

Abstract
Many of the gene mutations found in genetic disorders, including cancer, result in premature termination codons (PTCs) and the rapid degradation of their mRNAs by nonsense mediated RNA decay (NMD). We used virtual library screening (VLS) targeting a pocket in the SMG7 protein, a key component of the NMD mechanism, to identify compounds that disrupt the SMG7-UPF1 complex and inhibit NMD. Several of these compounds upregulated NMD targeted mRNAs at nanomolar concentrations with minimal toxicity in cell based assays. As expected, pharmacological NMD inhibition disrupted SMG7-UPF1 interactions. When used in cells with PTC mutated p53, pharmacological NMD inhibition combined with a PTC “read-through” drug led to restoration of full-length p53 protein, upregulation of p53 downstream transcripts, and cell death. These studies serve as proof-of-concept that pharmacological NMD inhibitors can restore mRNA integrity in the presence of PTC and be used as part of a strategy to restore full length protein in a variety of genetic diseases.
Keywords: nonsense mutations, nonsense mediated RNA decay, p53, tumor suppressor genes, gene expression
Introduction
Human genetic disorders are caused by diverse types of mutations. Many of these mutations, including nonsense mutations, frameshift mutations, and mutations that cause alternative splicing events, result in premature termination codons (PTCs). For example, approximately 15% of the mutations leading to Duchenne muscular dystophy (DMD), 10% of the mutations responsible for cystic fibrosis, and common β globin mutations responsible for thalassemia are PTC mutations (1–3). It has been estimated that up to 30% of all mutations resulting in human genetic disorders result in PTCs (4). In addition, many acquired mutations in cancer, including those that disable p53 and BRCA1, ATM, VHL, and NF1 and NF2, result in PTCs (5).
Many transcripts carrying a PTC are targeted for rapid degradation before they can be translated into protein through a multistep process termed nonsense mediated RNA decay (NMD). The molecular mechanism of NMD has not been fully delineated, though working models have been proposed. During the processing of mammalian pre mRNA, introns are excised and marked by a multi-protein exon junction complex (EJC) (6–8). When the translation complex pauses at a PTC that is upstream of an EJC, eukaryotic release factors recruit the RNA helicase UPF1, a vital component of the NMD mechanism (9). UPF1 then associates with UPF2, a component of the EJC, and is phosphorylated at several sites by SMG1 (10). Phosphorylated UPF1 recruits SMG-7 and SMG5, two 14-3-3 proteins which each bind to UPF1 and form a heterodimer, and results in the de-phosphorylation of UPF-1 by PP2A (11–12). The phosphorylation and de-phosphorylation of UPF1 are necessary steps prior to the transcript’s degradation by exonucleases (12–13).
One approach to treat genetic disorders and to restore tumor suppressor genes with PTC mutations is to pharmacologically promote ribosomes to read-through a PTC and produce a full-length protein (14). For example treating cells with the antibiotic gentamicin increases the full length expression of both PTC mutated β globin and PTC mutated p53 (15–16). Gentamicin treatment of cells with PTC mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) can increase cellular chloride transport (17–18) and patients with PTC mutations treated with intranasal gentamicin increase CFTR protein expression and improve the potential difference of nasal epithelial cells (19). While application of gentamicin or the closely related geneticin (G418) is not practical clinically, a recent high throughput screen identified a small molecule, Ataluren, which has similar properties but appears to be more potent and less toxic (20), and the use of antisense oligonucleotides to promote exon skipping are also being tested in muscular dystrophy (reviewed in (21)). The clinical activities of these approaches are likely to be enhanced with higher cellular concentrations of PTC mutated mRNA. Indeed, in vitro experiments have demonstrated that the expression of PTC mutated CFTR, as well as CFTR mediated chloride transport, are improved in gentamicin treated cells when UPF1 or UPF2 are also depleted (22).
Because translation is necessary for NMD (23), inhibitors of translation are effective inhibitors of NMD (24). Unfortunately these drugs are not candidates for clinical therapies because of general toxicity and, as translation is inhibited, the ultimate goal of restoring protein expression is not achieved. We have recently demonstrated that NMD inhibition can be achieved via other mechanisms. Specifically, we have shown that phosphorylation of the translation initiation factor eIF2α by a variety of cellular stresses inhibits NMD (5, 25–26). We have also determined that modest (80%) depletion of UPF1 or UPF2 can suppress NMD activity without diminishing the proliferation or survival of cells (26). These observations suggest that the pharmacological inhibition of NMD can be achieved with limited toxicity or adverse effects. We hypothesized that the combination of a drug that can bypass a PTC and/or promote exon skipping in combination with a drug that inhibits NMD would be a synergistic combination and serve as an effective platform to treat genetic diseases. To pursue this approach, we performed a virtual screen to identify commercially available compounds predicted to dock within a SMG7 pocket previously shown to be vital in the binding of UPF1. We then tested whether these compounds interfere with NMD, and using PTC mutated p53 as a proof of principle model, whether the pharmacological inhibition of NMD would make ribosomal read-through strategies more effective in producing full length proteins.
Materials and Methods
Virtual Library Screening
The crystallographic structure of SMG7 (PDB ID: 1ya0) was analyzed using ICM PocketFinder (Molsoft, LLC, La Jolla, CA). An appropriately sized pocket (based on criteria previously reported (27)) was selected as the target site for virtual library screening using ICM-VLS software (Molsoft, LLC, La Jolla CA). ICM-VLS uses global optimization with a biased probability Monte Carlo conformational search to rapidly dock fully flexible, full-atom models of the ligands to a set of grid potential maps calculated from the coordinates of the atoms in the protein receptor. Each ligand docking conformation is then evaluated with a scoring function. The ICM scoring function integrates van der Waals energy, electrostatics, hydrogen bonding, conformational entropy loss and solvation electrostatic energy change (28). ICM-VLS was used to dock the entire set of 436,115 compounds from Chembridge Express Library using default ICM-docking parameters on three 3.0-GHz Intel Xeon processors.
Cell lines and reagents
U2OS, Hela, 293, BJhTert, PBMC, MDA231, DU145, T98G and HCT116 cells were grown as previously described (29–30). N417 cells were grown in RPMI containing 10% FBS. Calu-6 (gift of E. Welch) and HDQP1 cell lines (DSMZ German Collection of Microorganism and Cell Cultures) were grown in DMEM with 10% Fetal Bovine Serum. Pools of β-Globin mRNA-expressing cells were generated as previously described (25). Compounds were obtained from Chembridge. UPF1 shRNA (26) and SMG7 shRNA (TRCN0000127998) were obtained from Sigma. Primer sequences for β-Globin, endogenous NMD targets, p53 and p53 downstream targets are available upon request.
Immunoblots and RNA assessment
Immunoblots were prepared as previously described (29) and membranes were probed with antibodies against p53 (Abcam Inc, ab26), SMG1 (Santa Cruz Biotechnology, Inc. 6-RE13), UPF1/Rent1 (SC H-300), SMG7 (A302-170A, Bethyl Laboratories, Inc). Immunoprecipitations were performed as described (10). Briefly, cells were lysed and incubated 30 mins at 4°C with buffer containing 50mM Tris-HCL at pH 7.4, 50mM NaCL, 0.05% Tween-20, 10mM tetrasodium pyrophosphate, 100mM NaF, 17.5 mM β-Glycerophosphate, 1mM PMSF and protease inhibitor (Roche). Lysates were clarified and incubated with 200μg/mL RNaseA (Qiagen) at room temperature for 15 mins with gentle rotation prior to 4°C overnight immunoprecipitation with the appropriate antibodies. Isolation of RNA for cDNA generation, real-time PCR, DRB treatment, RNA stability experiment, expression profiling and expression array data analysis were as described previously (25–26). For unbiased gene expression experiments, cells were treated with 50 μM NMDI14 for six hours, 100 μg/ml emetine for three hrs, or depleted of UPF1 and RNA was harvested, prepared, hybridized to Affymetrix HG-U133 Plus 2.0 GeneChips and analyzed as previously described (26).
Cell Viability Assay and Proliferation Assay
To assess viability cells were cultured in 6 well dishes and incubated with DMSO, G418, NMDI alone or G418 with NMDI together for the indicated hours. After incubations, cells and media were collected and cells viability was measured as described (31). To assess cell proliferation U2OS, Hela and BJ-htert cells were cultured in 6 well plates and, after 24hrs, treated with NMDI14 for 0, 24, 48 and 72hrs. The cells were collected and viable cells were counted by using the Countess Automated Cell Counter (Invitrogen).
Protein Synthesis (35S incorporation assay)
U2OS, Hela and BJ-hTERT cells were incubated with DMSO, NMDI14 or Emetine as indicated. The cells were labeled with 100 μCi/mL [35S] Methionine for 30 min and the protein were precipitated using standard TCA precipitation method as described previously (26). The precipitated protein was collected in glass fiber filters and [35S]Methionine incorporation was measured by using liquid scintillation counter (PerkinElmer, Inc.).
Results
Identification of potential nonsense mediated RNA decay inhibitors (NMDIs)
Prior studies have demonstrated that NMD activity is dependent on the formation of a UPF1-SMG7 complex (Fig 1A), which has been crystallographically resolved (11). Though UPF1 and SMG7 may have additional non-NMD roles, the interaction between the two appears to be unique for NMD. Thus the disruption of this complex is an ideal target for NMD inhibition while minimimzing the toxicity that might be seen with other known mechanism of NMD inhibitors, including translation inhibitors and inducers of eIF2α phosphorylation (24–25).
Fig 1.

Virtual library screening of SMG7 highlighting the interaction of SMG7 with UPF1. A. A model of the NMD. UPF1 interactions with the EJC results in recruitment of SMG7 and dephosphorylation of UPF1. B. Plot of SMG7 pocket sizes. 12 drug-binding pockets on the 3D structure of SMG7 were identified using the ICM-PocketFinder algorithm. The volume/areas of all twelve pockets are plotted with volume in cubed-angstroms (Å3) on the X-axis and pocket surface area in squared-angstroms (Å2) on the Y-axis. 15,000 drugs in clinical use have a range of volumes/areas represented approximately by the hashed blue square area, so a pocket with a volume/area within this region is suitable for VLS. The target pocket selected on SMG7 is outlined with a red box and falls within the suitable area. C. Image of the target pocket location on SMG7. The pocket is displayed as a gold-colored solid geometric object. The peptide backbone of SMG7 is depicted in ribbon diagram. D. Superimposition of the 31 compounds identified by VLS in their docked conformations within the target pocket on SMG7, which is at its interface with Upf1. The electrostatic molecular surface of SMG7 is depicted as a smooth contoured surface colored red for negative charge, blue for positive charge and white for hydrophobic areas.
We elected to take a 3D structure-activity approach to NMDI discovery by targeting this protein interface. Only one pocket on the 3D structure of SMG7 was both of suitable size and volume (between 150 and 550A3) and was lined with functionally sensitive amino acid side chains that mediate the interaction with UPF1 (11) (Fig 1B). This pocket was selected as the target site for virtual library screening of a collection of 315,102 compounds from the ChemBridge Express Library (San Diego, CA, USA). The resulting compounds achieving a docking score of <32 (see materials and methods) were further filtered by selecting those with extensive hydrogen bonds and van der Waals contacts, followed by hierarchical clustering for a diversity of scaffolds, resulting in the identification of 31 diverse compounds highly structurally and chemically compatible with the target SMG7 pocket (Fig 1C, 1D).
Identified compounds inhibit NMD in a cell based assay
To test these putative NMD inhibitors (NMDIs), we utilized a common and straight-forward assay for NMD. We first generated fibroblasts stably expressing either a wild-type β globin construct or a PTC 39 mutated β globin gene construct well established to be degraded by NMD (32). In our initial screen we treated cells expressing these genomic constructs with 50 μM of the 31 identified compounds for 6 hours, a condition which led to no gross toxicity in these cells. RNA was then harvested and expression of PTC β globin and wild-type β globin mRNA was determined using real time PCR.
We identified ten compounds that selectively increased the expression level of PTC 39 β globin greater than 2-fold, without affecting wild-type β globin expression, with a p value < 0.05 (Fig 2A). Because of rapid degradation by NMD, the steady-state mRNA expression of the PTC39 β globin was approximately 3% of the wild-type β globin expression. Treating cells with NMDI14 for 6 hours, for example, led to an increase of PTC 39 β globin to 12%, a relative four-fold increase that, if resulting in biologically active hemoglobin, would be sufficient to ameliorate the clinical symptoms of thalassemia (Fig 2B). When these ten compounds were tested at decreasing concentrations, we identified several NMDIs active at nanomolar concentrations, with some active over a wide range of concentrations and others exhibiting more narrow effective concentration which peaked at micromolar concentrations and displayed even a biphasic effect (Fig 2C and data not shown). These compounds, previously not reported to affect NMD, represent a diverse group of chemical scaffolds (Fig 2D and data not shown). We chose to focus the majority of our further studies on one of these compounds, ethyl 2-{[(6,7 --dimethyl--3-oxo-1,2,3,4-tetrahydro-2-quinoxalinyl)acetyl]amino}-4,5-dimethyl-3-thiophenecarboxylate (NMDI14), which demonstrated dose dependent activity, with significant activity at low concentrations.
Fig 2.
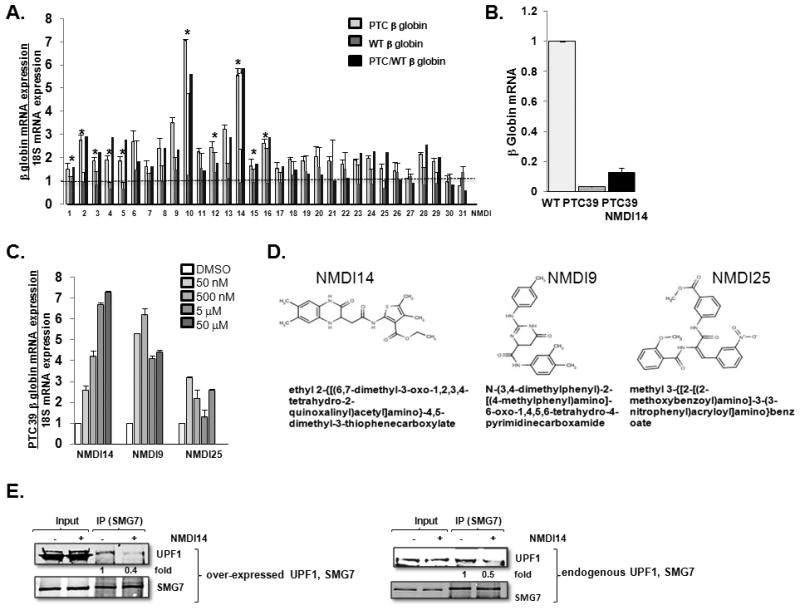
NMDIs increase expression and stability of NMD reporters. A. Wild type and PTC 39 β globin expressing U2OS cells were treated with 50 μM NMDIs for 6 hrs, and RNA was harvested and assessed for β globin expression by quantitative PCR. Black bars indicate the ratio of PTC to WT expression. * = p<0.05 by students t test. Experiments were biologically replicated five times (average ± SE displayed). B. Relative mRNA expression of wild-type β globin and PTC39 β globin in cells treated with DMSO, and PTC39 β globin in cells treated with NMDI14 for 6 hours. Experiments were repeated three times and average ± SE displayed. C. Induction of PTC 39 β globin mRNA after 6 hrs of treatment with various concentrations of selected drugs, ranging from 50 μM to 5 × 10−4 μM, normalized to expression with DMSO. Experiments were biologically replicated five times (average ± SE displayed). D. Structure of three NMDIs. E. UPF1 and SMG7 over-expressed 293T or control 293T cells were treated with DMSO or 50 μM of NMDI14 for 6 hours and, interaction of UPF1 and SMG7 was determined. Experiment is representative of three biological replicates, with fold change reflective of UPF1 immunoprecipitated/total UPF1 loaded.
Although the activity of our identified NMDIs does not depend on the mechanism of action, because these drugs were screened in silico to fit into the SMG7 pocket necessary to bind UPF1, we expected that NMDI14 would interfere with the interaction of UPF1 and SMG7. We immunoprecipitated SMG7, in the presence of RNAse and confirmed that both over-expressed (Fig 2E left) and endogenous (Fig 2E, right) SMG7 interacted with over-expressed and endogenous UPF1, respectively, and observed that this interaction was disrupted when cells were treated with NMDI14.
NMDI can be achieved with minimal cellular toxicity in multiple cell lines
NMD inhibition can result in the upregulation of non-mutated, mutated and alternatively spliced mRNAs (5). Thus NMD inhibition might be predicted to result in widespread biological effects. In fact, we have recently reported that the genetic inhibition of NMD subtly but significantly increases autophagy, presumably in an attempt to rid the cell of mutated and misfolded proteins that accumulate with NMD suppression (31). To determine the cellular toxicity of select NMDIs, we assessed viability in treated Hela, U2OS and Calu-6 cell lines, and in immortalized/non-transformed human fibroblasts. These cells were treated with 50 μM of NMDI14 and two other effective NMDIs, as an assessment of any class toxicity effects, for 48 hours, and viability was assessed. We noted almost no toxicity with NMDI14 and NMDI25, and minimal toxicity with NMDI9, suggesting the pharmacological inhibition of NMD is not unduly toxic in contrast to 24 hours of treatment with emetine which blocks NMD by inhibiting translation (Fig 3A).
Fig 3.

NMDIs are not unduly toxic. A. U2OS, Hela, Calu-6 and BJ-hTERT cells were treated with three NMDIs at 50 μM for 48 hours, and viability was assessed. Toxicity with translation/NMD inhibitor emetine is shown as control. Average of three readings shown. B. U2OS, Hela and BJ-hTERT cells were treated with NMDI14 at 5 μM. The cell numbers were determined at 0, 24, 48 and 72hrs. C. U2OS, Hela and BJ-hTERT cells were treated with 5 μM of NMDI14 for 6 and 24 hours. The cells were pulsed with S35 methionine (100 μCi/mL) for last one hour and protein synthesis was measured by S35 incorporation. The protein synthesis with translation/NMD inhibitor emetine is shown as control
To determine whether the effective pharmacological inhibition of NMD exerted a sublethal effect on proliferation, we assessed proliferation in NMDI14 treated transformed and non-transformed cell lines (Fig 3B). Three days of treatment resulted in no decrease in cell counts, demonstrating that the pharmacological inhibition of NMD can be achieved without subtle changes in proliferation.
Because suppression of translation is a potent mechanism to inhibit NMD, previously described inhibitors of NMD also inhibit translation. Although we did not predict this mechanism of action for our compounds, we tested protein synthesis after 6–24 hrs of NMDI14 treatment. During the last hour of treatment cells were pulsed with S35 methionine, and protein was then precipitated and the incorporated S35 assessed. NMDI14 had no effect on S35 incorporation (Fig 3C). Together these data suggest that the pharmacological inhibition of NMD can be achieved without cellular toxicity.
NMDI14 increases the expression of endogenous NMD targets
The effectiveness of NMDI14 in inhibiting NMD was originally determined by assessing the expression of a NMD degraded reporter transcript which harbors a premature stop codon (Fig 2). While NMD has been primarily appreciated as a mechanism to rapidly degrade mutated transcripts, we and others have demonstrated that a wide variety of non-mutated transcripts are also degraded by NMD which have several distinct motifs, as well as unknown features, which render them sensitive to NMD (25–26, 33). We noted that the steady state mRNA expression, reflective of both the rate of synthesis as well as the rate of decay, of seven non-mutated NMD targets which we have validated (26) increased in U2OS and/or Hela cells after treatment with NMDI14, whereas no such increase was seen in non-NMD targeted mRNAs (Fig 4A).
Fig 4.

NMDIs stabilize endogenous mRNA. A. U2OS Hela cells were treated with 50 μM of NMDI14 for 6 hrs, and endogenous non-mutated NMD target expression was assessed by real time PCR. Actin and eIF2B4 serve as negative controls. * = p<0.05 by student’s T test vs control. B. U2OS cells treated with NMDI14 at 50 μM for 6 hours. The whole gene expression was determined and compared with UPF1 knockdown and emetine treated U2OS cells. C. N417 cells, with a PTC mutated p53, were treated with 5 μM of NMDI14 for 24 hrs and p53 mRNA expression was assessed by real time PCR. Experiments were repeated three times and average ± standard error is displayed. * = p<0.05 by student’s T test vs DMSO. D. Relative p53 mRNA expression in U2OS cells and N417 cells treated with DMSO, and N417 cells treated with NMDI14 for 6 hours. Experiments were repeated three times and average ± SE displayed. E. p53 mRNA stability was determined, as described in text, in N417 and U2OS cells treated with DMSO or NMDI14 for six hours. Experiments were repeated twice, with average ± SE for each time point shown.
In order to assess potential off target effects of NMDI14, we used expression arrays to globally assess gene expression in U2OS cells treated with NMDI14, emetine, or with UPF1 depletion and compared this to gene expression in control U2OS cells. 941 genes were increased >1.5 fold with NMDI14 (Fig 4B). There was a significant overlap (22%, p=1.2E−25, Chitest) of genes upregulated by NMDI with genes upregulated by emetine treatment (which inhibits NMD potently but clearly has other effects), and with UPF1 depletion (11%). In comparison, 20% of the 5290 genes upregulated by emetine were also upregulated in UPF1 depleted cells. The overlap of genes regulated by NMDI14 and those regulated by either emetine or UPF1 depletion is significantly higher than what would be expected by chance (~3%). Genes upregulated by NMDI14 but not regulated by UPF1 or emetine, likely representative of off-target effects, were not particularly enriched in any specific functional group, and only demonstrated a greater than 2 fold enrichment in ossification, actin binding, and cytoskeletal binding, suggesting that NMDI14 may not have specific toxicities.
We next determined whether our NMDIs are effective in up-regulating mutated endogenously expressed mRNAs. The small cell lung cancer cell line N417 has a p53 tumor suppressor gene that is deleted on one allele and on the other allele carries a PTC mutation in the 298th codon that renders it a target for NMD (34). When treated with an NMDI we noted increased expression of the mutated p53 mRNA (Fig 4C). Similar effects were seen in the breast cancer HDQP-I cell line, which carries a PTC mutation in the 213th codon of the p53 gene (35), and the lung adenocarcinoma cell line Calu-6 which carries a codon 196 PTC mutated p53 (36). The treatment of N417 cells with NMDI14 for 6 hours led to a steady state expression of p53 similar to that seen in U2OS cells (Fig 4D). No such effect was seen in cells expressing wild-type p53 in two other cell lines (Fig 4C). To directly assess p53 mRNA stability, N417 and U2OS cells were treated with vehicle or NMDI14 for 6 hours, and the RNA polymerase II inhibitor 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) was added to stop mRNA synthesis. RNA was collected at 0, 1.5, and 3 hrs after addition of DRB, and expression of p53 mRNA was assessed by real time PCR. NMDI14 significantly increased the stability of PTC mutated p53 mRNA in N417 cells, without altering the stability of wild-type p53 in NMDI treated U2OS cells (Fig 4E).
NMDI, along with a PTC “read-through” drug, increases the protein expression of endogenous NMD targets
While NMDI14 increases the stability and expression of NMD targeted mRNAs, a clinically effective strategy demands the expression of a full length, biologically active protein. As noted previously, there are several drugs that can promote read-through of PTCs. Although G418 has a modest effect of suppressing NMD, the most effective agent at non-toxic concentrations, Ataluren, does not protect mRNAs from NMD (20). We reasoned that if there were more mutated mRNA present, PTC read-through drugs might be more effective.
We noted minimal expression of full length p53 in N417 cells treated with G418, but significantly more in cells treated with both G418 and NMDI14 (Fig 5A). This increase was seen over a variety of dose concentrations of G418, demonstrating that in the presence of NMDI14 higher full length p53 expression can be achieved at lower and potentially less toxic G418 concentrations (Fig 5B). Similarly, the HDQP-I and Calu-6 cell lines which carry p53 PTC mutations showed significant increased full length p53 expression with the combination of G418 and NMDI14 (Fig 5A). The expression of wild-type p53 in HCT116 cells was unaltered by these treatments (Fig 5A). The effectiveness of the two drugs varied amongst cell lines, which could either reflect variability in ribosomal read-through (which depends on the termination codon sequence (20)), or cell-type dependent NMDI14 metabolism. Consistent with an increase in full length/active p53 restoration, the combination of both drugs led to a synergistic increase in multiple p53 target genes mRNAs including p21, BAX and PUMA in N417 and cells (Fig 5C). This led to mRNAs levels of the pro-apoptotic BAX and PUMA similar to those seen in U2OS cells (Fig 5D). As expected by the lack of p53 mRNA induction in non-mutated p53 cells with NMDI14 (Fig 4C), in p53 wild-type cell lines HCT116 and U2OS NMDIs had no effect on p53 protein expression or p53 target gene expression over a wide range of concentrations (Fig 5A, C and data not shwon). Together, these data indicate that NMDIs can increase endogenous mutated mRNAs and, in combination with a PTC-bypass drug, can result in full-length protein production.
Fig 5.

Combination of read-through drug and NMDI restores full length PTC mutated p53 and leads to cell death. A. Cells were treated with DMSO, G418 (200 μg/mL), NMDI14 (5 μM) or both for 48 hrs, and, full length p53 was assessed in N417, HDQP-1 and Calu-6 cells or, as a control HCT116 cells. B. N417 cells were treated with DMSO, 5 μM of NMDI14 and, 150, 175 and 400 μg/mL of G418 or both for 48 hours. The full length p53 was determined and U2OS serves as a control. * = p<0.05 by student’s T test vs DMSO, # = p< 0.05 vs G418 alone. C. mRNA expression of p53 downstream targets were assessed in N417 cells, U2OS cells, or HCT 116 cells with 48 hrs treatment of DMSO, G418 (200 μg/mL), NMDI (5 μM), or both. D. mRNA expression of Bax and Puma were detected in DMSO and NMDI14 treated cells by real time PCR. Experiments were repeated three times and average ± standard error is displayed. E. N417, HDQP-1 and U2OS cell were treated with DMSO, 200 μg/mL of G418, 5 μM of NMDI or both for 48 hrs and viability was assessed. Experiments were repeated twice in duplicates, with average ± standard error displayed. * = p<0.05 by student’s T test vs DMSO, # = p< 0.05 vs G418 alone. F. Additional cell lines with p53 deletion or missense mutations were treated with DMSO, G418 (200 μg/mL), NMDI (5 μM) and viability was assessed. Experiments were repeated twice in duplicates, with average ± standard error displayed.
The restoration of full-length p53 with NMD inhibition and a “read-through” drug leads to cell death in cells with a PTC mutated p53
The frequently mutated p53 gene is a tumor suppressor which can induce apoptosis. The re-establishment of p53 in cancer cells, with or without chemo/radiation, has been a therapeutic strategy pursued in a wide variety of cancer for almost two decades, but this approach has been hampered by problems in delivery of wild-type p53 (37–38). Although NMDIs are relatively non-toxic, we hypothesized that the combination of NMDI with a “read through” drug would restore full-length p53 and result in a p53-mediated cell death. We treated N417 and HDQP-I cells with a combination of read through drug and a dose of NMDI14 that results in full length p53 expression. We noted a synergistic cell death only with the combination of drugs that led to full-length p53 expression (Fig 5E). No such death was seen when G418 and NMDI14 was used on cells with wild-type p53 including U2OS and primary peripheral blood mononuclear cells (PBMCs) (Fig 5E).
Although the combination of G418 and NMDI14 was not toxic in p53 wild-type cells, we examined a number of additional cell lines as controls (Fig 5F). These included HCT116 p53 wild-type and isogenic HCT116 p53 null cells, to determine whether the combination of NMDI14 and G418 is toxic only to cells without a functional p53. We also tested the breast cancer cell line MDA-231, the prostate cancer cell line DU145, and the glioblastoma cell line T98G, all of which carry single nucleotide p53 mutations which do not result in PTCs and are thus not NMD targets. As with other cell lines we examined, 48 hr treatment with NMDI14 alone was minimally toxic (<5% cell death) in these cell lines. G418 alone showed variable toxicity. However, similar to the p53 wild-type cells and in contrast to those cell lines with NMD provoking p53 mutations. In no case did the addition of NMDI14 and G418 lead to a synergistic cell death.
Discussion
We have used a virtual screening strategy to identify small compounds that inhibit NMD. The inhibition of NMD augments the expression of PTC mutated mRNAs and non-mutated mRNAs normally targeted by NMD. Several of these identified NMD inhibitors, representing diverse chemical backbones, are active at the nanomolar level even without further chemical modification to optimize their solubilities or pharmacokinetic properties. The few pharmacological inhibitors previously described have been useful in delineating the mechanism of NMD, but either inhibit translation, interfere with mRNA splicing, have not been demonstrated to upregulate enogenous NMD targets, have weak activity, do not synergize with read-through drugs, were identified through functional screening assays, and/or have unknown mechanisms of action (24, 39–41). The rational design of our screening makes it likely that our identified compounds are more specific with less off-target effects than these. Although several of our NMDIs do not appear excessively toxic in a variety of cell lines and have no effect on the expression of mRNAs that are not targeted by NMD, further work is needed to document and potentially improve the specificity, in vivo effectiveness, and off-target toxicity of these lead compounds.
Because of the important role NMD plays in degrading many mutated, alternatively spliced, and physiologically expressed mRNAs, even specific inhibition of NMD without any off-target effects might be theorized to be toxic. Indeed, the complete depletion of UPF2 in mice results in non-viability (42). However, we and others have demonstrated that the more modest depletion of UPF1 and UPF2, through shRNA, does not adversely affect cell proliferation or survival (26). In addition, inhibition of NMD by UPF2 deletion in murine hematopoietic cells results only in diminished hematopoietic progenitor cells with almost no effect on differentiated hematopoietic cells (42), and widespread constitutive expression of a dominant negative UPF1 only modestly affects the differentiating mouse thymus and spleen (43). Although the inhibition of NMD by these stresses normally occurs in growing tumors and in fact is necessary for the three-dimensional growth of cells in soft agar and as explants, the depletion of UPF1 from cells with shRNA does not increase their three dimensional growth, and thus we would not expect inhibition of NMD to necessarily augment tumorigenesis (26). The future clinical use of these or other NMD inhibitors, however, will obviously require further studies to determine the short and long-term effects of potent NMD inhibition.
Although NMD requires the pausing of a ribosome at a PTC, the use of the PTC read through drug Ataluren does not inhibit NMD, potentially because only a small percentage of mutated mRNAs are read through by Ataluren. Thus we have predicted that Ataluren, as well as other strategies, would be more effective in the presence of more substrate mRNA. We have validated this approach by demonstrating that the combination of a read through drug and NMD inhibitor results in the increased protein expression of a PTC mutated p53, resulting in improved p53 function. Many genetic disorders have mutations that either directly (non-sense) or indirectly (e.g. splicing mutations, additions or deletions that alter reading frame, alternations in poly-A processing) can render a mRNA sensitive to NMD. These include cancer (e.g. p53 and BRCA1). Because truncated proteins may be able to replicate the function of the full length protein, may be completely non-functional, or may act as dominant negatives against the full normal allele, the effects of inhibiting NMD will have to be carefully considered, particularly without a simultaneous consideration of restoring a full length protein with ribosome read though. However, based on our data the concomitant pharmacological inhibition of NMD remains an attractive therapeutic strategy for many genetic diseases, including cancer.
Acknowledgments
Financial Support: NIH and the European Community and the Children’s Cancer and Blood Foundation
Supported by RO1DK081641 (L.B.G), 1DP2OD004631 (TC), UL1 TR000038 from the National Center for the Advancement of Translational Science (NCATS), National Institutes of Health (L.B.G and T.C), R01HL102449 and FP7-HEALTH-2012-INNOVATION from the European Community and the Children’s Cancer and Blood Foundation (S.R.). L.B.G. is the Saul Farber Associate Professor of Medicine
Footnotes
The authors declare no conflicts of interest
References
- 1.Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 2.Maquat LE, Kinniburgh AJ, Rachmilewitz EA, Ross J. Unstable beta-globin mRNA in mRNA-deficient beta o thalassemia. Cell. 1981;27:543–53. doi: 10.1016/0092-8674(81)90396-2. [DOI] [PubMed] [Google Scholar]
- 3.Kan YW, Lee KY, Furbetta M, Angius A, Cao A. Polymorphism of DNA sequence in the beta-globin gene region. Application to prenatal diagnosis of beta 0 thalassemia in Sardinia. N Engl J Med. 1980;302:185–8. doi: 10.1056/NEJM198001243020401. [DOI] [PubMed] [Google Scholar]
- 4.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 5.Gardner LB. Nonsense-mediated RNA decay regulation by cellular stress: implications for tumorigenesis. Mol Cancer Res. 2010;8:295–308. doi: 10.1158/1541-7786.MCR-09-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lykke-Andersen J, Shu MD, Steitz JA. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell. 2000;103:1121–31. doi: 10.1016/s0092-8674(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 7.Lykke-Andersen J, Shu MD, Steitz JA. Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science. 2001;293:1836–9. doi: 10.1126/science.1062786. [DOI] [PubMed] [Google Scholar]
- 8.Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30:13–9. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 9.Czaplinski K, Ruiz-Echevarria MJ, Paushkin SV, Han X, Weng Y, Perlick HA, et al. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev. 1998;12:1665–77. doi: 10.1101/gad.12.11.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada-Katsuhata Y, Yamashita A, Kutsuzawa K, Izumi N, Hirahara F, Ohno S. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012;40:1251–66. doi: 10.1093/nar/gkr791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuhara N, Ebert J, Unterholzner L, Lindner D, Izaurralde E, Conti E. SMG7 is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol Cell. 2005;17:537–47. doi: 10.1016/j.molcel.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Ohnishi T, Yamashita A, Kashima I, Schell T, Anders KR, Grimson A, et al. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol Cell. 2003;12:1187–200. doi: 10.1016/s1097-2765(03)00443-x. [DOI] [PubMed] [Google Scholar]
- 13.Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, et al. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006;20:355–67. doi: 10.1101/gad.1389006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke JF, Mogg AE. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 1985;13:6265–72. doi: 10.1093/nar/13.17.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salvatori F, Breveglieri G, Zuccato C, Finotti A, Bianchi N, Borgatti M, et al. Production of beta-globin and adult hemoglobin following G418 treatment of erythroid precursor cells from homozygous beta(0)39 thalassemia patients. Am J Hematol. 2009;84:720–8. doi: 10.1002/ajh.21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Floquet C, Deforges J, Rousset JP, Bidou L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011;39:3350–62. doi: 10.1093/nar/gkq1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–9. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 18.Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–4. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- 19.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–41. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 20.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 21.Nelson SF, Crosbie RH, Miceli MC, Spencer MJ. Emerging genetic therapies to treat Duchenne muscular dystrophy. Curr Opin Neurol. 2009;22:532–8. doi: 10.1097/WCO.0b013e32832fd487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linde L, Boelz S, Nissim-Rafinia M, Oren YS, Wilschanski M, Yaacov Y, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117:683–92. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishigaki Y, Li X, Serin G, Maquat LE. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell. 2001;106:607–17. doi: 10.1016/s0092-8674(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 24.Dang Y, Low WK, Xu J, Gehring NH, Dietz HC, Romo D, et al. Inhibition of nonsense-mediated mRNA decay by the natural product pateamine A through eukaryotic initiation factor 4AIII. J Biol Chem. 2009 doi: 10.1074/jbc.M109.009985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol. 2008;28:3729–41. doi: 10.1128/MCB.02284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, et al. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol. 2011;31:3670–80. doi: 10.1128/MCB.05704-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cardozo T, Abagyan R. Druggability of SCF ubiquitin ligase-protein interfaces. Methods Enzymol. 2005;399:634–53. doi: 10.1016/S0076-6879(05)99042-3. [DOI] [PubMed] [Google Scholar]
- 28.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 29.Martin L, Kimball SR, Gardner LB. Regulation of the unfolded protein response by eif2bdelta isoforms. J Biol Chem. 2010;285:31944–53. doi: 10.1074/jbc.M110.153148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin L, Rainey M, Santocanale C, Gardner LB. Hypoxic activation of ATR and the suppression of the initiation of DNA replication through cdc6 degradation. Oncogene. 2012;31:4076–84. doi: 10.1038/onc.2011.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wengrod J, Martin L, Wang D, Frischmeyer-Guerrerio P, Dietz HC, Gardner LB. The inhibition of nonsense mediated RNA decay activates autophagy. Mol Cell Biol. 2013 doi: 10.1128/MCB.00174-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang JC, Kan YW. beta 0 thalassemia, a nonsense mutation in man. Proc Natl Acad Sci U S A. 1979;76:2886–9. doi: 10.1073/pnas.76.6.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–8. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- 34.Mitsudomi T, Steinberg SM, Nau MM, Carbone D, D’Amico D, Bodner S, et al. p53 gene mutations in non-small-cell lung cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene. 1992;7:171–80. [PubMed] [Google Scholar]
- 35.Wang CS, Goulet F, Lavoie J, Drouin R, Auger F, Champetier S, et al. Establishment and characterization of a new cell line derived from a human primary breast carcinoma. Cancer Genet Cytogenet. 2000;120:58–72. doi: 10.1016/s0165-4608(99)00253-8. [DOI] [PubMed] [Google Scholar]
- 36.Lehman TA, Bennett WP, Metcalf RA, Welsh JA, Ecker J, Modali RV, et al. p53 mutations, ras mutations, and p53-heat shock 70 protein complexes in human lung carcinoma cell lines. Cancer Res. 1991;51:4090–6. [PubMed] [Google Scholar]
- 37.Fujiwara T, Grimm EA, Mukhopadhyay T, Zhang WW, Owen-Schaub LB, Roth JA. Induction of chemosensitivity in human lung cancer cells in vivo by adenovirus-mediated transfer of the wild-type p53 gene. Cancer Res. 1994;54:2287–91. [PubMed] [Google Scholar]
- 38.Gabrilovich DI. INGN 201 (Advexin): adenoviral p53 gene therapy for cancer. Expert Opin Biol Ther. 2006;6:823–32. doi: 10.1517/14712598.6.8.823. [DOI] [PubMed] [Google Scholar]
- 39.Keeling KM, Wang D, Dai Y, Murugesan S, Chenna B, Clark J, et al. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE. 2013;8:e60478. doi: 10.1371/journal.pone.0060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Durand S, Cougot N, Mahuteau-Betzer F, Nguyen CH, Grierson DS, Bertrand E, et al. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J Cell Biol. 2007;178:1145–60. doi: 10.1083/jcb.200611086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Hilarion S, Beghyn T, Jia J, Debreuck N, Berte G, Mamchaoui K, et al. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012;7:58. doi: 10.1186/1750-1172-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, et al. NMD is essential for hematopoietic stem and progenitor cells and for eliminating byproducts of programmed DNA rearrangements. Genes Dev. 2008;22:1381–96. doi: 10.1101/gad.468808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frischmeyer-Guerrerio PA, Montgomery RA, Warren DS, Cooke SK, Lutz J, Sonnenday CJ, et al. Perturbation of thymocyte development in nonsense-mediated decay (NMD)-deficient mice. Proc Natl Acad Sci U S A. 2011;108:10638–43. doi: 10.1073/pnas.1019352108. [DOI] [PMC free article] [PubMed] [Google Scholar]