 library, which contains approximately 12,000 open reading frames (ORFs) encompassing more than 50% of the ORFs encoded in the human genome (26). The DB-PHOX2B (WT) and each of the DB-PHOX2B variant-transformed yeast cells were screened against the AD-ORFeome using a mating-based Y2H assay in a 96-well format (25). In a parallel screen, the reciprocal interactions between AD-tagged PHOX2B preys (WT and each of the mutants) and the DB-tagged ORFeome baits were tested, resulting in 270 potential Y2H hits that were verified through two additional rounds of pairwise mating in yeast cells. Among the verified interactors was the EF-hand neuronal calcium sensor protein, HPCAL1, which specifically bound to WT PHOX2B but only weakly to the 721del20 and 676delG frameshift mutants and not at all to the K155X truncation variant (Figure 1b). Interestingly, the homeodomain (HD) missense mutants R100L and R141G retained their binding capacity for HPCAL1, although to a reduced degree; while the CCHS-associated PHOX2B alanine expansion variant (AlaExp) showed the same robust binding as WT PHOX2B. Thus, the PHOX2B mutant proteins with the weakest capability to bind to HPCAL1 (K155X, 721del20 and 676delG) were those with significantly altered proximal C-terminal regions, caused by mutations present before the second polyalanine stretch (Figure 1a).
Figure 1.
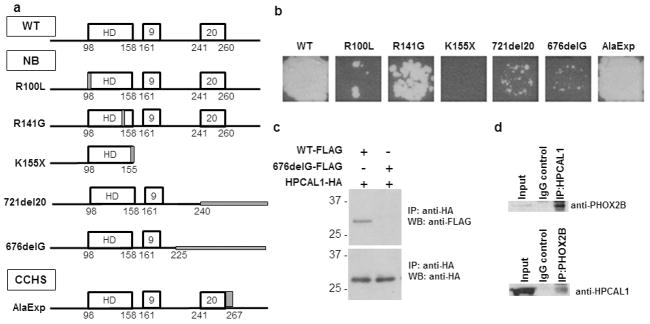
Neuroblastoma-associated PHOX2B variants exhibit impaired binding to HPCAL1. (a) Schema of wild-type (WT) and variant PHOX2B proteins used in the yeast two-hybrid (Y2H) screen. All but the AlaExp mutant (CCHS) are neuroblastoma-associated. The 721del20 variant was identified in the SK-N-SH cell line. Locations of the mutations are marked in gray. (b) Y2H analysis of interactions between WT and mutant PHOX2B proteins with HPCAL1 as determined by growth on histidine-negative media. (c) Verification of protein interactions by overexpression of FLAG-tagged WT (WT-FLAG) and mutant (676delG-FLAG) PHOX2B and HA-tagged HPCAL1 in 293T cells, followed by immunoprecipitation of the cell extracts with anti-HA antibody and immunoblotting with anti-FLAG and anti-HA antibodies. (d) Interaction between endogenously expressed PHOX2B and HPCAL1 in neuroblastoma cells. NGP neuroblastoma cell extracts were immunoprecipitated with HPCAL1 antibodies and immunoblotted with anti-PHOX2B antibodies (upper panel) and vice versa (lower panel).
To extend the verified Y2H screening results in mammalian cells, we overexpressed HA-tagged versions of HPCAL1 together with FLAG-tagged WT PHOX2B and a representative variant, 676delG, in 293T cells (which express neither PHOX2B nor the interacting proteins) and examined their binding potential through coimmunoprecipitation (co-IP) assays. Our results revealed that WT PHOX2B immunoprecipitated with HPCAL1, suggesting binding of this protein in these cells. The 676delG frameshift PHOX2B variant did not interact with HPCAL1, verifying the results of our Y2H screen (Figure 1c). To ensure that the interaction between WT PHOX2B and HPCAL1 was also present in human neuroblastoma cells, we performed co-IP experiments on cell lysates from NGP cells that endogenously express both proteins. Again, we noted that endogenously expressed PHOX2B formed a complex with HPCAL1 in these cells (Figure 1d). Together, these results show that HPCAL1 is a bona fide interactor of WT PHOX2B in neuroblastoma cells and that this interaction is lost or impaired with certain PHOX2B variants.
HPCAL1 is translocated to the nucleus upon binding to PHOX2B
We next determined whether the interaction between WT PHOX2B and HPCAL1 affects the cellular localization of the two proteins (Figure 2; Supplementary Figure S1). As with other classical transcription factors, WT PHOX2B is localized exclusively to the nucleus as determined by expression of a GFP-tagged WT PHOX2B construct in HeLa cells lacking endogenous PHOX2B (Figure 2a). We also determined the subcellular localization of HPCAL1 by expressing this protein fused to the mCherry fluorescent marker in HeLa cells, which also do not endogenously express this gene (RT-PCR analysis, data not shown). mCherry-HPCAL1 localized primarily to the cytoplasm (Figure 2a); but when coexpressed with GFP-tagged WT PHOX2B, its expression was more prominent in the nucleus, suggesting nuclear localization (Figure 2b). This nuclear translocation was not apparent when either the K155X truncation or the 676delG frameshift variants were coexpressed with mCherry-HPCAL1 (Figure 2b and Supplementary Figure S1). By contrast, overexpression of the R100L missense and the CCHS-associated AlaExp mutant allele led to nuclear localization of HPCAL1, similar to results with the WT allele (Figure 2b and Supplementary Figure S1). These data not only confirm the binding of WT PHOX2B to HPCAL1 and the altered binding of distinct neuroblastoma-associated PHOX2B mutants seen in our Y2H and co-IP assays, but also suggest that HPCAL1 may regulate PHOX2B function in the nucleus.
Figure 2.

The PHOX2B-HPCAL1 interaction results in nuclear translocation of HPCAL1. (a) GFP-tagged WT PHOX2B and mCherry-tagged HPCAL1 were transfected into HeLa cells. 48 hours later, the cells were fixed in 4% formaldehyde in PBS and mounted on ProLong Gold antifade reagent with DAPI (Invitrogen). Images were taken on a Nikon Eclipse Ti confocal microscope (Melville, NY) and processed with ImageJ software (40X magnification). (b) GFP-tagged WT and the indicated mutant GFP-tagged PHOX2B genes were cotransfected with HPCAL1-mCherry and imaged as in panel (a).
Interaction of PHOX2B with HPCAL1 is not calcium dependent
A subclass of EF-hand-containing calcium-binding proteins that includes HPCAL1 undergo post-translational N-terminal myristoylation. These proteins respond to elevations in cellular calcium (Ca2+) levels by exposing their myristoyl group to the aqueous environment, a conformational change that induces their translocation to specific membrane domains (27). To test whether the interaction between WT PHOX2B and HPCAL1 is Ca2+-dependent, we exposed HeLa cells expressing PHOX2B and HPCAL1 to either the Ca2+ chelator BAPTA-AM or calcium chloride (CaCl2) for 48 hours, to decrease or increase the intracellular Ca2+ concentrations, respectively. We observed that the PHOX2B-HPCAL1 interaction, as measured by co-IP, was not affected by changes in intracellular Ca2+, with robust binding at physiological, increased or decreased Ca2+ levels (Figure 3a). Next, we determined whether the nuclear translocation of HPCAL1 complexed with PHOX2B, depends on Ca2+ concentrations. Compared to untreated cells, the subcellular localization of the WT PHOX2B-HPCAL1 complex was not affected by changes in Ca2+ levels (Figure 3b). Moreover, varying the Ca2+ level did not alter the impaired nuclear translocation of HPCAL1 seen with the 676delG variant (Figure 3b). Thus, the interaction between WT PHOX2B and HPCAL1 is independent of the cellular Ca2+ concentration.
Figure 3.

The PHOX2B-HPCAL1 interaction is not calcium dependent. (a) PHOX2B-WT-FLAG and HPCAL1-HA constructs were transfected into 293T cells and harvested 48 hours later. Either BAPTA-AM [1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester)] (3 μM) or CaCl2 (2 μM) was added to the cell lysates at the indicated concentrations, immunoprecipitated with anti-HA antibodies and analyzed by western blotting with anti-HA and anti-FLAG antibodies. (b) HPCAL1-mCherry and PHOX2B-GFP (both WT and the 676delG mutant) constructs were transiently transfected into HeLa cells and either BAPTA-AM or CaCl2 was added to the media at the indicated concentrations. Cells were fixed and imaged 2 days after transfection as described for Figure 2.
The transactivation potential of PHOX2B is not altered by interaction with HPCAL1 in 293T cells
PHOX2B directly transactivates its own promoter as well as that of other transcriptional targets, such as the dopamine β-hydroxylase (DBH) gene (28, 29). All of the PHOX2B variants described to date have reduced transactivation potential (7, 14, 30). We therefore asked whether the disruption of HPCAL1 binding to PHOX2B might affect the latter protein’s ability to activate the regulatory regions of DBH. Using a dual-luciferase reporter assay, we analyzed the effect of ectopic expression of WT and mutant PHOX2B on the DBH promoter in the presence or absence of HPCAL1 overexpression (Supplementary Figure S2). Compared to WT PHOX2B, all of the PHOX2B variants demonstrated impaired transactivation of the DBH promoter, with K155X, 676delG and 721del20 showing the greatest deficits. Cotransfection with HPCAL1 did not significantly affect the reduced DBH promoter activation seen with the PHOX2B variants (Supplementary Figure S2). The transcription factor HAND2, implicated in autonomic neuron development, is another target of PHOX2B (29). To test the effect of the WT and variant PHOX2B proteins on the HAND2 promoter, we similarly cooverexpressed the 676delG and K155X mutations with the HAND2 regulatory region subcloned upstream of a luciferase reporter. Although compared to WT PHOX2B, these mutations led to decreased transactivation of the HAND2 promoter, there was no significant change induced by the presence of HPCAL1 (data not shown). Thus, overexpression of HPCAL1 does not alter the transactivation potential of WT or PHOX2B mutant proteins.
HPCAL1 binding is required for the differentiation of neuroblastoma cells
PHOX2B exerts strong antiproliferative activity in primary avian sympathetic (14) and human neuroblastoma cells (7). More importantly, it regulates sympathetic neuronal differentiation, its absence causing inhibition of terminal differentiation of sympathetic progenitors (14, 16). To investigate the functional role of the PHOX2B-HPCAL1 interaction in neuroblastoma cells and its role, if any, in cell growth and differentiation, we abrogated HPCAL1 expression in NGP cells using shRNA knockdown and examined the effects on cell growth and differentiation (Figure 4a, upper panel). Compared to findings in control shRNA-expressing cells, a deficit of HPCAL1 did not affect cell growth (Figure 4b). We next determined the effect of HPCAL1 knockdown on cell differentiation. NGP cells normally undergo spontaneous differentiation following incubation in serum-free media for 6 days as determined by increased neurite outgrowth (Figure 4c). When we abolished HPCAL1 expression in these cells, there was no evidence of neurite outgrowth, in fact, they appeared to take on a completely rounded shape (Figure 4c). The lack of neurite outgrowth seen with HPCAL1 knockdown appeared to be much more prominent than that seen with PHOX2B knockdown (Figure 4a, lower panel; Figure 4c). The impaired differentiation on HPCAL1 depletion was also seen in SH-SY5Y and LAN-5 neuroblastoma cells exposed to all-trans retinoic acid (Figure 4d, Supplementary Figure S3). Together, these results suggest that HPCAL1 contributes to spontaneous and exogenously induced differentiation in neuroblastoma cells expressing PHOX2B
Figure 4.

HPCAL1 knockdown inhibits the differentiation of NGP neuroblastoma cells. (a) Western blot analysis of cells in which HPCAL1 (upper panel) and PHOX2B (lower panel) expression was depleted by shRNA knockdown. NGP cells were infected with lentivirus expressing shRNAs against HPCAL1 or PHOX2B and selected in 0.5 μg/ml puromycin to obtain stable cell lines. C, control shRNA. (b) Growth curve of NGP cells in which HPCAL1 expression was knocked down. NGP cells stably expressing HPCAL1-shRNA were seeded into 96-well plates in triplicate, and cell growth analyzed by CellTiter-Glo assay every 2 days. Luminescence readings were normalized to day 1. Values represent the mean ± s.d. of at least 3 independent experiments. (c) Phase contrast micrographs of NGP cells stably expressing HPCAL1 and PHOX2B shRNAs as indicated. Cells were seeded into 6-well plates and incubated for 6 days in serum-free media. The same results were obtained for the additional shRNA KD depicted in (a). Scale bar, 25μm. (d) Phase contrast micrographs of untransfected, control and HPCAL1 shRNA-expressing SH-SY5Y and LAN-5 neuroblastoma cells treated with 10 μM of retinoic acid for 3 days. Scale bar, 25μm.
HPCAL1-PHOX2B interaction is linked to genetic programs mediating neuronal cell differentiation
To elucidate the effects of impaired PHOX2B-HPCAL1 interaction in neuroblastoma cells, we studied the expression profiles of NGP cells in which HPCAL1 expression was abrogated using shRNA knockdown compared with control cells transduced with shRNA against GFP (Figure 5). RNA from three independent biological replicates was processed and hybridized to Affymetrix GeneChip Human Gene 1.0 ST arrays. Supervised analysis revealed 205 probe sets that were differentially expressed on HPCAL1 knockdown, 121 of which were upregulated (corrected P <0.01) (Figure 5a). HPCAL1 was one of the most differentially expressed genes (Figure 5b), confirming adequate knockdown. Overrepresented gene ontology categories (GO) included those involved in the regulation of calcium ion transport, cAMP biosynthetic processes and differentiation of diverse cell types, including T cells, lymphocytes, leukocytes and lung goblet cells (data not shown).
Figure 5.

Depletion of HPCAL1 in neuroblastoma cells expressing WT PHOX2B causes differential regulation of genes involved in sympathetic neuronal differentiation. (a) Heat map representation of differentially regulated genes in NGP cells after HPCAL1 knockdown vs. control shRNA knockdown. (b) Histograms representing the relative expression of the indicated genes that were differentially regulated in cells with HPCAL1 depletion vs. those with intact HPCAL1 expression. Data are reported as means ± s.d. (*P<0.05, **P<0.01, ***P<0.001; n=3 for each subgroup).
Further scrutiny of the genes involved in cellular differentiation revealed significant downregulation of DBH, a major marker of terminal differentiation in sympathetic neuronal and neuroblastoma cells upon HPCAL1 knockdown (Figure 5b). Other downregulated genes included members of the alpha-2-adrenergic receptor family, ADRA2A and ADRA2C, which induce neuronal differentiation in PC12 cells (31). Interestingly, HPCAL1 knockdown also led to upregulation of the neural cell adhesion molecule, CNTN4, whose overexpression has been linked to inhibition of retinoic acid-induced differentiation of neuroblastoma cells (32) (Figure 5b). We also observed involvement of several genes that were present in a recently developed retinoic acid-induced neuroblastoma differentiation signature: namely, CRABP2, EGR1, ST6GAL1, PLEKHA6, MMP11 (upregulated) and SLC25A1, SLC36A1, SLC36A4 (downregulated) (33) (Figure 5b). These data suggest that disruption of the HPCAL1-PHOX2B interaction inhibits differentiation in sympathetic neuronal cells.
DISCUSSION
Using an unbiased yeast two-hybrid screen, we identified a direct interaction between WT PHOX2B and HPCAL1 that was confirmed in mammalian cells engineered to express both proteins and in human neuroblastoma cells endogenously expressing these two proteins. The interaction was absent or impaired in cells expressing the neuroblastoma-associated truncation and frameshift PHOX2B variants, but was retained in those expressing the missense and the CCHS-associated polyalanine expansion mutants. Moreover, HPCAL1 binding to PHOX2B was required for translocation of the calcium sensor protein to the nucleus. We also show that binding with HPCAL1 contributes to neurite outgrowth in neuroblastoma cells expressing PHOX2B, with disruption of the PHOX2B-HPCAL1 interaction leading to impaired differentiation of these cells. Thus, we report a protein-protein interaction that appears essential for the effects of PHOX2B on sympathetic neuron development, and whose loss may contribute to the neuroblastoma predisposition associated with a range of PHOX2B mutations.
Although PHOX2B mutations are seen mainly in hereditary neuroblastoma which accounts for only 1% of cases in this disease, our study provides another example of the newly evolved edgetic perturbation model proposed by Zhong and colleagues (18), in which distinct alleles of a single gene can have different effects on protein networks, thereby giving rise to varied phenotypes. HPCAL1 is one of the five proteins that form the highly conserved visinin-like (VSNL) subfamily of the EF-hand neuronal calcium sensor (NCS) proteins (34). It has a restricted expression pattern in the cerebellum, where it localizes to Purkinje and granule cells (35), as well as in sympathetic ganglia during development (Allen Brain Atlas; http:/www.brain-map.org). VSNL proteins perform specialized functions in membrane trafficking, signal transduction and differentiation in defined subsets of neurons, with each NCS protein having specificity for certain cell types and toward distinct receptors and signaling pathways (36). Importantly, NCS proteins modulate the function of transcription factors: S100B, which is overexpressed in many tumor cells including melanoma (37), interacts with the C-terminus of TP53 to inhibit both tetramerization and protein kinase C-dependent phosphorylation, resulting in its inactivation (38, 39). Similarly, the inability of the truncation and frameshift mutations to bind to HPCAL1 suggests that this region is necessary for the requisite protein-protein interaction. By modifying the C-terminus, these particular mutations may alter the protein folding of PHOX2B, accounting for the differences in protein-protein interactions between these neuroblastoma-associated variants and WT PHOX2B. The missense and polyalanine expansion mutations may not cause similar changes in protein conformation and the binding sites may be preserved. Additionally, the subcellular localization of HPCAL1 correlates with its binding to PHOX2B: both WT PHOX2B and the R100L and the alanine expansion mutants that retained their ability to bind to HPCAL1 caused it to be translocated to the nucleus, whereas HPCAL1 remained in the cytoplasm in the presence of the K155X or 676delG variants. The abrogation of nuclear transport seen with mutant forms of PHOX2B would be expected to have critical effects on PHOX2B function, a prediction that requires further investigation.
The presence of EF-hand calcium-binding motifs, together with a consensus N-terminal myristoylation sequence, enables VSNL proteins to translocate from the cytosolic to subcellular membrane compartments, particularly following elevation of cytosolic Ca2+ concentration (40, 41). By contrast, PHOX2B caused HPCAL1 to translocate to the nucleus in a Ca2+-independent manner, suggesting that the HPCAL1 motif which participates in PHOX2B binding is not affected by Ca2+-induced structural changes. HPCAL1 has been shown to localize to the Golgi in a Ca2+ independent manner (40). Its lower binding affinity for Ca2+ could also play a role in its Ca2+-independent activity (42). Alternatively, these results could also have been influenced by our use of fusion proteins that may have adversely affected Ca2+ affinities (34).
Interaction with HPCAL1 did not appreciably affect the transactivation potential of PHOX2B on either the DBH or HAND2 promoters, nor did the lack of binding with HPCAL1 further influence the already decreased transactivation potential of the neuroblastoma-associated PHOX2B variants in 293T cells. However, our expression microarray data clearly showed that endogenous DBH levels are significantly downregulated when HPCAL1 is depleted in neuroblastoma cells expressing both HPCAL1 and PHOX2B. These seemingly discordant results most likely reflect the cell-type specificity of PHOX2B – the former analysis was performed in non-neuronal human embryonic kidney cells while the latter was done in neuroblastoma cells. The inability of PHOX2A, a paralog of PHOX2B, to effectively stimulate transcription from the DBH promoter in non-neuronal cultures (43) also attests to the tissue specificity of homeobox proteins. Furthermore, the phenotypic expression of genes encoding catecholamine biosynthetic enzymes, such as DBH, is influenced not only by their transcriptional complexes, but also by cell type-specific signals such as growth factors and neurohormones (44), which are not likely to be present in 293T cells.
Many NCS proteins have roles in the differentiation of developing neurons (34). The prototype member of the VSNL family, VILIP-1, was identified in a screen for developmentally regulated proteins that were upregulated during terminal differentiation of the nervous system (45) Moreover, overexpression of VILIP-1 in SH-SY5Y neuroblastoma cells causes an increase in the number and length of neurites (46). We report that PHOX2B-expressing neuroblastoma cells in which HPCAL1 was depleted exhibited markedly decreased neurite outgrowth compared to control shRNA-expressing cells. This observation, together with the significantly downregulated expression of DBH, one of the major markers of terminal differentiation in sympathetic neuronal cells and neuroblastomas (5), suggests that HPCAL1 contributes to the role of PHOX2B in promoting sympathetic neuronal cell differentiation. This conclusion is reinforced by the transcriptional profiles of these cells, which showed differential regulation of genes involved in neuronal differentiation upon depletion of HPCAL1, such as CNTN4 (32).
In conclusion, our study demonstrates that a simple model of “gene-loss” is insufficient to explain complex genotype-phenotype relationships and that allele-specific perturbation of interactions need to be fully understood in the context of protein networks. The observations on the PHOX2B-HPCAL1 interaction described here help to elucidate the phenotypic consequences of PHOX2B mutations leading to the loss of protein-binding affinity and likely subsequent rewiring of the local PHOX2B interaction network as well. Since neuroblastoma tumors consist mainly of sympathetic progenitor cells that are either undifferentiated or poorly differentiated, our data suggest that aberrant interactions with HPCAL1, as seen with diverse PHOX2B mutants, could disrupt the normal differentiation of sympathoadrenal cells, which may predispose to neuroblastoma by increasing susceptibility to secondary transforming events.
Materials and Methods
Cell lines and antibodies
NGP, SH-SY5Y and LAN-5 neuroblastoma cells were maintained in RPMI-1640 supplemented with 10% FBS. 293T (human embryonic kidney, 293T) cells were cultured in DMEM medium with 10% FBS. Sources of antibodies: anti-FLAG antibody (Sigma-Aldrich, St. Louis, MO), mouse anti-HA antibody (Roche Applied Science, Indianapolis, IN), rabbit anti-HA antibody (Bethyl Laboratories, Inc., Montgomery, TX).
Construction of plasmids
PHOX2B mutant constructs were generated with the QuikChange II XL Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA). HPCAL1 cDNA was obtained from CCSB (Center for Cancer Systems Biology, Dana-Farber Cancer Institute, Boston, MA). FLAG and HA fusion constructs were generated by inserting the genes of interest into pcDNA3.1 vectors (Invitrogen, Grand Island, NY) modified with FLAG or HA sequences engineered C-terminal to the multiple cloning sites (MCS), respectively. GFP and mCherry fusion constructs were made by cloning genes of interest into pEGFP-C1 and pmCherry-C1 vectors (BD Biosciences, San Jose, CA), respectively. PHOX2B and its variants were cloned into Y2H destination vectors with GAL4 DNA binding domain (DB) or activation domain (AD) tags using Gateway cloning technology (Invitrogen, Grand Island, NY ).
Yeast two-hybrid screen
A pair-wise mating-based Y2H screen in 96-well high-throughput format was performed to screen yeast cells expressing the AD- or DB-fusion constructs in human ORFeome 3.1, containing ~12,000 open reading frames (ORFs). Briefly, the DB-PHOX2B bait-transformed yeast cells were individually mated with yeast cells expressing human AD-ORF preys; in the reverse screen, the reciprocal pairwise interactions between AD-PHOX2B preys and DB-ORFeome baits were tested. Positive clones from the primary screen were verified through two additional rounds of pairwise retesting in yeast cells to ensure reproducibility and to exclude experimental artifacts. The Y2H phenotypes of candidate Y2H pairs were verified by mating the matching individual MATα Y8930 DB-X yeast strains and MATa Y8800 AD-Y yeast strains on YEPD media. Growth of the diploid cells on synthetic media lacking histidine (-His) indicated activation of the GAL1-HIS3 reporter gene. To identify and exclude auto-activators that can spontaneously arise during the Y2H selection process, we used the pDEST-AD-CYH2 vector carrying the CYH2 counter-selectable marker, which allows for plasmid shuffling on cycloheximide (CHX)-containing media. Only pairs that gave rise to growth on synthetic media lacking histidine (-His) but not on synthetic media lacking histidine but containing 1mg/liter cycloheximide (-His/CHX) in 4 of 4 replicates were considered verified.
Transient transfection and lentiviral infection of shRNA in cultured cells
Transient DNA transfection was performed with the FuGENE 6 Transfection reagent (Roche Applied Science). Briefly, 1 μg plasmid DNA was transfected into cells in each well of a 6-well plate, incubated for 48 hours and harvested for protein extraction or fluorescence imaging. Lentiviral-based pLKO.1 shRNA constructs were obtained from the RNAi Consortium of the Broad Institute and MIT, Cambridge, MA. Virus was produced by transfecting pLKO.1 shRNA plasmids with packaging plasmid (pCMV-dR8.91) and envelope plasmid (VSV-G/pMD2.G) into 293T cells using Fugene6 (Promega Corp., Madison, WI) and Mirus Transfection Reagent (Mirus Bio LLC, Madison, WI), and harvesting the supernatant after 48 hours. For infection, lentivirus in growth medium containing 8 μg/ml polybrene was added to cells on 10-cm dishes and incubated overnight. A final concentration of 1 μg/ml puromycin in regular medium was added to the cells the next day for selection.
Co-immunoprecipitation
Plasmids were transfected into 293T cells and cultured for 48 hours before harvest. Cell pellets were lysed in RIPA lysis buffer supplemented with 1X protease inhibitor cocktail solution and PMSF (Roche Applied Science). 500 μg of total lysate and 1 μg of antibody were mixed and incubated at 4°C overnight, then 40 μl of protein G Agarose beads (EMD Millipore, Billerica, MA) were added and incubated for 2 hours. Beads were washed 3 times with 1X TBST buffer, resuspended in 40 μl Laemmli sample buffer, and boiled for 5 minutes before SDS-PAGE and immunoblot analysis.
Expression array and data analysis
Three independent batches of total RNAs were isolated with the Qiagen RNeasy kit from NGP cells stably infected with an HPCAL1 shRNA and a control GFP shRNA, as described above, and submitted for expression analysis with Affymetrix GeneChip Human Gene 1.0 ST arrays. Raw microarray data were normalized using Bioconductor, and the analysis performed using the limma (Linear Models for Microarray Data) and biomaRt (Interface to BioMart databases) packages. Gene lists that were normally upregulated and downregulated during cell growth and differentiation were retrieved from public databases. Genes that showed a significant differential effect (P<0.05) between control shRNA and HPCAL1 shRNA on cell growth and differentiation were retrieved.
Supplementary Material
Figure S1 The PHOX2B-HPCAL1 interaction results in nuclear translocation of HPCAL1. (a) GFP-tagged WT and the indicated mutant GFP-tagged PHOX2B genes were cotransfected with HPCAL1-mCherry into HeLa cells, fixed and confocal images acquired as described for Figure 2.
Figure S2 The PHOX2B-HPCAL1 interaction does not affect the transactivation potential of PHOX2B. Wild-type and mutant PHOX2B expression constructs were cotransfected with HPCAL1 or GFP as a negative control into 293T cells. A firefly luciferase reporter gene driven by the DBH promoter was used to evaluate the transactivation potential of PHOX2B; a CMV promoter-driven Renilla luciferase reporter served as the internal control. Luciferase activity was determined 48 hours after transfection using the Dual-Glo Luciferase Assay system (Promega, Madison WI). Relative luminescence was calculated by normalizing DBH-driven luciferase activity to Renilla luciferase activity. Values are reported as means ± s.d. of two independent experiments.
Figure S3 Phase contrast micrographs of untransfected, control and HPCAL1 shRNA-expressing LAN-5 neuroblastoma cells treated with 10 μM of retinoic acid for 6 days. Scale bar, 10 μm.
Acknowledgments
We thank Dr Christo Goridis for the generous gift of the PHOX2B antibody. We thank Stacey Frumm and Dr Kimberly Stegmaier for data regarding the neuroblastoma cell differentiation signature prior to publication.
Sources of support: Alex’s Lemonade Stand Foundation (REG), Abraham Research Fund (WW), National Cancer Institute grant R33CA132073 (MV), National Human Genome Research grants R01HG001715 and P50HG004233 (MV, DEH), and The Ellison Foundation (MV).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website.
References
- 1.Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362(23):2202–11. doi: 10.1056/NEJMra0804577. Epub 2010/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Pediatr Clin North Am. 2008;55(1):97–120. x. doi: 10.1016/j.pcl.2007.10.014. Epub 2008/02/05. [DOI] [PubMed] [Google Scholar]
- 3.Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399(6734):366–70. doi: 10.1038/20700. Epub 1999/06/09. [DOI] [PubMed] [Google Scholar]
- 4.Lo L, Morin X, Brunet JF, Anderson DJ. Specification of neurotransmitter identity by Phox2 proteins in neural crest stem cells. Neuron. 1999;22(4):693–705. doi: 10.1016/s0896-6273(00)80729-1. Epub 1999/05/07. [DOI] [PubMed] [Google Scholar]
- 5.Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399(6734):366–70. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- 6.Dubreuil V, Hirsch MR, Pattyn A, Brunet JF, Goridis C. The Phox2b transcription factor coordinately regulates neuronal cell cycle exit and identity. Development. 2000;127(23):5191–201. doi: 10.1242/dev.127.23.5191. [DOI] [PubMed] [Google Scholar]
- 7.Raabe EH, Laudenslager M, Winter C, Wasserman N, Cole K, LaQuaglia M, et al. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene. 2008;27(4):469–76. doi: 10.1038/sj.onc.1210659. [DOI] [PubMed] [Google Scholar]
- 8.Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Silvestri JM, Curran ME, et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. American journal of medical genetics Part A. 2003;123A(3):267–78. doi: 10.1002/ajmg.a.20527. Epub 2003/11/11. [DOI] [PubMed] [Google Scholar]
- 9.Amiel J, Laudier B, Attie-Bitach T, Trang H, de Pontual L, Gener B, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33(4):459–61. doi: 10.1038/ng1130. Epub 2003/03/18. [DOI] [PubMed] [Google Scholar]
- 10.Mosse YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. 2004;75(4):727–30. doi: 10.1086/424530. Epub 2004/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74(4):761–4. doi: 10.1086/383253. Epub 2004/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matera I, Bachetti T, Puppo F, Di Duca M, Morandi F, Casiraghi GM, et al. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset Central Hypoventilation syndrome. Journal of medical genetics. 2004;41(5):373–80. doi: 10.1136/jmg.2003.015412. Epub 2004/05/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Limpt V, Schramm A, van Lakeman A, Sluis P, Chan A, van Noesel M, et al. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene. 2004;23(57):9280–8. doi: 10.1038/sj.onc.1208157. Epub 2004/11/02. [DOI] [PubMed] [Google Scholar]
- 14.Reiff T, Tsarovina K, Majdazari A, Schmidt M, del Pino I, Rohrer H. Neuroblastoma phox2b variants stimulate proliferation and dedifferentiation of immature sympathetic neurons. J Neurosci. 2010;30(3):905–15. doi: 10.1523/JNEUROSCI.5368-09.2010. Epub 2010/01/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagashimada M, Ohta H, Li C, Nakao K, Uesaka T, Brunet JF, et al. Autonomic neurocristopathy-associated mutations in PHOX2B dysregulate Sox10 expression. J Clin Invest. 2012;122(9):3145–58. doi: 10.1172/JCI63401. Epub 2012/08/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pei D, Luther W, Wang W, Paw BH, Stewart RA, George RE. Neuroblastoma-associated PHOX2B mutations impair sympathetic neuronal differentiation. PLOS Genetics. 2013 doi: 10.1371/journal.pgen.1003533. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vidal M, Cusick ME, Barabasi AL. Interactome networks and human disease. Cell. 2011;144(6):986–98. doi: 10.1016/j.cell.2011.02.016. Epub 2011/03/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong Q, Simonis N, Li QR, Charloteaux B, Heuze F, Klitgord N, et al. Edgetic perturbation models of human inherited disorders. Molecular systems biology. 2009;5:321. doi: 10.1038/msb.2009.80. Epub 2009/11/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreze M, Charloteaux B, Milstein S, Vidalain PO, Yildirim MA, Zhong Q, et al. ‘Edgetic’ perturbation of a C. elegans BCL2 ortholog. Nature methods. 2009;6(11):843–9. doi: 10.1038/nmeth.1394. Epub 2009/10/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452(7188):713–8. doi: 10.1038/nature06731. Epub 2008/03/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu HT, Su YN, Hung CC, Hsieh WS, Wu KJ. Interaction between PHOX2B and CREBBP mediates synergistic activation: mechanistic implications of PHOX2B mutants. Hum Mutat. 2009;30(4):655–60. doi: 10.1002/humu.20929. Epub 2009/02/05. [DOI] [PubMed] [Google Scholar]
- 22.Hong SJ, Chae H, Lardaro T, Hong S, Kim KS. Trim11 increases expression of dopamine beta-hydroxylase gene by interacting with Phox2b. Biochem Biophys Res Commun. 2008;368(3):650–5. doi: 10.1016/j.bbrc.2008.01.165. Epub 2008/02/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Limpt V, Chan A, Schramm A, Eggert A, Versteeg R. Phox2B mutations and the Delta-Notch pathway in neuroblastoma. Cancer Lett. 2005;228(1–2):59–63. doi: 10.1016/j.canlet.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 24.Trang H, Laudier B, Trochet D, Munnich A, Lyonnet S, Gaultier C, et al. PHOX2B gene mutation in a patient with late-onset central hypoventilation. Pediatr Pulmonol. 2004;38(4):349–51. doi: 10.1002/ppul.20074. Epub 2004/08/31. [DOI] [PubMed] [Google Scholar]
- 25.Dreze M, Monachello D, Lurin C, Cusick ME, Hill DE, Vidal M, et al. High-quality binary interactome mapping. Methods in enzymology. 2010;470:281–315. doi: 10.1016/S0076-6879(10)70012-4. Epub 2010/10/16. [DOI] [PubMed] [Google Scholar]
- 26.Lamesch P, Li N, Milstein S, Fan C, Hao T, Szabo G, et al. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89(3):307–15. doi: 10.1016/j.ygeno.2006.11.012. Epub 2007/01/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haynes LP, Burgoyne RD. Unexpected tails of a Ca2+ sensor. Nat Chem Biol. 2008;4(2):90–1. doi: 10.1038/nchembio0208-90. Epub 2008/01/19. [DOI] [PubMed] [Google Scholar]
- 28.Yang C, Kim HS, Seo H, Kim CH, Brunet JF, Kim KS. Paired-like homeodomain proteins, Phox2a and Phox2b, are responsible for noradrenergic cell-specific transcription of the dopamine beta-hydroxylase gene. J Neurochem. 1998;71(5):1813–26. doi: 10.1046/j.1471-4159.1998.71051813.x. Epub 1998/11/03. [DOI] [PubMed] [Google Scholar]
- 29.Adachi M, Browne D, Lewis EJ. Paired-like homeodomain proteins Phox2a/Arix and Phox2b/NBPhox have similar genetic organization and independently regulate dopamine beta-hydroxylase gene transcription. DNA and cell biology. 2000;19(9):539–54. doi: 10.1089/104454900439773. Epub 2000/10/18. [DOI] [PubMed] [Google Scholar]
- 30.Bachetti T, Matera I, Borghini S, Di Duca M, Ravazzolo R, Ceccherini I. Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Hum Mol Genet. 2005;14(13):1815–24. doi: 10.1093/hmg/ddi188. Epub 2005/05/13. [DOI] [PubMed] [Google Scholar]
- 31.Taraviras S, Olli-Lahdesmaki T, Lymperopoulos A, Charitonidou D, Mavroidis M, Kallio J, et al. Subtype-specific neuronal differentiation of PC12 cells transfected with alpha2-adrenergic receptors. European journal of cell biology. 2002;81(6):363–74. doi: 10.1078/0171-9335-00250. Epub 2002/07/13. [DOI] [PubMed] [Google Scholar]
- 32.Hansford LM, Smith SA, Haber M, Norris MD, Cheung B, Marshall GM. Cloning and characterization of the human neural cell adhesion molecule, CNTN4 (alias BIG-2) Cytogenetic and genome research. 2003;101(1):17–23. doi: 10.1159/000073412. Epub 2003/10/23. [DOI] [PubMed] [Google Scholar]
- 33.Frumm S, Fan Z, Ross K, Duvall J, Gupta S, VerPlank L, et al. Selective HDAC/HDAC2 Inhibitors Induce Neuroblastoma Differentiation. Chemistry & Biology. 2013 doi: 10.1016/j.chembiol.2013.03.020. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braunewell KH, Klein-Szanto AJ. Visinin-like proteins (VSNLs): interaction partners and emerging functions in signal transduction of a subfamily of neuronal Ca2+-sensor proteins. Cell Tissue Res. 2009;335(2):301–16. doi: 10.1007/s00441-008-0716-3. Epub 2008/11/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paterlini M, Revilla V, Grant AL, Wisden W. Expression of the neuronal calcium sensor protein family in the rat brain. Neuroscience. 2000;99(2):205–16. doi: 10.1016/s0306-4522(00)00201-3. Epub 2000/08/12. [DOI] [PubMed] [Google Scholar]
- 36.Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nature reviews Neuroscience. 2007;8(3):182–93. doi: 10.1038/nrn2093. Epub 2007/02/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rustandi RR, Baldisseri DM, Weber DJ. Structure of the negative regulatory domain of p53 bound to S100B(betabeta) Nature structural biology. 2000;7(7):570–4. doi: 10.1038/76797. Epub 2000/07/06. [DOI] [PubMed] [Google Scholar]
- 38.Ikura M, Yap KL. Where cancer meets calcium--p53 crosstalk with EF-hands. Nature structural biology. 2000;7(7):525–7. doi: 10.1038/76721. Epub 2000/07/06. [DOI] [PubMed] [Google Scholar]
- 39.Ikura M, Osawa M, Ames JB. The role of calcium-binding proteins in the control of transcription: structure to function. BioEssays : news and reviews in molecular, cellular and developmental biology. 2002;24(7):625–36. doi: 10.1002/bies.10105. Epub 2002/07/12. [DOI] [PubMed] [Google Scholar]
- 40.Spilker C, Gundelfinger ED, Braunewell KH. Evidence for different functional properties of the neuronal calcium sensor proteins VILIP-1 and VILIP-3: from subcellular localization to cellular function. Biochimica et biophysica acta. 2002;1600(1–2):118–27. doi: 10.1016/s1570-9639(02)00452-1. Epub 2002/11/26. [DOI] [PubMed] [Google Scholar]
- 41.O’Callaghan DW, Tepikin AV, Burgoyne RD. Dynamics and calcium sensitivity of the Ca2+/myristoyl switch protein hippocalcin in living cells. J Cell Biol. 2003;163(4):715–21. doi: 10.1083/jcb.200306042. Epub 2003/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jheng FF, Wang L, Lee L, Chang LS. Functional contribution of Ca2+ and Mg2+ to the intermolecular interaction of visinin-like proteins. The protein journal. 2006;25(4):250–6. doi: 10.1007/s10930-006-9008-5. Epub 2006/05/17. [DOI] [PubMed] [Google Scholar]
- 43.Zellmer E, Zhang Z, Greco D, Rhodes J, Cassel S, Lewis EJ. A homeodomain protein selectively expressed in noradrenergic tissue regulates transcription of neurotransmitter biosynthetic genes. J Neurosci. 1995;15(12):8109–20. doi: 10.1523/JNEUROSCI.15-12-08109.1995. Epub 1995/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swanson DJ, Zellmer E, Lewis EJ. The homeodomain protein Arix interacts synergistically with cyclic AMP to regulate expression of neurotransmitter biosynthetic genes. J Biol Chem. 1997;272(43):27382–92. doi: 10.1074/jbc.272.43.27382. Epub 1997/10/27. [DOI] [PubMed] [Google Scholar]
- 45.Lenz SE, Henschel Y, Zopf D, Voss B, Gundelfinger ED. VILIP, a cognate protein of the retinal calcium binding proteins visinin and recoverin, is expressed in the developing chicken brain. Brain research Molecular brain research. 1992;15(1–2):133–40. doi: 10.1016/0169-328x(92)90160-d. Epub 1992/09/01. [DOI] [PubMed] [Google Scholar]
- 46.Braunewell KH, Dwary AD, Richter F, Trappe K, Zhao C, Giegling I, et al. Association of VSNL1 with schizophrenia, frontal cortical function, and biological significance for its gene product as a modulator of cAMP levels and neuronal morphology. Translational psychiatry. 2011;1:e22. doi: 10.1038/tp.2011.20. Epub 2011/01/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The PHOX2B-HPCAL1 interaction results in nuclear translocation of HPCAL1. (a) GFP-tagged WT and the indicated mutant GFP-tagged PHOX2B genes were cotransfected with HPCAL1-mCherry into HeLa cells, fixed and confocal images acquired as described for Figure 2.
Figure S2 The PHOX2B-HPCAL1 interaction does not affect the transactivation potential of PHOX2B. Wild-type and mutant PHOX2B expression constructs were cotransfected with HPCAL1 or GFP as a negative control into 293T cells. A firefly luciferase reporter gene driven by the DBH promoter was used to evaluate the transactivation potential of PHOX2B; a CMV promoter-driven Renilla luciferase reporter served as the internal control. Luciferase activity was determined 48 hours after transfection using the Dual-Glo Luciferase Assay system (Promega, Madison WI). Relative luminescence was calculated by normalizing DBH-driven luciferase activity to Renilla luciferase activity. Values are reported as means ± s.d. of two independent experiments.
Figure S3 Phase contrast micrographs of untransfected, control and HPCAL1 shRNA-expressing LAN-5 neuroblastoma cells treated with 10 μM of retinoic acid for 6 days. Scale bar, 10 μm.