

Abstract
A novel lead compound for inhibition of the antibacterial drug target, glutamate racemase, is optimized for both ligand efficiency and lipophilic efficiency. A previously developed hybrid MD-docking and scoring scheme, FERM-SMD, is utilized to predict relative potencies of potential derivatives prior to chemical synthesis. This scheme was successful in distinguishing between high and low affinity binders with minimal experimental structural information, saving time and resources in the process. In vitro potency is increased approximately 4-fold against glutamate racemase from the model organism, B. subtilis. Lead derivatives show 2- to 4-fold increased antimicrobial potency over the parent scaffold. In addition, specificity toward B. subtilis, over E. coli and S. aureus, show dependency on the chemical substituent added to the parent scaffold. Finally, insight is gained into the capacity for these compounds to reach the target enzyme in vivo using a bacterial cell wall lysis assay. The result of this study is a novel small molecule inhibitor of GR with the following characteristics: Ki = 2.5 μM, LE = 0.45 kcal/mol/atom, LiPE = 6.0, MIC50 = 260 μg/mL against B. subtilis, EC50,lysis = 520 μg/mL against B. subtilis
Keywords: Antibiotics, Fragment-Based Drug Discovery, Glutamate Racemase, Heterocyclic Aromatics, Inhibitors
Introduction
In an era of increasingly prolific multi- and total-drug resistant species of bacteria such as E. coli, M. tuberculosis and S. aureus, the need for rapid discovery of novel antibiotic classes is greater than any previous time in history. Compounding this problem is the pronounced dearth of both antimicrobial lead compounds as well as FDA-approved drugs emerging from the drug discovery enterprise, including both academia and industry[1]. An illuminating review by O’Shea and coworkers[2] reveals that no novel class of antibacterials was developed and approved between 1960 and 2001 despite exhaustive efforts and the parallel development of key techniques. Since the introduction of streptogramins and quinolones in the early 1960s, the growing need for antimicrobials has outpaced the rate of approval of novel drugs. Passage down the pipeline of drug discovery is complicated by the requirement that any antimicrobial target must be essential within a class of bacteria as well as non-essential or absent in humans; or possess significant structural distinction from any human homologues. Additionally, inhibitors must satisfy stringent physico-chemical requirements that ensure bioavailability, minimal toxicity, and efficacy. Recent reviews of the current state of affairs in drug discovery have revealed that lack of chemical diversity in HTS- and genomics-based drug discovery campaigns has been a significant culprit in the failure to obtain novel antimicrobial lead compounds[1, 3].
Bacteria require a number of D-amino acids for the biosynthesis of the peptidoglycan cell wall. It has been well established that improper peptidoglycan cross-linking is the basis for a number of known antimicrobial drugs, including the β-lactam class and vancomycins, which act to promote osmotic lysis[4]. In addition to preventing cross-linking itself, inhibiting enzymes that catalyze the formation of D-amino acids also leads to lysis due to high internal osmotic pressure. The two ubiquitous D-amino acids in bacterial cell walls are D-alanine and D-glutamate, which are biosynthesized by alanine and glutamate racemases, respectively. The natural product D-cycloserine is a mechanism based inhibitor of alanine racemase, a PLP- containing enzyme, and has been shown to kill bacteria by making them osmotically sensitive[5]. Similar studies have also been carried out on glutamate racemase (abbrev. GR; EC 5.1.1.3) inhibitors, establishing a mode of action involving damage to the maturation of the peptidoglycan cell wall[6]. Unlike AR, GR is a cofactor-independent racemase, which catalyzes a step-wise proton abstraction/donation via two cysteines acting in a general acid/base mechanism[7]. The general α/β-fold forms two domains, which enclose a relatively small buried active site that is saturated with polar residues. Not surprisingly, GR knockout studies on several pathogenic organisms resulted in D-glutamate auxotrophs[8]. Thus, the strategy of attenuating the pool of D-amino acids is an attractive option for the development of novel antimicrobial agents. However, the only compound in this class that is approved for clinical use is the natural product D-cycloserine, and only in combination with other antibiotics, due to its undesirable side effects.
To date, only a handful of potent inhibitors have been discovered for bacterial glutamate racemases. A SAR approach produced a 4S-substituted D-glutamate analog, which had low-micromolar potency against glutamate racemase from S. pneumoniae, but suffered from species specificity due to steric clashing with a species-variable valine bridge to a hydrophobic pocket proximal to the binding cleft[9]. Later, a high-throughput screening campaign of nearly 400,000 compounds resulted in the serendipitous discovery of an uncompetitive inhibitor, which binds to a species-specific allosteric site[6]. More recently, a virtual screening campaign targeting a transition-state-like model of the target enzyme produced several low-micromolar, competitive inhibitors[10]. A trend in the molecular makeup of these inhibitors was clear: aromatic or cyclic compounds containing sulfonic acid moieties. This isn’t surprising considering previous work that supports the presence of a cyclic carbanion/aci-dienolate transition in the glutamate racemase reaction, which places significant negative charge density in the back of the active site[11]. The superiority of these sulfonic acids over carboxylates (such as that in the natural substrate) could be due to the more dispersed partial negative charge in the sulfonate, compared to the SP2 hybridization of a carboxylate. Most recently, a unique ensemble docking scheme was applied to GR from B. subtilis to successfully rank several sulfonate-containing aromatic compounds with potencies ranging from low micromolar to high millimolar[12]. The best of these compounds, 1H-benzimidazole-2-sulfonic acid (Ki = 9 μM) is the subject of this study.
We present here a fragment-based approach to optimization of the previously mentioned lead compound using entirely in silico methods for derivative ranking prior to synthesis and experimental testing. Fragment-based methods offer a number of distinct advantages in drug discovery, particularly the optimization of ligand efficiency (LE) and lipophilic efficiency (LiPE) while maintaining potency[13]. Placement and subsequent scoring of potential derivative compounds was achieved via ensemble docking with a unique scoring scheme described in Whalen and coworkers.[12] In the current study, thirty-three derivatives of the lead compound were docked to an ensemble of conformations generated using steered molecular dynamics and ranked using a modified binding energy score. Six derivatives were synthesized and assayed experimentally, resulting in the discovery of two competitive inhibitors with increased inhibitory potency, as well as excellent ligand and lipophilic efficiencies. Additionally, compounds were assayed for bacterial growth inhibition as well as induction of cell wall lysis, ultimately establishing that this class of GR inhibitors targets bacterial cell wall synthesis in vivo.
Results and Discussion
BISA, a Scaffold for Optimization
Compound 1 (Figure 4, 4-hydroxy-1,3-benzenedisulfonic acid) was discovered in a virtual screening campaign against GR using the Chemical Computing Group Lead-like library (~1 million compounds)[10]. The inhibitory constant against GR from B. subtilis was 58 ± 13 μM. Scaffold hopping to compound 2 (Figure 4, 1H-benzimidazole-2-sulfonic acid) increases affinity against this target to 9 ± 2 μM. Compound 2 also shows equal potency against two isozymes of GR from B. anthracis (RacE1 and RacE2) as well as GR from F. tularensis (MurI), two bacterial species currently considered as Tier 1 Biological Select Agents by the US government (Figure 1). The high ligand efficiency of this fragment, coupled with its cross-species activity made compound 2 an ideal candidate for optimization.
Figure 4.
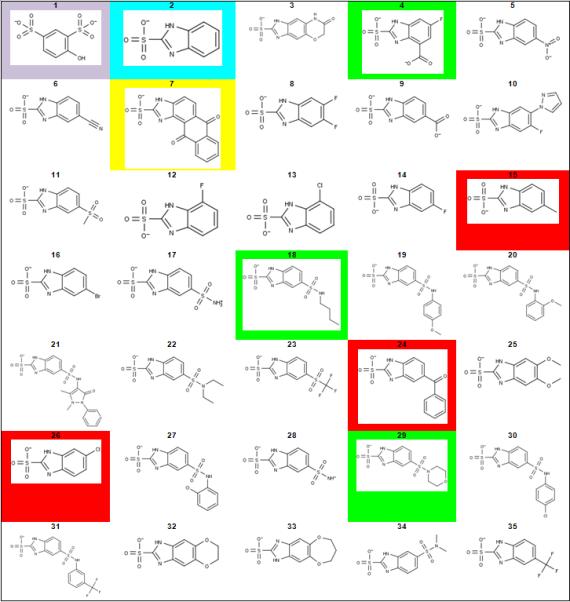
Parent scaffolds and lead derivatives considered in this study. Compounds of interest are highlighted accordinglylavender = original virtual screening hit; cyan = parent scaffold; green = compounds tested with high predicted affinity; red = compounds tested with low predicted affinity; yellow = compound synthesis attempted, yet unsuccessful.
Figure 1.
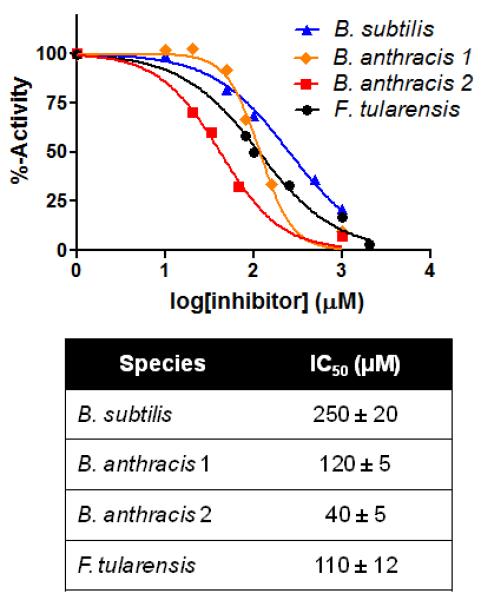
IC50 curves for the parent compound 2 against a range of GR isozymes isolated from the indicated bacterial species. Indicated IC50 values (in micromolar) acquired via fitting to a dose-response curve.
In order to generate a basis for rational lead optimization, a basic understanding of the physicochemical components of binding between ligand and receptor is required. As an alternative to x-ray crystallography or NMR, virtual docking was used to generate structural information regarding the interaction of GR and compound 2. Compound 2 was docked in silico to GR using a previously solved crystal structure (PDB: 1ZUW) as the receptor. The result of docking shows compound 2 with its sulfonic acid situated in the most buried region of the active site, between the catalytic cysteines (Figure 2). The sulfonate moiety is seen participating in several hydrogen bonding interactions with Asn75, Thr186, and Cys185. Additionally, the benzene moiety is interacting with Ser11 via an O-H--pi interaction. These moieties both appear to contribute to recognition of compound 2, and thus the optimization strategy focused on the addition of substituents that would produce additional interactions while preserving the original contacts. As seen by their solvent exposure and protein proximity (symbolized with light blue shading or a grey dotted line, respectively, in Figure 2), carbons 4, 5, and 6 within the benzene ring could serve as starting points to build on additional chemical groups without encountering steric clash from active site residues. Depending on their size, substituents added at these positions have the capacity to reach additional binding pockets proximal to the main substrate binding cleft.
Figure 2.
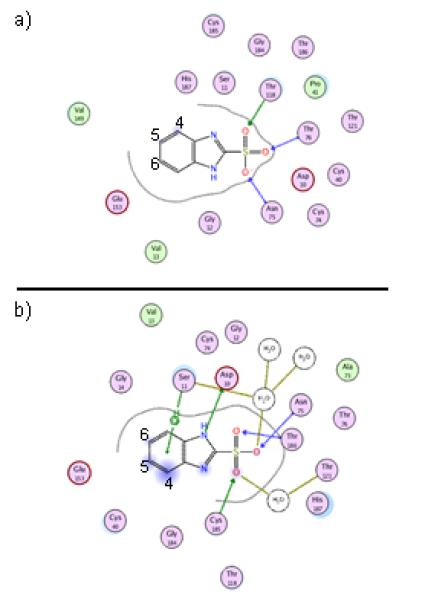
Top-ranked binding pose for compound 2 bound to GR from B. subtilis, as predicted by docking (a). Also, binging pose for compound 2 after 4 nsec of MD simulation with explicit water (b).
Derivative Selection and Synthesis
An in silico library containing compounds 3 through 35 (Figure 4) was developed based on a previously established two-step synthetic process (Scheme 1) and the commercially-available 1,2-phenylenediamine derivatives. This synthetic scheme was chosen for its relative ease, while additional chemistry may be attempted in the future to further grow fragments out of the substrate-binding cleft. Before any compound was synthesized, the library was subjected to a hybrid ensemble docking scheme, referred to as the Flexible Enzyme Receptor Method by Steered Molecular Dynamics (aka. FERM-SMD), previously described by Whalen and coworkers[12]. Figure 3 details how unique conformations of the protein target were generated using steered molecular dynamics simulations to emulate the substrate unbinding trajectory. Starting with a crystal structure of D-glutamate bound to glutamate racemase, D-glutamate is pulled from the active site over the course of the simulation. In the process, the enzyme alters its structural conformation to allow substrate passage from the buried binding cleft. Three snapshots were chosen to represent three distinct structural states, distinguished by the entrance to the binding cleft: closed, partially open, and fully open (Figure 3). Compounds are docked to all three structures and their predicted binding affinities are adjusted according to the respective protein solvation energy (these varied greatly, and affected the accuracy of the binding affinity calculation) and weighted to indicate relative binding specificity to one of the three receptors. Previous studies have shown that the final score produced by FERM-SMD, deemed FERMscore, shares a high correlation with experimental binding affinities, particularly for congeneric ligands of GR. On a set of 17 ligands, FERM-SMD has a predictive accuracy of ± 1 kcal/mol[12].
Scheme 1.
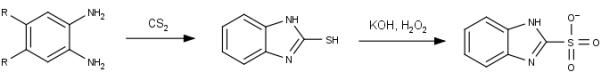
Scheme for synthesis of 1H-benzimidazole-2-sulfonic acid derivatives using differing phenylenediamine starting points.
Figure 3.
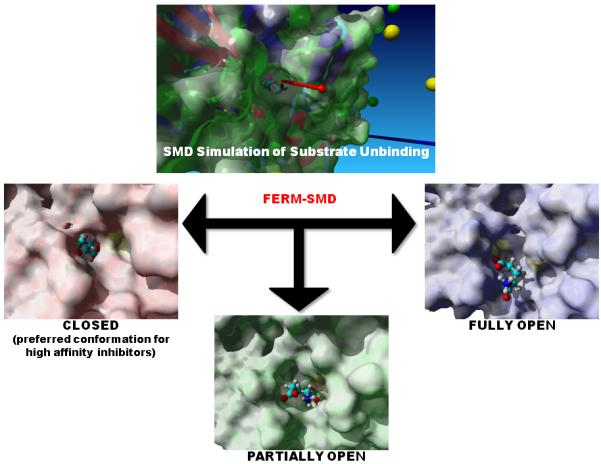
A steered molecular dynamics simulation was conducted on the glutamate-bound crystal structure of glutamate racemase from B. subtilis. A force was applied on the bound substrate along the vector indicated in the top picture (red arrow). Structures were obtained along the unbinding trajectory that correspond approximately to the following states: closed, partially open, and fully open. With substrate removed, these structures then provide the receptors for ensemble docking of the derivative library. Previous results indicate that the highest-affinity inhibitors will bind preferably to the closed conformation, over the partially and fully open conformations.
FERMscores for the library of interest, spanning from 0.4 to 13.7, are indicated in Table 1. The parent compound, 2, scored the third highest FERMscore. The two compounds giving higher calculated FERMscores (compounds 18, and 29) in addition to a third compound, 4, that possessed a FERMscore in the top 15% of all derivatives were synthesized under contract by Enamine Ltd. (see Materials and Methods) and all compounds were heretofore synthetically novel. In addition to the compounds predicted to have improved binding affinity, three immediately available compounds (15, 24, and 26) were acquired to test the predictive capacity of the employed scoring method. Compound 7 had an intermediate FERMscore, but distinct chemotype, which had not been heretofore tested on any GR and was thusly chosen for testing. Unfortunately, several attempts to synthesize compound 7 were unsuccessful, and it was eventually abandoned. The experimental results are detailed below.
Table 1.
FERMscore assignments as predicted by FERM-SMD, ranked from highest to lowest score, corresponding to highest to lowest predicted binding affinity.
| Compound [a] | FERMscore | Compound | FERMscore |
|---|---|---|---|
| 29 | 13.7 | 26 | 1.20 |
| 18 | 9.25 | 7 | 1.15 |
| 2 | 8.28 | 6 | 1.10 |
| 5 | 4.92 | 23 | 1.03 |
| 4 | 4.45 | 15 | 0.969 |
| 34 | 3.75 | 20 | 0.902 |
| 14 | 3.23 | 33 | 0.792 |
| 13 | 2.99 | 16 | 0.781 |
| 12 | 2.98 | 32 | 0.756 |
| 9 | 2.97 | 28 | 0.756 |
| 8 | 2.33 | 35 | 0.751 |
| 22 | 1.76 | 25 | 0.750 |
| 17 | 1.64 | 3 | 0.732 |
| 21 | 1.44 | 10 | 0.723 |
| 11 | 1.31 | 24 | 0.643 |
| 19 | 1.29 | 31 | 0.534 |
| 30 | 1.28 | 27 | 0.406 |
Compounds of interest are highlighted accordingly: green = compounds tested with high predicted affinity; cyan = parent scaffold; red = compounds tested with low predicted affinity; yellow = compound synthesis attempted, yet unsuccessful.
In vitro Testing of Derivatives
Inhibition constants (Ki) were acquired for all derivatives against purified GR from B. subtilis (Figure 5, Table 2). Compounds 15, 24 and 26, all possessing predicted FERMscores lower than the parent compound, gave Ki values greater than or within error of that of the parent compound. Compound 24 suffered from a nearly 100-fold loss in binding affinity, which was well predicted by FERM-SMD, as it possessed the lowest FERMscore of the compounds tested. Of the compounds predicted to be higher affinity binders by FERMscore, compound 18 and 29 have Ki values within error of the parent compound, although the Ki of 29 is improved, 6.4 μM versus 9 μM. This result was not surprising considering the FERMscores only vary by 5.4 units between the parent scaffold and the highest scoring derivative. Compound 4 is also predicted to be high affinity, and shows 4-fold improved affinity over the parent compound, with a Ki of 2.5 μM. This is the most potent non-glutamate-based, competitive inhibitor of glutamate racemase to date. Overall, FERM-SMD was successful in distinguishing between tight binding derivatives (Ki between 2.5 to 12 μM) and weaker binding derivatives (Ki between 13 and 830 μM).
Figure 5.

In vitro inhibition data used to acquire Ki values for each derivative.
Table 2.
In vitro results for derivatives of the parent scaffold, 2.
| Compound | Ki (uM) | LE[a] | LiPE[b] |
|---|---|---|---|
| 2 | 9.0 ± 2.0 | 0.53 | 5.3 |
| 4 | 2.5 ± 0.4 | 0.45 | 6.0 |
| 15 | 21 ± 5.0 | 0.46 | 4.4 |
| 18 | 12 ± 3.6 | 0.30 | 5.0 |
| 24 | 830 ± 75 | 0.20 | 1.9 |
| 26 | 13 ± 2.4 | 0.48 | 4.5 |
| 29 | 6.4 ± 3.5 | 0.34 | 6.6 |
Ligand efficiencies (LE) determined by converting Ki values to binding energies and dividing by the number of non-hydrogen atoms. Units = kcal/mol/atom.
Lipophilic efficiences (LiPE) determined by subtracting logP values from the log(Ki) for each compound.
Ligand efficiency and lipophilic efficiency were calculated for each derivative (Table 2). Ligand efficiency is a method of normalizing binding affinities for compounds of differing molecular weights[14]. Additionally, several studies have shown that fragment-based drug discovery is more successful if high ligand-efficiency is maintained through lead optimization[14]. This practice lowers the occurrence of so-called “molecular obesity” as compounds are modified to achieve higher potency and favorable pharmacokinetic/pharmacodynamic profiles[14b]. With the exception of compound 15, each assayed derivative maintained high ligand efficiency ( > 0.3 kcal/mol/atom). Of the compounds predicted to be high affinity by the FERM-SMD method, compound 4 and 29 exhibited higher efficiency than compound 18 (0.45, 0.34 and 0.30, respectively). Lipophilic efficiency is another measure that is indicative of successful passage down the drug development pipeline, where affinity values are normalized for the partition coefficient (logP) of the inhibitor[15]. Compound 4 and 29 benefit from an improved lipophilic efficiency (6.0 and 6.6, respectively) over the parent scaffold (5.3). Ligand efficiency and lipophilicity efficiency values equal to or greater than 0.3 kcal/mol/atom and 6.0, respectively, are in the desirable range for further study and optimization[14a, 15].
Additionally, novel compounds were tested for formation of colloidal aggregates, a common cause of false positive results. Previous studies have revealed that glutamate racemase is susceptible to inhibition by colloidal aggregates in a non-drug-like fashion. In order to distinguish between inhibition via colloidal aggregation and true binding, enzyme activity is measured in the presence of the inhibitor in question, as well as a sub-micellular concentration of detergent, 0.01% Triton X-100. In the case that a compound is inhibiting an enzyme via a colloidal aggregation, the apparent inhibition will be completely relieved in the presence of detergent. Colloidal aggregates must be abandoned due to their non-drug-like mechanism. All novel compounds tested in this study proved not to operate via the colloidal aggregate mechanism (Figure S10).
In vivo Testing of Biological Activity
In order to assess the capacity of these compounds to reach the enzyme target in vivo, inhibition of bacterial growth as well as capacity to induce cell lysis was assayed with several species of bacteria. B. subtilis was investigated, as the isozyme of GR from this species was the model for all in silico predictions. Additionally, E. coli and S. aureus were investigated as each provides a unique challenge for inhibitor compounds: an additional physical barrier to entry in the case of Gram-negative E. coli and an abundance of efflux pumps in the case of S. aureus[16]. All tested derivatives of compound 2 show increased potency with regards to growth inhibition for B. subtilis (Figure 6). MIC50 values are increased 2- to 3-fold over the parent scaffold (Table 3). Surprisingly, the least potent compound in vitro, 24, shows the greatest potency in vivo. This result suggests that factors other than enzyme binding affinity may complicate the overall efficacy of this chemotype of antimicrobial compounds.
Figure 6.

MIC50 curves for the parent scaffold and the two most potent derivatives against Bacillus subtilis.
Table 3.
MIC50[a] values for tested derivatives.
| Compound | B. subtilis | E. coli | S. aureus |
|---|---|---|---|
| 2 | 0.72 ± 0.06 | - | - |
| 4 | 0.26 ± 0.11 | 1.6 ± 0.20 | 1.0 ± 0.25 |
| 15 | > 3 | - | - |
| 18 | 0.36 ± 0.02 | > 3 | > 3 |
| 24 | 0.14 ± 0.02 | - | - |
| 26 | > 3 | - | - |
| 29 | 0.32 ± 0.01 | > 3 | > 3 |
in mg/mL
Compounds 4, 18, and 29 were also tested against E. coli and S. aureus (Figure 7). Compounds 18 and 29 were both highly specific for B. subtilis, showing no significant growth inhibition at concentrations below 3 mg/mL for E. coli and S. aureus (Table 3). On the contrary, compound 4 shows growth inhibition of both E. coli and S. aureus at concentrations approximately 2-fold higher than the MIC50 against B. subtilis (Table 3). Examination of their respective chemical structures yields one possible rationale for this distinction (see Figure 4 for structures). Compound 4 possesses a more compact chemical shape, most likely making contacts specific to the most buried, and most highly conserved, region of the GR active site. Whereas compounds 18 and 29 contain larger chemical additions to the benzene ring, which may clash with the outer region of the GR active site, which is more structurally diverse region. These in vitro (and in silico) results support the fragment-based strategy of growing the scaffold out of the highly buried active site without sacrificing the original contacts.
Figure 7.

MIC50 curves for highest-ranked derivatives, by FERMscore, comparing species specificity between E. coli, B. subtilis, and S. aureus.
In order to hone in on the mechanism of action for the derivatives assayed in this study, a commercially-available cytotoxicity assay, CytoTox-Glo™ (Promega), marketed for use with mammalian cells, was adapted for use with the examined bacterial species. The relationship between cytotoxicity, specifically cell lysis, and the readout (luminescence) is outlined in Figure 8. If the compounds here reach the glutamate racemase target and inhibit the production of D-glutamate, the lack of this key component will result in an overall break-down in peptidoglycan synthesis, and subsequent cell lysis caused by osmotic stress. Lysed cells will leak intracellular proteases into the surrounding media. The CytoTox-Glo™ reagent is composed of a pro-luciferin substrate, that once cleaved by proteases, can be acted upon by a supplied luciferase to produce the luminescent readout. Controls using FDA-approved antibiotics with known mechanisms of action were conducted to optimize the provided reagents (Figure 9a). S. aureus was exposed to varying concentrations of ampicillin (a transpeptidase inhibitor) or tetracycline (a microbial ribosome inhibitor) for 24 hours, and then incubated with the CytoTox-Glo™ reagent. As expected, ampicillin yields a dose-dependent increase in luminescence , while tetracycline elicits no increase in luminescence at concentrations up to 100-times the published MIC50 (Figure 9a). This optimized assay was then applied to cells treated with our inhibitor derivatives.
Figure 8.
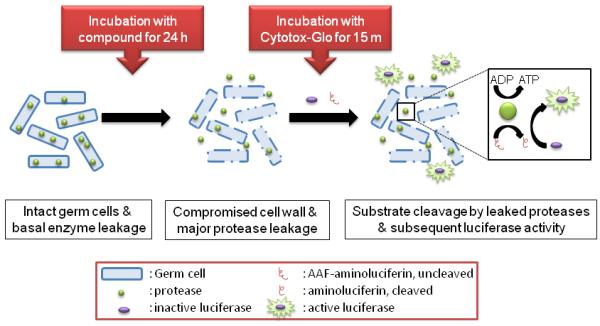
Schematic of treatment of bacterial cells with CytoTox-Glo™ assay. The CytoTox-Glo™ reagent contains both the protease substrate, AAF-luciferin, as well as luciferase. Bacterial cells contain intracellular enzymes capable of cleaving AAF-luciferin, releasing luciferin, the substrate of luciferase.
Figure 9.

Controls for a commercially-available cytotoxicity assay that monitors cell wall lysis with a luminescent readout (a). Bacteria treated with two antibiotics, only one utilizing a mechanism of action that interferes with peptidoglycan synthesis, shows distinct luminescent dose responses. Cell lysis data for S aureus and B. subtilis cultures treated for 24 hours with compound 4 (b). Luminescence is measured in arbitrary units, corrected for the cell density of the sample (variable depending on growth inhibition), and presented as a percentage of untreated cell luminescence.
The modified CytoTox-Glo™ assay confirmed 4 as acting via an inhibitory mechanism that effects the peptidoglycan with both S. aureus and B. subtilis (Figure 9b). Luminescence increases concurrently with increased dosing of compound 4 for S. aureus. While the assayed concentrations do not span the entire MIC50, due to solubility limitations, an approximately 40% increase in luminescence was observed in the millimolar range of inhibitor. For B. subtilis, the species for which compound 4 elicits greater growth inhibition, we observe a dose-dependent increase of 400% in luminescence in the low-millimolar range. The observed EC50 for lysis occurs at 520 μg/mL, a concentration only slightly above that of the MIC50 for growth inhibition (260 μg/mL). The proximity of these two values is supportive of cell lysis as being the main cause of cell death. Considering the many barriers an antibacterial compound must overcome, both physical (peptidoglycan and efflux) and chemical (metabolism), in order to reach the desired target protein, the ability of compound 4 to cause cell lysis is excellent support for its further development as an antibacterial therapeutic.
Conclusion
This study summarizes the successful utilization of a novel in silico docking and scoring scheme for selection of derivatives of a lead scaffold via rank-ordering of binding affinity. The model system used here employs the antibacterial target, glutamate racemase, and a low-micromolar competitive inhibitor, 1H-benzimidazole-2-sulfonic acid (compound 2), as the lead compound. This platform can potentially be used for optimization of lead compounds for other flexible drug targets. In current lead optimization campaigns, the rate-limiting step is often acquisition of high-resolution structural data, particularly for flexible enzymes, on the enzyme-drug complex. Binding pose predictions made by docking software have been largely validated as correct in many cases via comparison with experimental data, specifically several programs place ligands within 2 Å RMSD of the crystallographically determined pose for over 90% of the assessed ligands[17]. The perennial problem lies in the ability of the scoring functions to accurately rank-order docked ligands across a variety of targets[17]. By developing an in silico method of derivative ranking based on the docked complex of the parent scaffold with a predictive error of only ± 1 kcal/mol for binding energy, we can remove the need for time- and resource-consuming NMR or x-ray crystallography experiments. Additionally, the ability to predict the binding potency of potential derivatives, renders the chemical synthesis of weak binders unnecessary. Here we show that the described scheme guides optimization of a lead compound, with a minimized resource and time investment.
The goal of this exercise was to modify the existing chemical scaffold in order to increase binding affinity to the target enzyme, while also maintaining favorable physicochemical properties (here, ligand efficiency and lipophilic efficiency) and biological activity. A docked complex of compound 2 and glutamate racemase was used to assess the most optimal locations for substituent addition. Based on that analysis, a library of 33 derivatives of compound 2 were subjected to a hybrid ensemble docking scheme, FERM-SMD, in order to rank their potential binding potencies. Of the 33 derivatives, 6 compounds were tested experimentally producing the final selection of compound 4. Compound 4 has increased binding affinity for the purified enzyme: 2.5 μM versus 9 μM, high ligand efficiency: 0.45 kcal/mol/atom, high lipohilic efficiency: 6.0, increased growth inhibition of B. subtilis: MIC50 = 260 μg/mL versus 720 μg/mL, and finally, effective bacterial cell lysis: EC50,lysis = 520 μg/mL. The FERM-SMD methodology has afforded a facile optimization from a high-LE hit with relatively low synthetic cost by precisely identify binding rank-ordering. Future studies will focus these techniques on increasingly more complex derivative libraries, in order to achieve even greater in vitro and biological activity.
Experimental Section
Docking and FERM-SMD: Virtual Screening
The original FERM-SMD method is described in great detail by Whalen and coworkers[12] BISA derivatives were prepared in silico using MOE v2011.10[18] (Chemical Computing Group). An ensemble of GR structures was generated using steered molecular dynamics simulation. Three structures were chosen at approximately the most closed (corresponding to 0 ps of simulation time), partially open (13.9 ps, simulation time), and fully open (20 ps, simulation time). Docking to the ensemble of GR structures was achieved using YASARA v9.11.9[19], which utilizes an optimized version of AutoDock 4[20]. Simulation cells were centered around the active site and expanded to include the residues surrounding the cleft entrance. Simulations cells had the following dimensions (in Å): “0 ps” receptor = 18.75 × 20.19 × 19.29; “13.9 ps” receptor = 19.08 × 21.56 × 18.07; and “20 ps” receptor = 18.94 × 21.31 × 18.93. Receptor-ligand docking combinations that resulted in more than one high-ranking pose were visually assessed by the author, and the pose that placed the core scaffold in a position most similar to the parent scaffold position was chosen as the “true” pose. Resulting binding energies and affinities for docking to all three receptors were then imported into Excel (Microsoft Office) and adjusted for receptor-protein solvation, producing a final FERMscore[12].
Compound Synthesis and Acquisition
Compound 1 (catalog #BAS 00124393, >98% purity) was acquired from Asinex, Ltd. (Moscow, Russia). Compounds 2 (catalog # 530646, 98% purity) was acquired from Sigma-Aldrich (St. Louis, MO, USA). Compound 15 (catalog # 5648649, 100% purity) was acquired from ChemBridge Corp. (San Diego, CA, USA). Compound 24 (catalog # STK695918, 98% purity) was acquired from Vitas-M Laboratories, Ltd. (Moscow, Russia). Compound 26 (catalog # Z57080960, 95% purity) was acquired from Enamine Ltd. (Kiev, Ukraine). Compounds 4, 18, and 29 were synthesized for the first time in published literature by collaborators at Enamine Ltd. (Kiev, Ukraine) following the synthetic process outlined in Scheme 1. 1H NMR spectra for each synthesized compound is provided in the Supplemental Information (Figures S3-S5). HPLC data showing purity analysis is available in the Supplemental Information for each newly synthesized compound (Figures S6-S8)
Protein Expression and Purification
Genes of glutamate racemase were isolated from B. subtilis, B. anthracis (two isozymes), and F. tularensis, and expressed in E. coli and purified using a protocol previously described by Whalen and coworkers[12] Briefly, hexa-histidine-tagged recombinant proteins were purified via a two-step process composed of cobalt-affinity (His-Select Affinity Resin, Sigma-Aldrich) and anion exchange (UNO Q Continuous Bed column, BioRad). Proteins were stored in buffer containing 100 mM NaCl, 50 mM Tris, 0.2 mM DTT, pH 8.0 at a concentration of 7-10 mg/mL. Molecular weight was confirmed via SDS-PAGE analysis (Figure S1), and protein foldedness was assessed using circular dichroism (Figure S2).
In Vitro Inhibition of Enzyme Activity
Steady-state kinetics for D to L racemization was measured using circular dicroism on a JASCO J-715 Spectropolarimeter. All compound stocks were made up in 50 mM potassium borate buffer pH 8.0, at concentrations varying from 25-100 mM, depending on compound solubility. Reactions were carried out at 25°C in 50 mM potassium borate buffer pH 8.0 with 1 μM purified enzyme. CD signal (mdeg) was measured continuously at 220nm for 10 min. Plots of CD versus time were fit linearly to obtain initial velocity. Substrate was varied from 0.25 to 5 mM. For Ki determination, three Michaelis-Menten curves were obtained for each inhibitor: one in the presence of no inhibitor, and two in varying concentrations of the inhibitor. A single data set, composed of three curves, was fit to a competitive inhibition model using GraphPad Prizm v.5.0[21], and the Ki is obtained as a best-fit value. For IC50 determination, reactions are supplemented with varying concentrations of the inhibitor, and the observed V0 in nmol/sec is normalized to a %-activity value based on an uninhibited reation. %-Activity values are plot versus the log of the inhibitor concentration. The data set is then fit to a log[inhibitor] versus response model (with variable slope) to calculate the IC50, using GraphPad Prizm v.5.0[21]. For LiPE calculations, compound logP values for the ionic species were calculated using MarvinSketch (ChemAxon).
In Vivo Inhibition of Bacterial Growth
A 5 mL culture of bacteria (B. subtilis DB104, E. coli Acella, or S. aureus ATCC 12600) was incubated overnight at 37 °C in Tryptic Soy Broth one day prior to the assay. 96-well plates were prepared with 2X media, phosphate-buffered saline, the compound of interest and a 20 uL inoculum of bacteria, totally 200 uL per well. Compound stocks were prepared in phosphate-buffered saline at a concentration of 10, 12.5 or 25 mM, depending on compound solubility. A serial dilution ranging from 0.1 to 3000 micromolar for the compound of interest was assayed. The overnight culture, at an optical density of approximately 2.0, was diluted 20-fold in water prior to inoculation, such that initial optical densities were approximately 0.01. A table of reagent volumes and diagram of plate layout can be found in Figure S9 of the Supplemental Information. Plates were mixed and incubated at 37 °C for 24 hr. Absorbance at 600 nm was measured on a GloMax-Mutli Detection System. MIC50 values were determined by fitting data to a log[inhibitor] versus response model using GraphPad Prism v.5.0[21]. The bottom and top values are constrained to 20% and 100%, respectively.
Cell Wall Lysis Assay
Bacterial cell wall lysis was assayed using a modification of the CytoTox-Glo assay[22] (Promega Corp.). 100 μL of the contents of a 96-well plate treated as described above were moved to a white, round-bottom, 96-well plate. 25 μL of the CytoTox-Glo reagent buffer was added to each well, mixed and incubated at room temperature in the dark for 15 min. Luminescence was measured on a GloMax-Multi Detection System. Luminescent values were normalized based on the absorbance value at 600 nm for each respective well. Data was then fit to a log[agonist] versus response model, with no upper or lower constraints, using GraphPad Prism v5.0[21].
Supplementary Material
Acknowledgements
This work was supported by the NIH in the form of grant numbers R01 GM097373-01A1 (M. A. S.) and AI076830 (M. A. S.), and the Chemical Biology Training Grant at UIUC (K. L. W.). A special thanks to Irina Yavnyuk and Tatiana Galushka for facilitating our fruitful collaboration with Enamine Ltd.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
References
- [1].Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Nat. Rev. Drug Discovery. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- [2].O’Shea R, Moser HE. J. Med. Chem. 2008;51:2891–2878. [Google Scholar]
- [3].Li JW-H, Vederas JC. Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- [4].Bugg TD, Walsh CT. Nat. Prod. Rep. 1992;9:199–215. doi: 10.1039/np9920900199. [DOI] [PubMed] [Google Scholar]
- [5] a).Belanger AE, Porter JC, Hatfull GF. J. Bacteriol. 2000;182:6854–6856. doi: 10.1128/jb.182.23.6854-6856.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) de Roubin MR, Mengin-Lecreulx D, van Heijenoort J. J. Gen. Microbiol. 1992;138(Pt 8):1751–1757. doi: 10.1099/00221287-138-8-1751. [DOI] [PubMed] [Google Scholar]; c) Hebeler BH, Young FE. J. Bacteriol. 1975;122:385–392. doi: 10.1128/jb.122.2.385-392.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lundqvist T, Fisher SL, Kern G, Folmer RH, Xue Y, Newton DT, Keating TA, Alm RA, de Jonge BL. Nature. 2007;447:817–822. doi: 10.1038/nature05689. [DOI] [PubMed] [Google Scholar]
- [7].Conti P, Tamborini L, Pinto A, Blondel A, Minoprio P, Mozzarelli A, De Micheli C. Chem. Rev. 2011;111:6919–6946. doi: 10.1021/cr2000702. [DOI] [PubMed] [Google Scholar]
- [8] a).LoVullo ED, Molins-Schneekloth CR, Schweizer HP, Pavelka MS., Jr. Microbiology. 2009;155:1152–1163. doi: 10.1099/mic.0.022491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shatalin KY, Neyfakh AA. FEMS Microbiol. Lett. 2005;245:315–319. doi: 10.1016/j.femsle.2005.03.029. [DOI] [PubMed] [Google Scholar]
- [9] a).de Dios A, Prieto L, Martin JA, Rubio A, Ezquerra J, Tebbe M, Lopez de Uralde B, Martin J, Sanchez A, LeTourneau DL, McGee JE, Boylan C, Parr TR, Jr., Smith MC. J. Med. Chem. 2002;45:4559–4570. doi: 10.1021/jm020901d. [DOI] [PubMed] [Google Scholar]; b) May M, Mehboob S, Mulhearn DC, Wang Z, Yu H, Thatcher GR, Santarsiero BD, Johnson ME, Mesecar AD. J. Mol. Biol. 2007;371:1219–1237. doi: 10.1016/j.jmb.2007.05.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Whalen KL, Pankow KL, Blanke SR, Spies MA. ACS Med. Chem. Lett. 2010;8:9–13. doi: 10.1021/ml900005b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Spies MA, Reese JG, Dodd D, Pankow KL, Blanke SR, Baudry J. J. Am. Chem. Soc. 2009;131:5274–5284. doi: 10.1021/ja809660g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Whalen KL, Chang KM, Spies MA. Mol. Inf. 2011;30:459–471. doi: 10.1002/minf.201100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Murray CW, Rees DC. Nat. Chem. 2009;1:187–192. doi: 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- [14] a).Hopkins AL, Groom CR, Alex A. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]; b) Hann M. Med. Chem. Commun. 2011;2:349–355. [Google Scholar]
- [15].Leeson PD, Springthorpe B. Nat. Rev. Drug Discovery. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- [16].Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. Lancet. 2001;357:1225–1240. doi: 10.1016/s0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- [17].Warren GL, Peishoff CE, Head MS. Computational and structural approaches to drug discovery : ligand-protein interactions. 1 ed Vol. 1. RSC Publishing; Cambridge: 2008. [Google Scholar]
- [18].1010 Sherbooke St. West, Suite #910. 2011.10 ed. Chemical Computing Group Inc.; Montreal, QC, Canada: 2011. H3A 2R7. [Google Scholar]
- [19] a).Krieger E. YASARA Biosciences GmbH. 12.4.1 ed Vienna, Austria: 2011. [Google Scholar]; b) Krieger E, Koraimann G, Vriend G. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- [20].Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Software G, editor. GraphPadPrizm. 5 ed San Diego, California, USA: 2007. [Google Scholar]
- [22].Niles AL, Moravec RA, Eric Hesselberth P, Scurria MA, Daily WJ, Riss TL. Anal. Biochem. 2007;366:197–206. doi: 10.1016/j.ab.2007.04.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.