

Abstract
The conserved oligomeric Golgi (COG) complex is a soluble hetero-octamer associated with the cytoplasmic surface of the Golgi. Mammalian somatic cell mutants lacking the Cog1 (ldlB) or Cog2 (ldlC) subunits exhibit pleiotropic defects in Golgi-associated glycoprotein and glycolipid processing that suggest COG is involved in the localization, transport, and/or function of multiple Golgi processing proteins. We have identified a set of COG-sensitive, integral membrane Golgi proteins called GEARs (mannosidase II, GOS-28, GS15, GPP130, CASP, giantin, and golgin-84) whose abundances were reduced in the mutant cells and, in some cases, increased in COG-overexpressing cells. In the mutants, some GEARs were abnormally localized in the endoplasmic reticulum and were degraded by proteasomes. The distributions of the GEARs were altered by small interfering RNA depletion of ε-COP in wild-type cells under conditions in which COG-insensitive proteins were unaffected. Furthermore, synthetic phenotypes arose in mutants deficient in both ε-COP and either Cog1 or Cog2. COG and COPI may work in concert to ensure the proper retention or retrieval of a subset of proteins in the Golgi, and COG helps prevent the endoplasmic reticulum accumulation and degradation of some GEARs.
INTRODUCTION
Several multisubunit peripheral membrane protein complexes are thought to play key roles in controlling Golgi-associated membrane trafficking and glycoconjugate processing (Waters et al., 1991; Barlowe et al., 2002; Schekman and Orci, 1996; Sacher et al., 1998; Siniossoglou and Pelham, 2001; Conibear et al., 2003; Storrie and Nilsson 2002; Roth, 2002). One of these is the conserved oligomeric Golgi (COG) complex (Krieger et al., 1981; Kingsley et al., 1986; Podos et al., 1994; Wuestehube et al., 1996; VanRheenen et al., 1998, 1999; Walter et al., 1998; Chatterton et al., 1999; Kim et al., 1999, 2001; Spelbrink and Nothwehr, 1999; Whyte and Munro, 2001; Loh and Hong, 2002; Ram et al., 2002; Suvorova et al., 2002; Ungar et al., 2002; Farkas et al., 2003). Mutations in COG subunits (Cog1–8) have been shown to affect the structure and function of the Golgi in yeast, Drosophila melanogaster sperm, and mammalian somatic cells. Compromising COG function can cause defects in glycoconjugate synthesis, intracellular protein sorting, protein secretion and, in some cases, cell growth. For example, in mammalian recessive, null, Chinese hamster ovary (CHO) cell mutants called ldlB (Cog1-negative) and ldlC (Cog2-negative), multiple cisternae are dilated (Ungar et al., 2002), and there are pleiotropic defects in a number of medial- and trans-Golgi–associated reactions affecting virtually all N-linked, O-linked, and lipid-linked glycoconjugates (Kingsley et al., 1986). These glycoconjugate defects are somewhat reminiscent of those in brefeldin A-treated cells, in which the Golgi disintegrates and early Golgi processing enzymes are relocated to the endoplasmic reticulum (ER) (Doms et al., 1989; Perkel et al., 1989; van Echten et al., 1990; Sampath et al., 1992). The diversity and heterogeneity (e.g., partial N-linked sugar endoglycosidase H sensitivity) of these defects suggested that the COG mutations affect the regulation, compartmentalization, or activity of multiple Golgi glycosylation enzymes and/or their substrate transporters without substantially disrupting secretion or endocytosis (Kingsley et al., 1986; Reddy and Krieger, 1989). The activities of these proteins depend on their proper intra-Golgi localization and appropriate intralumenal environments (e.g., pH) (Harris and Waters, 1996; Skrincosky et al., 1997; Axelsson et al., 2001; Martínez-Menarguez et al., 2001; Mironov et al., 2001; Opat et al., 2001; Berger, 2002; Puri et al., 2002; Roth, 2002; Zerfaoui et al., 2002). Thus, COG might play a role directly or indirectly in resident Golgi proteins' transport to, retention at, or retrieval to appropriate sites, or otherwise determine the Golgi's structure and/or lumenal environment (Kingsley et al., 1986).
A direct role for COG in controlling anterograde or retrograde membrane trafficking was suggested by its ability to stimulate an in vitro intra-Golgi transport and glycosylation assay (Walter et al., 1998) and by genetic studies in yeast that have identified a large number of COG-interacting genes that encode proteins implicated in Golgi trafficking (VanRheenen et al., 1998, 1999; Kim et al., 1999, 2001; Spelbrink and Nothwehr, 1999; Whyte and Munro, 2001; Ram et al., 2002; Suvorova et al., 2002). Yeast COG has been proposed to function as a vesicle tethering factor in anterograde ER-to-Golgi trafficking (VanRheenen et al., 1998, 1999) or in COPI-mediated retrograde trafficking (Ram et al., 2002; Suvorova et al., 2002), or in cargo sorting during exit from the ER (Morsomme and Riezman, 2002; Morsomme et al., 2003).
Here, we have identified seven COG mutation-sensitive Golgi resident integral membrane proteins (GEARs) among a wide variety of mammalian secretory pathway-related proteins. Their abundances were reduced in COG-deficient mutant cells and, in some cases, increased in COG-overexpressing cells. In the mutant cells, some GEARs were abnormally accumulated in or mislocalized to the ER and were degraded by proteasomes. Thus, COG selectively influences trafficking between the Golgi and the ER. Short interfering RNA (siRNA)-mediated ε-COP depletion established that COG and ε-COP, at least in part, contribute to similar functions necessary for normal Golgi organization (e.g., establishing the proper distribution of GEARs). The results, including the generation of synthetic phenotypes in cells deficient in ε-COP and either Cog1 or Cog2, raise the possibility that COG functions in concert with COPI to ensure the proper trafficking and localization of GEARs in the Golgi.
MATERIALS AND METHODS
Materials and Antibodies
Reagents and sources were as follows: ECL Plus detection kit (Amersham Biosciences, Piscataway, NJ); methionine- and cysteine-free Ham's F-12 medium (Invitrogen, Carlsbad, CA); Expre35S35S protein labeling mix (PerkinElmer Life Sciences, Boston, MA); MG132 and lactacystin (Calbiochem, San Diego, CA); and stock solutions (10 mM) prepared in dimethyl sulfoxide (DMSO), cycloheximide (Sigma-Aldrich, St. Louis, MO). The COG subunit expression plasmids (pLDLB-1 for Cog1, pLDLC-1 for Cog2, and pHM6-COG7 for hemagglutinin (HA)-tagged Cog7) used for establishing the COG-overexpressing cell lines were described previously (Podos et al., 1994; Chatterton et al., 1999; Ungar et al., 2002).
Antibodies used for immunoblot (IB) and immunofluorescence (IF) studies were obtained from standard commercial sources and as gifts from generous individual investigators or generated by us (see below). Antibodies (and their dilutions) were as follows: rabbit polyclonal antibodies: affinity-purified anti-Cog1 antibody (IB, 1:2000; Ungar et al., 2002), affinity-purified anti-Cog2 antibody (IB, 1:1000; Podos et al., 1994; Ungar et al., 2002), affinity-purified anti-Cog3 antibody (IB, 1:10,000; Suvorova et al., 2001), anti-Cog4 antiserum (IB, 1:2000), affinity-purified anti-Cog5 antibody (IB, 1:500; Walter et al., 1998), anti-Cog6 antiserum (IB, 1:1000), anti-Cog7 antiserum (IB, 1:2000), anti-Cog8 antiserum (IB, 1:2000), anti-mannosidase II antiserum (IB, 1:2000; IF, 1:1000; a gift from Kelley Moremen, University of Georgia, Athens, GA), anti-GM130 antibody (IF 1:150; from Nobuhiro Nakamura, Kanazawa University, Ishikawa, Japan), anti-ERp72 antibody (IF, 1:500; Stressgen Biotechnologies, San Diego, CA), anti-GPP130 antibody (IB, 1:1000; IF, 1:500; Covance), anti-giantin antibody (IB, 1:10,000; IF, 1:2000; Covance, Berkeley, CA), anti-CASP antiserum (IB, 1:1000; IF, 1:200; from Sean Munro, MRC, Cambridge, United Kingdom), anti-p115 antibody (IB, 1:400; from Gerry Waters, Princeton University, Princeton, NJ), anti-syntaxin 5 antibody (IB, 1:500; from James Rothman, Memorial Sloan-Kettering Cancer Center, New York, NY and William Balch, The Scripps Research Institute, La Jolla, CA), anti-α-COP antibody (IB, 1:1000; Affinity Bioreagents, Golden, CO), anti-β-COP antibody (IF, 1:200; Affinity Bioreagents), anti-rab6 antibody C-19 (IB, 1:250; Santa Cruz Biotechnology, Santa Cruz, CA), anti-ε-COP antiserum [#310] (IB, 1:5000; IF, 1:1500; Guo et al., 1996), anti-Sec61β antibody (IF, 1:4000; Upstate Biotechnology, Lake Placid, NY); and murine monoclonal antibodies: anti-GM130 antibody (IB, 1:250; IF, 1:150; BD Biosciences, San Jose, CA), anti-Cog1/ldlBp antibody (IF, 1:100; BD Biosciences), anti-GOS-28 antibody (IB, 1:3000; IF, 1:1000; BD Biosciences), anti-GS15 antibody (IB, 1:250, IF; 1:150; BD Biosciences), anti-golgin-84 antibody (IB, 1:250, IF, 1:150; BD Biosciences), anti-membrin antibody (IB, 1:1000; IF, 1:250; Stressgen Biotechnologies), anti-β-COP antibody [maD] (IB, 1:1000; Sigma-Aldrich).
Cell Culture and Transfection
Wild-type CHO, ldlB, ldlC, ldlB[COG1], and ldlC[COG2] cells were maintained and incubated for experiments as described previously (Podos et al., 1994; Chatterton et al., 1999) at 37°C unless otherwise noted. Wild-type CHO and ldlB cells were transfected with the indicated expression plasmids by using FuGENE 6 (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions. The transfected cells were selected in medium A (Ham's F-12 supplemented with 2 mM glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin) containing 5% fetal bovine serum (medium B) and 0.3 mg/ml active Geneticin (Invitrogen) (medium C), and individual colonies were harvested and grown to mass culture. The relative levels of expression of the proteins of interest were determined using immunoblotting (see below), and those cell lines overexpressing the proteins were used in subsequent experiments.
Construction of the GS15 Expression Plasmid
The 780-base pair EcoRV/ApaI DNA fragment, containing the entire open reading frame for murine GS15, from the expressed sequence tag (EST) clone (IMAGE 4164150; Invitrogen) was cloned into the EcoRV/ApaI sites of pcDNA3.1 (Invitrogen) to construct the pGS15-1 plasmid.
Radioactive Labeling and Immunoprecipitation
Metabolic labeling of cells with Expre35S35S (a mixture of [35S]methionine and [35S]cysteine) in methionine- and cysteine-free medium A supplemented with 5% (vol/vol) dialyzed fetal bovine serum was performed as described previously (Kozarsky et al., 1986).
Immunoprecipitation of GOS-28 from radioactive cell lysates by using monoclonal anti-GOS-28 antibody was carried out as described previously (Oka et al., 1991).
Transfection of the siRNA Duplex for ε-COP
Hamster ε-COP was targeted with a siRNA duplex (sense, AGAUGAGGAUGCCACUCUCAC; antisense, GAGAGUGGCAUCCUCAUCUUG; Dharmacon Research, Lafayette, CO). Preparation and transfection of the siRNA duplex were performed as described previously (Novina et al., 2002). A second hamster ε-COP siRNA duplex (sense, GAAGCUGCAAGAAGCCUACUA; antisense, GUAGGCUUCUUGCAGCUUCUC) was also used. On day 0, cells (∼20,000–30,000 cells/well) were set in medium B in 12-well dishes. On day 1, cells were transfected with the siRNA duplex (0.1 μM) by using OligofectAMINE (Invitrogen) in medium B according to the manufacturer's instruction. After incubation at 34°C overnight, an equal volume of fresh medium was added, and cells were further incubated at the same temperature. At 48 h after transfection, cells were fixed with 2% (wt/vol) paraformaldehyde before immunofluorescence analysis.
Immunoblotting and Immunofluorescence Microscopy
Cells were grown at 37°C in medium B or medium C until nearly confluent, harvested, and solubilized by addition of buffer A (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol) with scraping, and heated at 95°C for 5–10 min. The lysates (20 μg of protein) were then fractionated by SDS-polyacrylamide (between 5 and 12%) gel electrophoresis, and the proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA) and immunoblotted with the indicated antibodies using the ECL Plus detection kit (Amersham Biosciences) according to the manufacturer's instructions.
Cells grown in medium B or medium C until subconfluent were fixed, stained, and analyzed by immunofluorescence microscopy as described previously (Ungar et al., 2002). Images were obtained with an LSM510 confocal microscope Carl Zeiss, Thornwood, NY).
Preparation of Antibodies against Cog4, 6, 7, and 8
To prepare a recombinant fragment of the Cog6 protein, a segment of the COG6 cDNA, coding for amino acids 427–605, was cloned into the expression vector pQE31 (QIAGEN, Valencia, CA). The His-tagged recombinant protein was expressed in Escherichia coli, purified over a Ni2+-NTA column (QIAGEN) and then dialyzed against phosphate-buffered saline buffer. To prepare Cog4, 7, and 8 antigens, a cDNA encoding each full-length protein was cloned into the expression vector pET28a (Novagen, Madison, WI). The proteins were expressed in E. coli, and the resulting inclusion bodies were solubilized in 6 M guanidinium hydrochloride. The solubilized proteins were loaded onto Ni2+-NTA columns that were washed with 8 M urea buffers according to the manufacturer's instructions. Bound proteins were eluted by boiling in 1% SDS in the presence of 5 mM EDTA, and then dialyzed against PBS buffer.
Rabbits were injected with 1 mg of a purified antigen. Injections were performed once a month, and sera were collected every 2–4 wk starting at 6 wk after the first injection. Specificity of the antisera was tested by immunoblotting against both partially purified bovine brain COG complex and CHO cell lysates (Ungar et al., 2002).
RESULTS
COG Sensitivity of a Set of Golgi-resident Proteins Called GEARs
To study the function of COG, we surveyed the effects of the COG mutations in ldlB and ldlC mutants on a diverse group of Golgi and ER-associated proteins. Wild-type CHO cells, the ldlB and ldlC mutants, and stable revertants of these mutants generated by transfection with expression plasmids for the corresponding cDNAs (ldlB[COG1], ldlC[COG2]) were studied. We initially screened with a collection of antibodies by using immunoblotting and/or by immunofluorescence microscopy to assess their intracellular distributions and relative abundances.
Figure 1 illustrates representative immunoblots (A) and immunofluorescence micrographs (B) from this survey with the results summarized in Table 1. Of the 32 proteins examined, seven were COG sensitive, in that they exhibited a significant reduction in their steady-state levels in the Cog1negative ldlB and Cog2-negative ldlC mutants. They were mannosidase II (Man II), GPP130, CASP, giantin, golgin-84, GOS-28, and GS15. We refer to these COG-sensitive proteins as “GEARs.” All seven GEARs are resident Golgi transmembrane proteins (Linstedt and Hauri, 1993; Nagahama et al., 1996; Subramaniam et al., 1996; Linstedt et al., 1997; Lowe et al., 1997; Hay et al., 1998; Xu et al., 1997; Bascom et al., 1999; Orci et al., 2000; Gillingham et al., 2002; Moremen, 2002) and are located virtually exclusively in a perinuclear Golgi ribbon-like distribution in wild-type cells (Figure 1B). In contrast, the COG-insensitive Golgi integral membrane proteins examined to date—membrin (Figure 1B), vti1a, syntaxin-5, and TGN38 — could also be detected, at least in part, in distinct punctate structures. Other COG-insensitive proteins that were identified (Table 1; also see Figure 1) included Golgi-associated peripheral proteins p115 and GM130 (Figure 1B), the lipid-anchored GRASP55, the COPII coat protein mSec23, and the ER luminal proteins ERp72 and Bip. It is worth noting that several of the COG-insensitive proteins can form complexes with the COG-sensitive GEARs, e.g., p115 and GM130 with giantin (Sönnichsen et al., 1998), syntaxin-5 and mYkt6 with GOS-28 and GS15 (Shorter et al., 2002; Xu et al., 2002), and rab1 with golgin-84 (Diao et al., 2003; Satoh et al., 2003).
Figure 1.

Immunoblotting and immunofluorescence localization of Golgi-associated proteins in wild-type CHO, mutant ldlB and ldlC, and transfected ldlB[COG1] and ldlC[COG2] cells. (A) Total cell lysates (20 μg of protein) were prepared from parental wild-type CHO, ldlB, ldlB[COG1] (ldlB cells transfected with COG1 cDNA, Cog1 overexpressed), ldlC and ldlC[COG2] (ldlC cells transfected with COG2 cDNA, Cog2 overexpressed) cells. Lysates were subjected to SDS-PAGE and immunoblotting by using antibodies to the indicated Golgi-associated proteins. Solid and open arrowheads indicated a 115-kDa band of GPP130 and two isoforms (42 and 35 kDa) of syntaxin-5, respectively. (B) The indicated cells (left) were grown at 37°C (except those stained with anti-CASP antibody, which were grown at 34°C), fixed with paraformaldehyde, and stained with antibodies to the indicated proteins. To permit comparison of signal intensities among the images in the same column, the images were collected with a fixed signal gain by confocal microscopy. Bar, 10 μm.
Table 1.
Summary of proteins tested in the COG mutants
| COG-sensitive proteins (GEARs) | COG-insensitive proteins | |
|---|---|---|
| SNAREs | GOS-28 | Membrin |
| GS15 | mYkt6 | |
| Syntaxin-5 | ||
| Syntaxin-6 | ||
| Vtila | ||
| Golgins | CASP | GRASP55 |
| Giantin | GM130 | |
| Golgin-84 | Golgin-97 | |
| Golgin-245 | ||
| Small GTPases | Rab1 | |
| Rab6 | ||
| Rab7a | ||
| Rab8 | ||
| Rab11 | ||
| RalA | ||
| Others | Mannosidase II | Bip |
| GPP130 | ERp72a | |
| mSec23a | ||
| p115 | ||
| RabGDI | ||
| Sec8 | ||
| Sec61α and β | ||
| TRAPα | ||
| TGN38a |
Determined by immunofluorescence only.
The GEARs are members of several distinct classes of Golgi-associated proteins. Of the seven golgins tested (CASP, golgins-84, -97, -245, GM130, GRASP55, and giantin), only those three that are integral membrane proteins (CASP, golgin-84, and giantin) (Linstedt and Hauri, 1993; Bascom et al., 1999; Gillingham et al., 2002) are GEARs (Figure 1 and Table 1). Of the seven SNAREs tested, the only ones that seem to be exclusively Golgi localized, GOS-28 and GS15 (Nagahama et al., 1996; Subramaniam et al., 1996; Xu et al., 1997; Orci et al., 2000; Xu et al., 2002) are GEARs. None of the small GTPases (e.g., rabs) examined are GEARs. It was somewhat surprising to find that only one of the GEAR genes, GOS-28, is orthologous to a yeast gene GOS1 that genetically interacts with COG and this interaction is rather weak (Kim et al., 1999), whereas several mammalian COG-insensitive proteins are orthologues of proteins encoded by genes that strongly interact with yeast COG, including the SNAREs Ykt6p (mYkt6) and Sed5p (syntaxin-5), Ypt1p (rab1), and Uso1p (p115).
The relatively low steady-state levels of the GEARs in the ldlB and ldlC mutants were due to the absence of the Cog1 or Cog2 subunits, because higher level expression was observed in the ldlB[COG1] and ldlC[COG2] transfectants (Figure 1). In the cases of GPP130, CASP, giantin and golgin-84, the levels in the transfectants were about the same as those in wild-type cells. Strikingly, the steady-state levels of GOS-28, GS15 and Man II were greater in the transfectants than in wild-type cells (also see Podos et al., 1994). Because the levels of these three GEARs were reduced in the mutants and elevated in COG-overexpressing transfectants (see below), it seems likely that their abundances are directly influenced by COG function.
Only two of the seven GEARs are glycoproteins. One, GPP130, is a protein of unknown function (Linstedt et al., 1997) whose localization in the Golgi is sensitive to changes in intralumenal pH (Linstedt et al., 1997; Puri et al., 2002). The other, Man II, is a glycosidase required for normal N-linked oligosaccharide processing (Moremen, 2002). In the ldlB and ldlC mutants, reductions in the amounts of Man II might contribute to the global defects observed in N-linked oligosaccharide synthesis (Kingsley et al., 1986). As additional reagents for other glycoconjugate processing enzymes become available, it will be possible to determine whether some of them are also GEARs. As expected because of the global glycosylation defects in ldlB and ldlC cells (Kingsley et al., 1986), the electrophoretic mobilities of the GPP130 and Man II glycoproteins in the mutants were increased, indicating abnormal glycosylation (Figure 1A). It is possible that some of the COG sensitivities of these proteins might be a secondary consequence of their abnormal glycosylation. However, similarities in the COG sensitivities of Man II to the unglycosylated GS15 and GOS-28 suggest that, at least in the case of Man II, such secondary effects do not account fully for the COG sensitivities.
Stabilities and Intracellular Distributions of the Golgi SNAREs GOS-28 and GS15 in ldlB and ldlC Mutants
To examine the mechanism(s) underlying the COG sensitivity of the GEARs, we focused on the unglycosylated SNAREs GOS-28 and GS15 so that any secondary effects due to changes in the GEARs' protein glycosylation would not confound the analysis. In the metabolic labeling, pulse-chase experiment shown in Figure 2A (top), newly synthesized GOS-28 in wild-type CHO cells was stable throughout the first 12 h of the chase, and there was only a small loss (∼25%) of the protein between 12 and 24 h (Figure 2A, top). In the ldlB and ldlC mutants, the rate of GOS-28 synthesis (0-h chase) seemed to be only a little lower than that in the wild-type cells. However, the rates of loss of GOS-28 during the chase were substantially higher in the mutants than in the wild-type cells. Approximately 50% of the labeled protein was lost by 12 h of chase and ∼10% remained at 24 h in the ldlB and ldlC mutants (half-lives of ∼11–12 h in the mutants vs. >24 h in wild-type CHO cells). The abnormally rapid degradation of GOS-28 in the mutant cells was not observed in ldlB[COG1] transfectants (Figure 2A, bottom), indicating that the instability was a consequence of COG deficiency. Similar results were obtained when the stability of pulse-labeled GS15 was examined.
Figure 2.

Stability and intracellular distributions of GOS-28 in wild-type CHO, ldlB, and ldlC cells. (A) Cells were metabolically labeled with [35S]methionine/cysteine for 30 min (pulse), and then either chased for the indicated times in medium B containing unlabeled methionine and cysteine (top) or incubated for 24 h with the same medium plus 25 μM of the proteasome inhibitors MG132 or lactacystin (bottom). Cell lysates were subjected to immunoprecipitation with an anti-GOS-28 antibody. The immunoprecipitates were fractionated by SDS-PAGE, and the bands were visualized by autoradiography. (B) Cells were fixed and stained with antibodies to GOS-28 (red) and the ER marker ERp72 (green). To visualize the distribution of GOS-28 by using confocal microscopy, the images from ldlB and ldlC cells (weaker signals) were collected with higher signal gains than those from CHO cells. Note that because of the location of the section, some of the confocal images of the mutants do not show the perinuclear Golgi ribbon staining that is present (e.g., see Figure 1B). Bar, 10 μm.
The mechanism underlying the rapid degradation of GOS-28 in the mutants was examined by determining with immunofluorescence confocal microscopy GOS-28's intracellular distribution in wild-type and mutant cells. To visualize the detailed distribution of GOS-28, which is present at lower levels in ldlB and ldlC cells, images of mutant cells were collected with higher signal gains than those of wild-type cells (Figures 2, 3, and 7). As shown in Figure 2B, in wild-type cells the classic perinuclear Golgi ribbon distribution of GOS-28 was observed as previously described (Subramaniam et al., 1995), and did not overlap with the reticular distribution of the ER luminal protein ERp72 (Mazzarella et al., 1990). A similar result was observed using coimmunofluorescence with the ER membrane protein Sec61β (Hartmann et al., 1994). In contrast, in the ldlB and ldlC mutants, a significant fraction of GOS-28 could be seen in a reticular staining that was dispersed throughout the cytoplasm in a distribution consistent with it being mislocalized into the ER. The residual perinuclear Golgi-ribbon distribution of GOS-28 in the mutants is not seen as clearly as in Figures 1B or 3A, because of the relative positions of some of the confocal sections shown in Figure 2B (especially that for the ldlC cells). Indeed, this mislocalized protein colocalized with the ER markers ERp72 and Sec61β. Thus, we conclude that a substantial portion of the reticular pattern of GOS-28 staining in the mutants was due to its location in the ER. In the ldlB[COG1] and ldlC[COG2] cells, the distribution of GOS-28 was the same as in CHO cells, exclusively located in the Golgi region. The mislocalization of GS15 to the ERp72-positive reticular compartment in ldlB cells that overexpress this protein (ldlB[GS15] (Supplemental Figure 1, A and B) was similar to that seen for endogenous GOS-28 in ldlB cells (Figure 2B). Thus, these GEARs exhibited COG sensitivity of both their steady-state levels and intracellular localizations.
Figure 3.

Immunofluorescence localization of GOS-28 and GM130 in CHO, ldlB, and ldlC cells treated without and with lactacystin. (A) Cells were incubated for 8 h without (none) or with (+Lact) 25 μM lactacystin, fixed, and stained with antibodies to GOS-28 (red) and GM130 (green). To visualize more clearly the distribution of GOS-28 by using confocal microscopy, the images from ldlB and ldlC cells (weaker signals) were collected with higher signal gains than those from CHO cells. However, a fixed signal gain was used to collect images from any given cell type when comparing the effects of incubation without or with lactacystin. (B) The ldlB cells were preincubated with cycloheximide (+CHX, 140 μg/ml) for 1 h and then further incubated for 8 h without or with (+Lact) 25 μM lactacystin in the presence of the cycloheximide. Bar, 10 μm.
Figure 7.

Effects of siRNA depletion of ε-COP in ldlB and ldlC cells on the immunofluorescence of GOS-28, GM130, and ε-COP. ldlB and ldlC mutants were transfected without (control) or with (+siRNA) ε-COP–specific siRNA. Cells were incubated at 34°C for 48 h and then fixed and stained with antibodies to either GOS-28 (red) or GM130 (red) and ε-COP (green). To facilitate visualization of the distribution of GOS-28 by using confocal microscopy, the images from ldlB and ldlC cells (weaker signals) were collected with relatively high signal gains. Arrowheads indicate cells in which there was a loss or reduction the signal intensity for ε-COP. Bar, 10 μm.
Effects of Proteasome Inhibitors on the Stabilities of GOS-28 and GS15
The rapid degradation of GOS-28 and GS15 and their abnormal distribution in the ER raised the possibility that dislocation (retrotranslocation) and proteasomal degradation (Kostova and Wolf, 2003; McCracken and Brodsky, 2003) might be responsible for the instability of some GEARs in the mutants. Therefore, we added the proteasome inhibitors MG132 (25 μM) or lactacystin (25 μM) in DMSO to the chase media of a pulse-chase experiment to examine their effects on GOS-28 stability. Both inhibitors prevented the abnormally rapid degradation of GOS-28 that occurred in the mutants (Figure 2A, bottom). Neither inhibitor affected GOS-28's stability in wild-type CHO or ldlB[COG1] cells. Similar results were observed when the effects of these inhibitors on GS15 stability in the mutants were examined. The effects of these proteasome inhibitors on the steady-state levels of expression were confirmed by immunoblotting analysis. The inhibitors restored the steady state levels of GOS-28 in the ldlB and ldlC mutants to those seen in the wild-type CHO and transfected control cells (Supplemental Figure 2A).
Prolonged incubation of cells with high concentrations of proteasome inhibitors can induce caspase activation (Drexler et al., 2000) and apoptosis-associated fragmentation of the Golgi (Mancini et al., 2000; Lane et al., 2002; Chiu et al., 2002), and thus might possibly indirectly alter the stability of the GEAR proteins. To ensure that the effects of these inhibitors were independent of apoptosis, we examined the dose dependence of lactacystin on the steady-state level of GOS-28 after only 4 h of incubation with the inhibitor. Lactacystin significantly increased the levels of GOS-28, even at concentrations as low as 1 μM (Supplemental Figure 2B). In control experiments, we measured activation of caspase-3 as a measure of apoptosis and observed no evidence that lactacystin-induced apoptosis at concentrations of 1–25 μM after incubations with the inhibitor for 4 h. This finding is consistent with the report that lactacystin induction of caspase-3 processing is relatively slow (∼8 h) compared with its rapid inhibition of proteasomal activity (Kim et al., 2003). In contrast to the proteasome inhibitors, bafilomycin A (∼10 nM), an inhibitor of lysosomal protein degradation (Yoshimori et al., 1991), did not significantly affect the stabilities of GOS-28 and GS15 in the ldlB and ldlC cells. Thus, even though we were unable to detect polyubiquitinated intermediates (Bonifacino and Weissman, 1998), the abnormal degradation of GOS-28 and GS15 in ldlB and ldlC mutants was apparently due to proteasome-mediated hydrolysis.
Effects of Proteasome Inhibitors on GOS-28 Accumulation in Reticular Structures
We next explored the relationship between the abnormal localization of GOS-28 in the ER and its proteasomal degradation in ldlB and ldlC mutants. In some cases, undegraded dislocated (retrotranslocated) proteins can be trapped in the cytoplasm by inhibiting proteasome activity (Wiertz et al., 1996a,b). In others, dislocation and proteasomal degradation are tightly coupled and treatment with proteasome inhibitors results in accumulation of the proteins within the ER (Johnston et al., 1998). We used immunofluorescence microscopy to determine the effects of lactacystin treatment (25 μM for 4 or 8 h) of wild-type and mutant cells on the intracellular distribution of GOS-28, along with the COG-insensitive Golgi marker GM130 (Nakamura et al., 1995).
Figure 3A compares confocal immunofluorescence images of lactacystin-treated and untreated cells. Because of the signal gain and the z-section used to collect the confocal signals, the relatively weak reticular patterns of GOS-28 staining in the untreated ldlB and ldlC mutants (Figure 2B) are difficult to see in these images. Even after incubation with lactacystin for 8 h, most of the GM130 signal in wild-type and mutant cells exhibited a perinuclear Golgi distribution (middle). There was some fragmentation of the GM130 staining pattern induced by lactacystin treatment; however, this was unlikely to be due to apoptosis, because some fragmentation was also observed at 4 h when there was no caspase-3 activation. The GOS-28 distribution (Figure 3A, left) in wild-type CHO cells was virtually identical to that of GM130 with or without lactacystin treatment, whereas in both ldlB and ldlC cells lactacystin induced a significant increase in the ER-like reticular distribution of GOS-28. Coimmunofluorescence staining with antibodies to GOS-28 and the ER marker Sec61β established that a substantial amount of the lactacystin-induced increase in reticular staining was located in the ER. In wild-type and mutant cells, the distribution of the COG-insensitive Golgi SNARE membrin was, like GM130, not substantially affected by lactacystin treatment. To determine whether new protein synthesis was required for lactacystin-induced accumulation of these GEARs in the ER, we pretreated cells with 140 μg/ml cycloheximide for 1 h before adding 25 μM lactacystin and then incubated the cells for an additional 8 h. Protein synthesis was dramatically inhibited by this cycloheximide treatment (>90% inhibition after the 1-h preincubation and >95% inhibition by the end of the 8-h incubation). The intracellular distributions of GOS-28 and GM130 in wild-type cells with or without lactacystin treatment were not altered by the cycloheximide treatment. Figure 3B shows that inhibition of protein synthesis by cycloheximide in the ldlB cells also did not prevent lactacystin-induced accumulation of GOS-28 in the ER of these COG deficient cells, although the intensity of the ER staining was somewhat reduced by the cycloheximide treatment. Thus, transport of preexisting GOS-28 to the ER contributes in part to the lactacystin-induced accumulation.
Together with the stabilizing effects of lactacystin on GOS-28 and GS15, the results suggest that proteasomal hydrolysis accounts for the rapid degradation of these proteins and their reduced steady-state levels in the mutant cells and that inhibition of proteasomes blocks both GOS-28 degradation and its dislocation from the ER, but not its abnormal localization in the ER. Thus, it seems that abnormal ER localization of these GEARs, probably due at least in part to retrograde trafficking from the Golgi, contributes to their reduced steady-state levels.
Effects of Overexpressing Individual COG Subunits on the GEARs GOS-28 and GS15 and on the Other COG Subunits
In both wild-type and mutant cells, overexpression of the Cog1, Cog2, or HA-Cog7 COG subunits (ldlB[COG1], ldlC[COG2], CHO[COG1], CHO[COG2], or CHO[HA-COG7]) resulted in elevated steady-state levels of some of the GEARs, GOS-28, GS15, and Man II, relative to wild-type CHO cells (Figures 1A and 4). The levels of the COG-insensitive protein GM130 were not affected by the transfected COG genes (Figure 4, bottom). It seems likely that by mass action the excess Cog1, Cog 2, or Cog7 subunits induced the formation of a greater than normal steady-state concentration of COG complexes in the transfectants and thus resulted in a gain-of-function, or hypermorphic-like, response of some GEARs. We therefore used immunoblotting to compare the steady-state levels of the COG subunits in the wild-type, mutant, and transfectant cells. Figure 4 shows that in ldlB cells, in which there is no detectable Cog1, there was very little change in the levels of Cogs 2–4, whereas there was a substantial reduction in the amounts of Cogs 5–8. This is consistent with the proposal that Cogs 5–8 may form a subcomplex within the intact COG complex (Ungar et al., 2002, also see Whyte and Munro, 2001). Strikingly, the patterns of protein levels for all of the COG subunits in the overexpressing transfectants were similar to those of GOS-28 and GS15: increased levels relative to untransfected wild-type CHO controls, with somewhat higher levels in the ldlB[COG1] and CHO[COG1] cells than in the CHO[COG2] and CHO[HA-COG7] cells. Thus, overexpression of any one of at least three of the COG subunits induces increased levels of the others, suggesting that the steady-state levels of the GEARs GOS-28, GS15, and Man II are proportional to the amounts of the COG complex (reduced in the mutants, increased in the transfectants).
Figure 4.
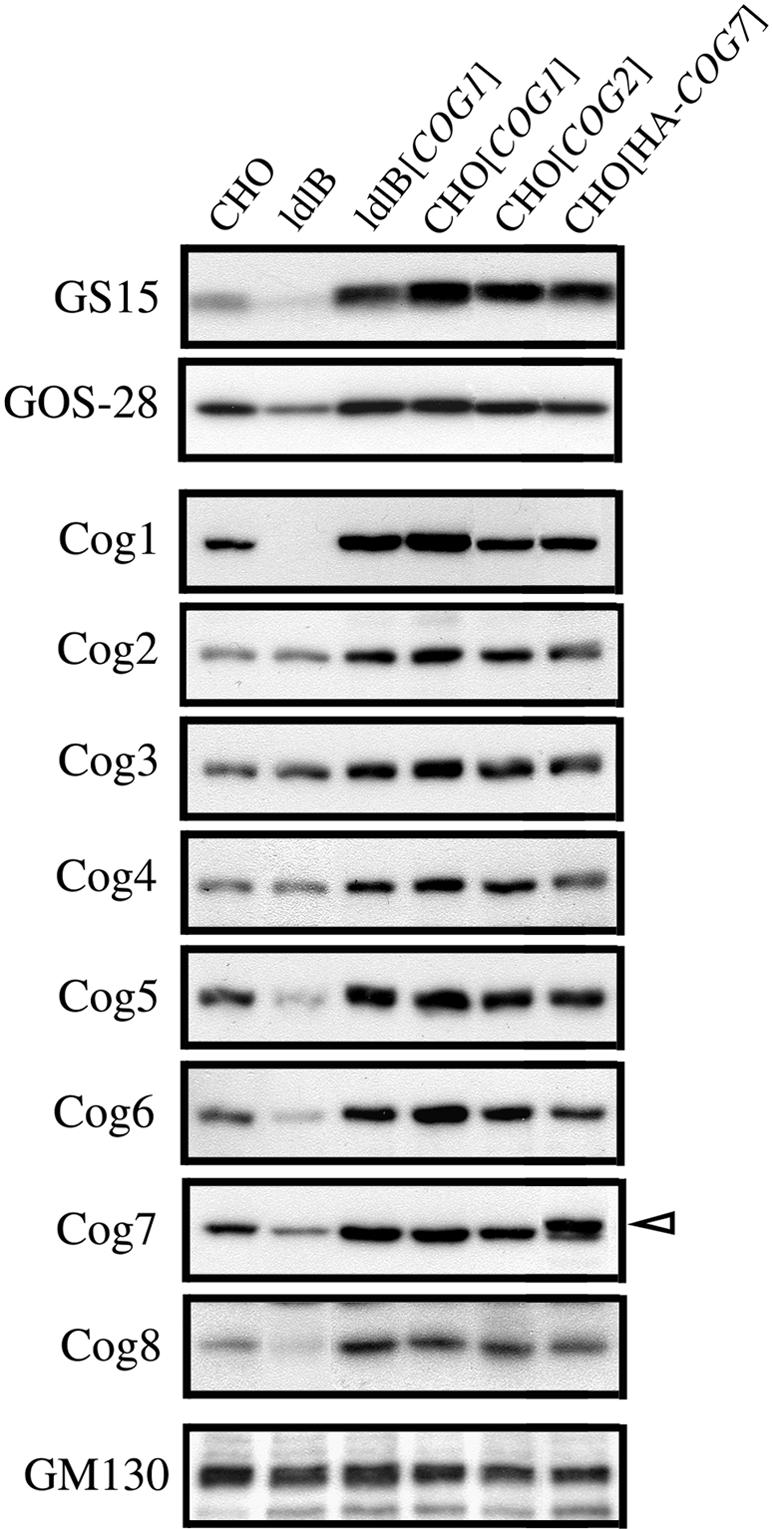
Steady-state levels of GOS-28, GS15, the subunits of COG and GM130 in untransfected and COG subunit-overexpressing CHO and ldlB cells. Total cell lysates from the CHO, ldlB, and ldlB[COG1] cells, as well as transfected CHO cells that overexpress exogenous Cog1, Cog2, or HA-tagged Cog7 (CHO[COG1], CHO[COG2] and CHO[HA-COG7]) were subjected to SDS-PAGE and immunoblotting. Open arrowhead indicates the HA-tagged Cog7 protein whose electrophoretic mobility is slightly less than that of the endogenous Cog7.
Interactions between COG and COPI
Previous studies in yeast have suggested a functional and physical relationship between COG and the vesicle coat complex COPI (Kim et al., 2001; Ram et al., 2002; Suvorova et al., 2002). Therefore, we determined the effects of the COG1 and COG2 mutations in the ldlB and ldlC mutants on the amounts and intracellular distributions of several COPI subunits. The amounts of the COPI α, β, and ε subunits in the mutants were unchanged relative to those in wild-type and transfected cells (Figure 5A, similar results for β-COP were previously reported by Ungar et al., 2002). However, immunofluorescence microscopy (Figure 5B) showed that the distribution of the β subunit was altered in the mutants. In wild-type CHO and transfected cells, β-COP staining exhibited the typical, perinuclear Golgi pattern combined with only a relatively low-intensity, punctate signal seen distributed throughout the cytoplasm (also see Allan and Kreis, 1986). In the ldlB and ldlC mutants, the cytoplasmic β-COP staining increased, whereas the perinuclear staining decreased. Similar results were observed for ε-COP (Figures 6 and 7). Thus, the intracellular distribution of COPI is altered by mutations in COG1 and COG2.
Figure 5.

Immunoblotting and immunofluorescence localization of the α-, β-, and ε-COP subunits of COPI. (A) Lysates from the indicated cells were analyzed by immunoblotting with antibodies to α-, β-, or ε-COP. (B) The indicated cells were fixed and stained with an anti-β-COP antibody, and images were collected by confocal microscopy as described in Figure 1. Bar, 10 μm.
Figure 6.

Effects of siRNA depletion of ε-COP in wild-type CHO cells on the immunofluorescence of Cog1, GOS-28, GM130, and β-COP. Wild-type CHO cells were transiently transfected without (control) or with (+siRNA) a 21-base pair siRNA duplex specific for hamster ε-COP. After a 46.5-h incubation at 34°C, the cells were further incubated for 90 min at either 34°C (where the loss of ε-COP had no apparent phenotype) or 39.5°C and then fixed and stained with antibodies to either ε-COP or β-COP (green) together with antibodies (red) to either Cog1, GM130, or GOS-28. Arrowheads indicate the cells in which there was a loss or major reduction in the signal intensity for either ε-COP (A and B) or GOS-28 (C), which indicated effective knockdown of ε-COP expression. Bars, 10 μm.
We assessed the effects of alterations in the expression of ε-COP on both COG and GEARs by using RNA interference (Elbashir et al., 2001) to knock down ε-COP expression in wild-type CHO cells.1 Cells were transfected with a siRNA duplex specific for hamster ε-COP, maintained in culture for 46.5 h at 34°C and then either shifted to 39.5°C or kept at 34°C for 90 min before immunofluorescence analysis of the intracellular distribution of a variety of Golgi-associated proteins. Previous studies in yeast have suggested that ε-COP is not required at the normal growth temperature but is indispensable for cellular function at high temperature (Duden et al., 1998; Kimata et al., 1999). Figure 6A shows that there was a complete loss or major reduction in the amounts of detectable ε-COP protein in a subset of the siRNA-treated CHO cells at both 34 and 39.5°C (arrowheads). At 34°C, the ε-COP–deficient cells seemed to be viable and healthy, and we detected no changes in the intracellular distribution of GOS-28, GM130 (Figure 6, A and B), GS15, golgin-84 (Supplemental Figure 3, A and B), Man II, GPP130, membrin, vti1a, or β-COP. We also observed no changes in the intracellular distribution of COG (Cog1) in the ε-COP-depleted cells at 34°C. However, there was a significant reduction in the perinuclear staining of Cog1 in the ε-COP–depleted cells after incubation for 90 min at 39.5°C (Figure 6A). This was not due to a general breakdown of the structure of the Golgi apparatus, because there was no disruption in the normal perinuclear distribution of several other Golgi markers, including GM130 (Figure 6A; see below). Thus, the proper distribution of COG at 39.5°C requires ε-COP activity.
Interactions between COPI and GEARs
Ater incubation for 90 min at 39.5°C, those CHO cells in which ε-COP was depleted by introduction of the siRNA also lost their normal perinuclear Golgi distributions of the six GEARs examined (CASP distribution was not analyzed) (Figure 6B and Supplemental Figure 3). In contrast, there was no apparent effect of the siRNA on the distributions of GM130 (Figure 6A), membrin, or vti1a after incubation for 90 min at 39.5°C. One possible explanation for these observations is that the ε-COP activity is indispensable for COG function at 39.5°C. In ε-COP–depleted cells identified by alteration in the GOS-28 signal, the Golgi distribution of a different COPI subunit, β-COP, was unaffected after incubation at 39.5°C for 90 min (Figure 6C), even though COG and GEARs were affected. As an additional control, a second independent siRNA, corresponding to a different portion of the ε-COP coding sequence, was introduced into the cells, and its effects were similar to those of the first siRNA. Furthermore, the siRNA for a distinct gene, GFP (Novina et al., 2002), was introduced into the cells and had no effect on the intensity of staining or the distributions of GOS-28 or ε-COP, indicating that the effects of the ε-COP siRNA were ε-COP specific. Thus, under conditions where there is apparently some initial temperature-dependent loss of COPI function due to ε-COP-depletion, but not complete breakdown of the Golgi's structure or even dissociation of some COPI subunits, the COG-sensitive, but not-insensitive, proteins were mislocalized and possibly in some cases degraded. These findings suggest that COG and ε-COP, at least in part, contribute to similar functions necessary for normal Golgi organization. They also suggest that ε-COP, presumably together with α and β′-COPs in a COPI subcomplex (Lowe and Kreis, 1995; Hoffman et al., 2003), is not required for the association of β-COP with the Golgi (Gomez et al., 2000).
Generation of Synthetic Phenotypic Defects by Simultaneously Disabling COG and COPI
To explore the genetic interactions between COG and COPI in mammalian cells, we introduced the ε-COP siRNA into ldlB and ldlC mutants. Unlike the case with wild-type CHO or ldlB[COG1] cells, a substantial fraction of the mutants transfected with ε-COP siRNA exhibited abnormal morphologies when incubated at 34°C during the 2 d after transfection. Cell rounding and presumably cell death made it difficult to assess Golgi protein distributions in many of the transfectants. Immunofluorescence analysis of those ε-COP–depleted mutants that retained a reasonably well-spread morphology revealed various abnormalities in the distributions of Golgi markers, including an abnormally concentrated juxtanuclear distribution of both the COG-sensitive GOS-28 and the COG-insensitive GM130 (Figure 7). This suggested a substantial change in the structure (compaction) of at least portions of the Golgi under these conditions. We were unable to determine whether the abundances of these Golgi proteins were altered under these conditions. We also noted that in some of these cells there was a significant reduction in the reticular staining of GOS-28. This raises the possibility that ER accumulation of GOS-28 in the mutants might be dependent on the intracellular levels of ε-COP and that COPI function might contribute to the ER localization of GEARs in the COG-deficient mutants. However, the defects in the structure of the Golgi in these transiently transfected cells and the apparent toxicity of ε-COP depletion in COG-deficient mutants suggest that caution must be exercised in drawing conclusions about the role of COPI in the ER accumulation of GOS-28 in COG-deficient mutants. Nonetheless, the distinctive phenotypes of the ε-COP–depleted mutants were not observed in ε-COP-depleted ldlB[COG1] cells. The synthetic phenotypes (altered distribution of Golgi proteins at 34°C, apparent toxicity) generated by simultaneously disabling Cog1/Cog2 and ε-COP in CHO cells both suggest some redundancy in the functions of the COG and COPI complexes and are reminiscent of the synthetic lethality in yeast generated by combining specific mutations in COG and COPI (Kim et al., 2001; Ram et al., 2002; Suvorova et al., 2002).
DISCUSSION
COG Controls the Abundance and Intracellular Distribution of a Distinct Set of Resident Golgi Proteins, the GEARs
The current study of the mammalian COG complex helps to further define the function of this peripheral Golgi-associated hetero-octameric protein complex. We have characterized the consequences of the loss of COG function on the steady-state levels and localization of a diverse set of 32 secretory pathway-related proteins that includes seven SNAREs, five rabs, seven golgins, and others. Seven of these exhibited abnormally low steady-state levels in the mammalian somatic cell mutants ldlB (Cog1 negative) and ldlC (Cog2 negative). We call these COG-sensitive proteins GEARs. The GEARs identified to date include two SNAREs (GOS-28 and GS15), three golgins (CASP, giantin, and golgin-84), a glycosidase (Man II), and GPP130, a phosphorylated glycoprotein of unknown function. These GEARs are all Golgi-resident type II transmembrane proteins that are normally virtually exclusively located in a perinuclear Golgi ribbon-like distribution. This is not the case for any of the COG-insensitive proteins examined in this study.
Although the structural features responsible for the COG sensitivity of the GEARs have not been defined, it is noteworthy that the three golgins among the GEARs (CASP, giantin, and golgin-84) all have highly conserved tyrosine and histidine residues within their transmembrane domains (Gillingham et al., 2002). These residues are functionally important in yeast CASP (Gillingham et al., 2002) and may contribute to their COG sensitivities. It seems likely that additional GEARs remain to be identified and that some of these will be involved in glycoconjugate processing (e.g., glycosidases, glycosyl transferases, and substrate transporters). Defects in their localization (Harris and Waters, 1996; Skrincosky et al., 1997; Axelsson et al., 2001; Martínez-Menarguez et al., 2001; Mironov et al., 2001; Opat et al., 2001; Berger, 2002; Puri et al., 2002; Roth, 2002; Zerfaoui et al., 2002) and steady-state levels could alter their activities and would provide an attractive explanation for the pleiotropic glycosylation defects observed in ldlB and ldlC cells (Kingsley et al., 1986).
We focused our detailed analysis of GEARs in the ldlB and ldlC mutants on the SNARE proteins GOS-28 and GS15, because they are not glycosylated. Thus, their COG sensitivities could not simply be consequences of changes in their structures due to the previously described glycoconjugate processing defects in these mutants. The steady-state levels of these SNAREs, as well as that of the GEAR Man II, were not only decreased in mutants with defective COG but also elevated in cells containing increased levels of COG. This suggests that COG controls the levels of at least some GEARs directly. In the mutants, but not in wild-type cells, GOS-28 and GS15 could be readily detected in the ER as well as the perinuclear Golgi ribbon, and they were abnormally rapidly degraded. This degradation was apparently a consequence of mislocalization/accumulation in the ER and directly coupled to dislocation/retrotranslocation from the ER and subsequent proteosomal degradation, because lactacystin inhibition of proteasomes blocked degradation and increased GOS-28's accumulation in the ER. A substantial amount of the lactacystin-induced accumulation of GOS-28 in the ER was independent of new protein synthesis. Thus, it is likely that transport from other compartments, most likely retrograde traffic from the Golgi, accounted for at least some of the lactacystin-induced accumulation of these GEARs in the ER.
These results suggest that COG might influence the distribution of these GEARs in several ways. First, COG might control intra-Golgi transport of the GEARs such that they are either directed by COG to their appropriate sites of residence or prevented by COG from inappropriate retrograde trafficking to the ER. A role for COG in directing GEAR transport to specific Golgi sites rather than blocking a transport step is supported by the observation that COG stimulates an in vitro intra-Golgi transport assay (Walter et al., 1998). Second, COG might be required for efficient ER exit of some GEARs that are either newly synthesized (Morsomme and Riezman, 2002; Morsomme et al., 2003) or constitutively recycling (Storrie et al., 1998; Girod et al., 1999; Zaal et al., 1999; Miles et al., 2001; Ward et al., 2001; Stroud et al., 2003). These processes involve Golgi-to-ER retrograde transport, which has been reported in numerous studies (Lippincott-Schwartz et al., 1989; Letourneur et al., 1994; Harris and Waters, 1996; Gaynor and Emr, 1997; Gaynor et al., 1998; Glick and Malhotra, 1998; Pelham and Rothman, 2000; Todorow et al., 2000; Opat et al., 2001) of many normally Golgi-associated proteins, including the GEARs giantin and Man II (Storrie et al., 1998; Girod et al., 1999; Zaal et al., 1999; Lippincott-Schwartz et al., 2000; Seemann et al., 2000; Storrie et al., 2000; Miles et al., 2001; Ward et al., 2001; Shorter and Warren, 2002; Storrie and Nilsson 2002; Stroud et al., 2003).
Relationship of COG with COPI and the Function of COG
COPI-coated transport vesicles have been proposed to play a key role in retrograde transport and the retrieval of Golgi resident proteins (Letourneur et al., 1994; Gaynor and Emr, 1997; Gaynor et al., 1998; Glick and Malhotra, 1998; Pelham and Rothman, 2000; Opat et al., 2001; Duden, 2003). The differential effects on transport of blocking COPI activity by injection of anti-COPI antibodies have suggested that Golgi-to-ER retrograde transport can be either COPI dependent or COPI independent (Storrie et al., 1998, 2000; Girod et al., 1999; Storrie and Nilsson, 2002). A significant body of evidence supports the hypothesis that COG and COPI collaborate in controlling the correct localization and abundance of GEARs. In mammalian cells four GEARs (GOS-28, GS15, Man II, and giantin) can physically associate with COPI (Nagahama et al., 1996; Sönnichsen et al., 1998; Orci et al., 2000; Martínez-Menarguez et al., 2001; Xu et al., 2002). Two other GEARs (GPP130 and golgin-84) are concentrated on vesicles and the lateral edges of the Golgi where COPI resides (Yuan et al., 1987; Diao et al., 2003; Satoh et al., 2003). The kinetics of brefeldin A-induced dissociation from the Golgi of Cog2 and the COPI subunit β-COP are similar (Donaldson et al., 1990; Podos et al., 1994). In yeast, COG and COPI interact genetically and physically (Kim et al., 2001; Ram et al., 2002; Suvorova et al., 2002).
Here, we report additional evidence for a functional relationship between mammalian COG and COPI. COPI was partially dispersed throughout the cytoplasm in the ldlB and ldlC mutants, whereas COG's distribution was altered at 39.5°C in wild-type CHO cells in which ε-COP expression was suppressed by siRNA. Thus, the normal distribution of COPI was dependent on COG and that of COG was dependent on COPI. Furthermore, the GEARs were sensitive to alterations in COPI as well as being sensitive to mutations in COG. Disrupting intact COPI in wild-type CHO cells with ε-COP siRNA at 39.5°C altered the distributions of GEARs without affecting COG-insensitive proteins. Thus, although COG and COPI play distinct roles in membrane trafficking, they contribute, at least in part, to similar functions necessary for normal Golgi organization (e.g., establishing the proper distribution of GEARs). The changes in COG and GEARs induced by depleting cells of ε-COP were temperature sensitive (observed only after a temperature shift from 34 to 39.5°C). This is reminiscent of the previously reported temperature-sensitive growth of ε-COP null yeast mutants (Duden et al., 1998; Kimata et al., 1999). Finally, simultaneously disabling COG and COPI by introducing ε-COP siRNA into ldlB and ldlC mutants generated a synthetic phenotypic defect: disruption of normal Golgi morphology at 34°C. This indicates that COG and COPI may function in parallel to establish or maintain normal Golgi organization. In addition, the abnormal accumulation of GOS-28 in the ER of ldlB and ldlC mutants was in some cases reduced by ε-COP depletion. These findings raise the possibility that COPI vesicles might participate in the redistribution to the ER of at least one of the GEAR proteins when COG function is compromised. Despite the interactions between COG and COPI and their influence on the intracellular distribution of GEARs, it is possible that the retrograde movement of some GEARs to the ER in COG-deficient cells might be COPI independent, as in the systems studied by Girod et al. (1999). However, COPI-independent mechanisms may not play a major role in the ER accumulation of GEARs in COG-deficient cells.
The identification and characterization of GEARs and their relationships to COG and COPI provide additional insights into the function of COG. It is possible that COG influences the distributions and/or abundances of GEARs primarily by regulating their exit from the ER (Morsomme and Riezman, 2002; Morsomme et al., 2003). Thus, the ER accumulation of some GEARs in COG-deficient cells might be similar to the ER accumulation of several Golgi glycosylation proteins when ER exit is blocked by a dominant negative Sar1 protein (Aridor et al., 1995; Storrie et al., 1998; Girod et al., 1999; Seemann et al., 2000; Miles at el. 2001; Ward et al., 2001; Storrie and Nilsson 2002; Stroud et al., 2003). Another possibility is that COG influences the distributions and/or abundances of GEARs less directly by affecting the Golgi's structure and/or lumenal environment (Kingsley et al., 1986; Linstedt et al., 1997; Puri et al., 2002; Ungar et al., 2002). Because GEARs are normally exclusively Golgi localized, retrograde vesicular traffic (COPI dependent and/or independent) is probably required for their mislocalization in COG mutant cells. It seems likely that a major function of COG is to work in concert with COPI to control retrograde trafficking (retrieval), thereby ensuring the proper intra-Golgi distribution and abundance of the GEARs. Thus, in the absence of normal COG function in the mutants, some GEARs are abnormally transported by retrograde traffic from the Golgi to the ER. The effects of COG on retrograde trafficking and proteasomal degradation of some GEARs may provide insights into the broader issue of how the steady-state levels of Golgi-resident proteins are controlled. The GEAR/COG/COPI system provides new tools to study both this relatively unexplored area of membrane cell biology as well as other aspects of the structure and function of the Golgi apparatus.
Supplementary Material
Acknowledgments
We are grateful to Gerry Waters, Kelley Moremen, James Rothman, William Balch, Sean Munro, Wanjin Hong, Yoh Wada, Masamitsu Futai, and Nobuhiro Nakamura for providing antibodies and to Chris Kaiser for helpful discussions. We thank H.R. Horvitz for providing access to the confocal microscope, Tom Rapoport for suggestions regarding the ER marker proteins, and Carl Novina and Philip Sharp for advice regarding RNA interference. This work was supported National Institutes of Health grants GM-59115 to M.K. and GM-59280 to F.M.H. D.U. was supported by a postdoctoral fellowship from the American Heart Association and is a fellow of the AHA New Jersey Board of Directors.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03-09-0699. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03–09–0699.
Online version of this article contains supporting material. Online version is available at www.molbiolcell.org.
Footnotes
We also examined the levels of Cog1 and Cog2 in the temperature-sensitive ε-COP mutant ldlF. In this mutant, a point mutation in ε-COP renders it unstable at the nonpermissive temperature of 39.5°C (Guo et al., 1994, 1996). At both the permissive temperature (34°C) and after 12 h at the nonpermissive temperature when the structure of the Golgi is disrupted (Guo et al., 1994), there were no obvious changes in the levels of Cog1 and Cog2 in ldlF cells, even though the distribution of these Cogs in ldlF at the nonpermissive temperature was dramatically altered concomitantly with other Golgi markers.
References
- Allan, V.J., and Kreis, T.E. (1986). A microtubule-binding protein associated with membranes of the Golgi apparatus. J. Cell Biol. 103, 2229–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor, M., Bannykh, S.I., Rowe, T., and Balch, W.E. (1995). Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 131, 875–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson, M.A., Karlsson, N.G., Steel, D.M., Ouwendijk, J., Nilsson, T., and Hansson, G.C. (2001). Neutralization of pH in the Golgi apparatus causes redistribution of glycosyltransferases and changes in the O-glycosylation of mucins. Glycobiology 11, 633–644. [DOI] [PubMed] [Google Scholar]
- Bonifacino, J.S., and Weissman, A.M. (1998). Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol. 14, 19–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe, C. (2002). COPII-dependent transport from the endoplasmic reticulum. Curr. Opin. Cell Biol. 417–422. [DOI] [PubMed]
- Bascom, R.A., Srinivasan, S., and Nussbaum, R.L. (1999). Identification and characterization of golgin-84, a novel Golgi integral membrane protein with a cytoplasmic coiled-coil domain. J. Biol. Chem. 274, 2953–2962. [DOI] [PubMed] [Google Scholar]
- Berger, E.G. (2002). Ectopic localizations of Golgi glycosyltransferases. Glycobiology 12, 29R–36R [DOI] [PubMed] [Google Scholar]
- Chatterton, J.E., Hirsch, D., Schwartz, J.J., Bickel, P.E., Rosenberg, R.D., Lodish, H.F., and Krieger, M. (1999). Expression cloning of LDLB, a gene essential for normal Golgi function and assembly of the ldlCp complex. Proc. Natl. Acad. Sci. USA 96, 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, R., Novikov, L., Mukherjee, S., and Shields, D. (2002). A caspase cleavage fragment of p115 induces fragmentation of the Golgi apparatus and apoptosis. J. Cell Biol. 159, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conibear, E., Cleck, J.N., and Stevens, T.H. (2003). Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol. Biol. Cell 14, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao, A., Rahman, D., Pappin, D.J., Lucocq, J., and Lowe, M. (2003). The coiled-coil membrane protein golgin-84 is a novel rab effector required for Golgi ribbon formation. J. Cell Biol. 160, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms, R.W., Russ, G., and Yewdell, J.W. (1989). Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J. Cell Biol. 109, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson, J.G., Lippincott-Schwartz, J., Bloom, G.S., Kreis, T.E., and Klausner, R.D. (1990). Dissociation of a 110-kD peripheral membrane protein from the Golgi apparatus is an early event in brefeldin A action. J. Cell Biol. 111, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler, H.C., Risau, W., and Konerding, M.A. (2000). Inhibition of proteasome function induces programmed cell death in proliferating endothelial cells. FASEB J. 14, 65–77. [DOI] [PubMed] [Google Scholar]
- Duden, R., Kajikawa, L., Wuestehube, L., and Schekman, R. (1998). ε-COP is a structural component of coatomer that functions to stabilize α-COP. EMBO J. 17, 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duden, R. (2003). ER-to-Golgi transport: COP I and COP II function. Mol. Membr. Biol. 20, 197–207. [DOI] [PubMed] [Google Scholar]
- Elbashir, S.M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K., and Tuschl, T. (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Farkas, R.M., Giansanti, M.G., Gatti, M., and Fuller, M.T. (2003). The Drosophila Cog5 homologue is required for cytokinesis, cell elongation, and assembly of specialized Golgi architecture during spermatogenesis. Mol. Biol. Cell 14, 190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor, E.C., and Emr, S.D. (1997). COPI-independent anterograde transport: cargo-selective ER to Golgi protein transport in yeast COPI mutants. J. Cell Biol. 136, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor, E.C., Graham, T.R., and Emr, S.D. (1998). COPI in ER/Golgi and intra-Golgi transport: do yeast COPI mutants point the way? Biochim. Biophys. Acta 1404, 33–51. [DOI] [PubMed] [Google Scholar]
- Gillingham, A.K., Pfeifer, A.C., and Munro, S. (2002). CASP, the alternatively spliced product of the gene encoding the CCAAT-displacement protein transcription factor, is a Golgi membrane protein related to giantin. Mol. Biol. Cell 13, 3761–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod, A., Storrie, B., Simpson, J.C., Johannes, L., Goud, B., Roberts, L.M., Lord, J.M., Nilsson, T., and Pepperkok, R. (1999). Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat. Cell Biol. 1999 1, 423–430. [DOI] [PubMed] [Google Scholar]
- Glick, B.S., and Malhotra, V. (1998). The curious status of the Golgi apparatus. Cell 95, 883–889. [DOI] [PubMed] [Google Scholar]
- Gomez, M., Scales, S.J., Kreis, T.E., and Perez, F. (2000). Membrane recruitment of coatomer and binding to dilysine signals are separate events. J. Biol. Chem. 275, 29162–29169. [DOI] [PubMed] [Google Scholar]
- Guo, Q., Vasile, E., and Krieger, M. (1994). Disruptions in Golgi structure and membrane traffic in a conditional lethal mammalian cell mutant are corrected by epsilon-COP. J. Cell Biol. 125, 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Q., Penman, M., Trigatti, B.L., and Krieger, M. (1996). A single point mutation in epsilon-COP results in temperature-sensitive, lethal defects in membrane transport in a Chinese hamster ovary cell mutant. J. Biol. Chem. 271, 11191–11196. [DOI] [PubMed] [Google Scholar]
- Harris, S.L., and Waters, M.G. (1996). Localization of a yeast early Golgi mannosyltransferase, Och1p, involves retrograde transport. J. Cell Biol. 132, 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, E., Sommer, T., Prehn, S., Gorlich, D., Jentsch, S., and Rapoport, T.A. (1994). Evolutionary conservation of components of the protein translocation complex. Nature 367, 654–657. [DOI] [PubMed] [Google Scholar]
- Hay, J.C., Klumperman, J., Oorschot, V., Steegmaier, M., Kuo, C.S., and Scheller, R.H. (1998). Localization, dynamics, and protein interactions reveal distinct roles for ER and Golgi SNAREs. J. Cell Biol. 141, 1489–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, G.R., Rahl, P.B., Collins, R.N., and Cerione, R.A. (2003). Conserved structural motifs in intracellular trafficking pathways: structure of the gamma-COP appendage domain. Mol. Cell 12, 615–625. [DOI] [PubMed] [Google Scholar]
- Johnston, J.A., Ward, C.L., and Kopito, R.R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.-W., Massey, T., Sacher, M., Pypaert, M., and Ferro-Novick, S. (2001). Sgf1p, a new component of the Sec34p/Sec35p complex. Traffic 2, 820–830. [DOI] [PubMed] [Google Scholar]
- Kim, D.W., Sacher, M., Scarpa, A., Quinn, A.M., and Ferro-Novick, S. (1999). High-copy suppressor analysis reveals a physical interaction between Sec34p and Sec35p, a protein implicated in vesicle docking. Mol. Biol. Cell. 10, 3317–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.-M., et al. (2003). Early mitochondrial hyperpolarization and intracellular alkalinization in lactacystin-induced apoptosis of retinal pigment epithelial cells. J. Pharmacol. Exp. Ther. 305, 474–481. [DOI] [PubMed] [Google Scholar]
- Kimata, Y., Lim, C.R., Kiriyama, T., Nara, A., Hirata, A., and Kohno, K. (1999). Mutation of the yeast epsilon-COP gene ANU2 causes abnormal nuclear morphology and defects in intracellular vesicular transport. Cell Struct. Funct. 24, 197–208. [DOI] [PubMed] [Google Scholar]
- Kostova, Z., and Wolf, D.H. (2003). For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 22, 2309–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley, D.M., Kozarsky, K.F., Segal, M., and Krieger, M. (1986). Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 102, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarsky, K.F., Brush, H.A., and Krieger, M. (1986). Unusual forms of low density lipoprotein receptors in hamster cell mutants with defects in the receptor structural gene. J. Cell Biol. 102, 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger, M., Brown, M.S., and Goldstein, J.L. (1981). Isolation of Chinese hamster cell mutants defective in the receptor-mediated endocytosis of low density lipoprotein. J. Mol. Biol. 150, 167–184. [DOI] [PubMed] [Google Scholar]
- Lane, J.D., Lucocq, J., Pryde, J., Barr, F.A., Woodman, P.G., Allan, V.J., and Lowe, M. (2001). Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J. Cell Biol. 156, 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur, F., Gaynor, E.C., Hennecke, S., Demolliere, C., Duden, R., Emr, S.D., Riezman, H., and Cosson, P. (1994). Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell 79, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Linstedt, A.D., and Hauri, H.P. (1993). Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell 4, 679–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt, A.D., Mehta, A., Suhan, J., Reggio, H., and Hauri, H.P. (1997). Sequence and overexpression of GPP130/GIMPc: evidence for saturable pH-sensitive targeting of a type II early Golgi membrane protein. Mol. Biol. Cell 8, 1073–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., Yuan, L.C., Bonifacino, J.S., and Klausner, R.D. (1989). Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., Roberts, T.H., and Hirschberg, K. (2000). Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. Cell Dev. Biol. 16, 557–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh, E., and Hong, W. (2002). Sec34 is implicated in traffic from the endoplasmic reticulum to the Golgi and exists in a complex with GTC-90 and ldlBp. J. Biol. Chem. 277, 21955–21961. [DOI] [PubMed] [Google Scholar]
- Lowe, M., and Kreis, T.E. (1995). In vitro assembly and disassembly of coatomer. J. Biol. Chem. 270, 31364–31371. [DOI] [PubMed] [Google Scholar]
- Lowe, S.L., Peter, F., Subramaniam, V.N., Wong, S.H., and Hong, W. (1997). A SNARE involved in protein transport through the Golgi apparatus. Nature 389, 881–884. [DOI] [PubMed] [Google Scholar]
- Mancini, M., Machamer, C.E., Roy, S., Nicholson, D.W., Thornberry, N.A., Casciola-Rosen, L.A., and Rosen, A. (2000). Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J. Cell Biol. 149, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Menárguez, J.A., Prekeris, R., Oorschot, V.M., Scheller, R., Slot, J.W., Geuze, H.J., and Klumperman, J. (2001). Peri-Golgi vesicles contain retrograde but not anterograde proteins consistent with the cisternal progression model of intra-Golgi transport. J. Cell Biol. 155, 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarella, R.A., Srinivasan, M., Haugejorden, S.M., and Green, M. (1990). ERp72, an abundant luminal endoplasmic reticulum protein, contains three copies of the active site sequences of protein disulfide isomerase. J. Biol. Chem. 265, 1094–1101. [PubMed] [Google Scholar]
- McCracken, A.A., and Brodsky, J.L. (2003). Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD). Bioessays 25, 868–877. [DOI] [PubMed] [Google Scholar]
- Miles, S., McManus, H., Forsten, K.E., and Storrie, B. (2001). Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J. Cell Biol. 155, 543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov, A.A., et al. (2001). Small cargo proteins and large aggregates can traverse the Golgi by a common mechanism without leaving the lumen of cisternae. J. Cell Biol. 155, 1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsomme, P., Prescianotto-Baschong, C., and Riezman, H. (2003). The ER v-SNAREs are required for GPI-anchored protein sorting from other secretory proteins upon exit from the ER. J. Cell Biol. 162, 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsomme, P., and Riezman, H. (2002). The Rab GTPase Ypt1p and tethering factors couple protein sorting at the ER to vesicle targeting to the Golgi apparatus. Dev. Cell 2, 307–317. [DOI] [PubMed] [Google Scholar]
- Moremen, K.W. (2002). Golgi alpha-mannosidase II deficiency in vertebrate systems: implications for asparagine-linked oligosaccharide processing in mammals. Biochim. Biophys. Acta 1573, 225–235. [DOI] [PubMed] [Google Scholar]
- Nagahama, M., Orci, L., Ravazzola, M., Amherdt, M., Lacomis, L., Tempst, P., Rothman, J.E., and Söllner, T.H. (1996). A v-SNARE implicated in intra-Golgi transport. J. Cell Biol. 133, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, N., Rabouille, C., Watson, R., Nilsson, T., Hui, N., Slusarewicz, P., Kreis, T.E., and Warren, G. (1995). Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131, 1715–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novina, C.D., Murray, M.F., Dykxhoorn, D.M., Beresford, P.J., Riess, J., Lee, S.K., Collman, R.G., Lieberman, J., Shankar, P., and Sharp, P.A. (2002). siRNA-directed inhibition of HIV-1 infection. Nat. Med. 8, 681–686. [DOI] [PubMed] [Google Scholar]
- Oka, T., Nishikawa, S., and Nakano, A. (1991). Reconstitution of GTP-binding Sar1 protein function in ER to Golgi transport. J. Cell Biol. 114, 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opat, A.S., van Vliet, C., and Gleeson, P.A. (2001). Trafficking and localisation of resident Golgi glycosylation enzymes. Biochimie 83, 763–773. [DOI] [PubMed] [Google Scholar]
- Orci, L., Ravazzola, M., Volchuk, A., Engel, T., Gmachl, M., Amherdt, M., Perrelet, A., Söllner, T.H., and Rothman, J.E. (2000). Anterograde flow of cargo across the Golgi stack potentially mediated via bidirectional “percolating” COPI vesicles. Proc. Natl. Acad. Sci. 97, 10400–10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham, H.R., and Rothman, J.E. (2000). The debate about transport in the Golgi-two sides of the same coin? Cell 102, 713–719. [DOI] [PubMed] [Google Scholar]
- Perkel, V. S., Y. Miura, and J. A. Magner. (1989) Brefeldin A inhibits oligosaccharide processing of glycoproteins in mouse hypothyroid pituitary tissue at several subcellular sites. Proc. Soc. Exp. Biol. Med. 190, 286–293. [DOI] [PubMed] [Google Scholar]
- Podos, S.D., Reddy, P., Ashkenas, J., and Krieger, M. (1994). LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. J. Cell Biol. 127, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri, S., Bachert, C., Fimmel, C.J., and Linstedt, A.D. (2002). Cycling of early Golgi proteins via the cell surface and endosomes upon lumenal pH disruption. Traffic 3, 641–653. [DOI] [PubMed] [Google Scholar]
- Ram, R.J., Li, B., and Kaiser, C.A. (2002). Identification of Sec36p, Sec37p and Sec38p: components of the yeast complex that contains Sec34p and Sec35p. Mol. Biol. Cell. 13, 1484–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, P., and Krieger, M. (1989). Isolation and characterization of an extragenic suppressor of the low-density lipoprotein receptor-deficient phenotype of a Chinese hamster ovary cell mutant. Mol. Cell. Biol. 9, 4799–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, J. (2002). Protein N-glycosylation along the secretory pathway: relationship to organelle topography and function, protein quality control, and cell interactions. Chem. Rev. 102, 285–303. [DOI] [PubMed] [Google Scholar]
- Sacher, M., Jiang, Y., Barrowman, J., Scarpa, A., Burston, J., Zhang, L., Schieltz, D., Yates J.R. 3rd, Abeliovich, H., and Ferro-Novick, S. (1998). TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO J. 17, 2494–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath, D., Varki, A., and Freeze, H.H. (1992). The spectrum of incomplete N-linked oligosaccharides synthesized by endothelial cells in the presence of brefeldin A. J. Biol. Chem. 267, 4440–4455. [PubMed] [Google Scholar]
- Satoh, A., Wang, Y., Malsam, J., Beard, M.B., and Warren, G. (2003). Golgin-84 is a rab1 binding partner involved in Golgi structure. Traffic 4, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schekman, R., and Orci, L. (1996). Coat proteins and vesicle budding. Science 271, 1526–1533. [DOI] [PubMed] [Google Scholar]
- Seemann, J., E. Jokitalo, M, Pypaert, and G. Warren. 2000. Matrix proteins can generate the higher order architecture of the Golgi apparatus. Nature 407, 1022–1026. [DOI] [PubMed] [Google Scholar]
- Shorter, J., Beard, M.B., Seemann, J., Dirac-Svejstrup, A.B., and Warren, G. (2002). Sequential tethering of golgins and catalysis of SNAREpin assembly by the vesicle-tethering protein p115. J. Cell Biol. 157, 45–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter, J., and Warren, G. (2002). Golgi architecture and inheritance. Annu. Rev. Cell Dev. Biol. 18, 379–420. [DOI] [PubMed] [Google Scholar]
- Siniossoglou, S., and Pelham, H.R. (2001). An effector of Ypt6p binds the SNARE Tlg1p and mediates selective fusion of vesicles with late Golgi membranes. EMBO J. 20, 5991–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrincosky, D., Kain, R., El-Battari, A., Exner, M., Kerjaschki, D., and Fukuda, M. (1997). Altered Golgi localization of core 2 beta-1,6-N-acetylglucosaminyltransferase leads to decreased synthesis of branched O-glycans. J. Biol. Chem. 272, 22695–22702. [DOI] [PubMed] [Google Scholar]
- Sönnichsen, B., Lowe, M., Levine, T., Jamsa, E., Dirac-Svejstrup, B., and Warren, G. (1998). A role for giantin in docking COPI vesicles to Golgi membranes. J. Cell Biol. 140, 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelbrink, R.G., and Nothwehr, S.F. (1999). The yeast GRD20 gene Is required for protein sorting in the trans-Golgi network/endosomal system and for polarization of the actin cytoskeleton. Mol. Biol. Cell. 10, 4263–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storrie, B., White, J., Röttger, S., Stelzer, E.H., Suganuma, T., and Nilsson, T. (1998). Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J. Cell Biol. 143, 1505–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storrie, B., Pepperkok, R., and Nilsson, T. (2000). Breaking the COPI monopoly on Golgi recycling. Trends Cell Biol. 10, 385–391. [DOI] [PubMed] [Google Scholar]
- Storrie, B., and Nilsson, T. (2002). The Golgi apparatus: balancing new with old. Traffic 3, 521–529. [DOI] [PubMed] [Google Scholar]
- Stroud, W.J., Jiang, S., Jack, G., and Storrie, B. (2003). Persistence of Golgi matrix distribution exhibits the same dependence on Sar1p activity as a Golgi glycosyltransferase. Traffic 4, 631–641. [DOI] [PubMed] [Google Scholar]
- Subramaniam, V.N., Krijnse-Locker, J., Tang, B.L., Ericsson, M., Yusoff, A.R., Griffiths, G., and Hong, W. (1995). Monoclonal antibody HFD9 identifies a novel 28 kDa integral membrane protein on the cis-Golgi. J. Cell Sci. 108, 5–14. [DOI] [PubMed] [Google Scholar]
- Subramaniam, V.N., Peter, F., Philp, R., Wong, S.H., and Hong, W. (1996). GS28, a 28-kilodalton Golgi SNARE that participates in ER-Golgi transport. Science 272, 1161–1163. [DOI] [PubMed] [Google Scholar]
- Suvorova, E.S., Kurten, R.C., and Lupashin, V.V. (2001). Identification of a human orthologue of Sec34p as a component of the cis-Golgi vesicle tethering machinery. J. Biol. Chem. 276, 22810–22818. [DOI] [PubMed] [Google Scholar]
- Suvorova, E.S., Duden, R., and Lupashin, V.V. (2002). The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J. Cell Biol. 157, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorow, Z., Spang, A., Carmack, E., Yates, J., and Schekman, R. (2000). Active recycling of yeast Golgi mannosyltransferase complexes through the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 97, 13643–13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar, D., Oka, T., Brittle, E.E., Vasile, E., Lupashin, V.V., Chatterton, J.E., Heuser, J.E., Krieger, M., and Waters, M.G. (2002). Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 157, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Echten, G., Iber, H., Stotz, H., Takatsuki, A., and Sandhoff, K. (1990). Uncoupling of ganglioside biosynthesis by brefeldin A. Eur. J. Cell Biol. 51, 135–139. [PubMed] [Google Scholar]
- VanRheenen, S.M., Cao, X., Lupashin, V.V., Barlowe, C., and Waters, M.G. (1998). Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J. Cell Biol. 141, 1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen, S.M., Cao, X., Sapperstein, S.K., Chiang, E.C., Lupashin, V.V., Barlowe, C., and Waters, M.G. (1999). Sec34p and Sec35p are components of a protein complex required for vesicle tethering the yeast Golgi. J. Cell Biol. 147, 729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, D.M., Paul, K.S., and Waters, M.G. (1998). Purification and characterization of a novel 13 S hetero-oligomeric protein complex that stimulates in vitro Golgi transport. J. Biol. Chem. 273, 29565–29576. [DOI] [PubMed] [Google Scholar]
- Ward, T.H., Polishchuk, R.S., Caplan, S., Hirschberg, K., and Lippincott-Schwartz, J. (2001). Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 155, 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, M.G., Serafini, T., and Rothman, J.E. (1991). `Coatomer': a cytosolic protein complex containing subunits of non-clathrin-coated Golgi transport vesicles. Nature 349, 248–251. [DOI] [PubMed] [Google Scholar]
- Whyte, J.R.C., and Munro, S. (2001). The Sec34/35 Golgi Transport Complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev. Cell 1, 527–537. [DOI] [PubMed] [Google Scholar]
- Wiertz, E.J., Jones, T.R., Sun, L., Bogyo, M., Geuze, H.J., and Ploegh, H.L. (1996b). The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769–779. [DOI] [PubMed] [Google Scholar]
- Wiertz, E.J., Tortorella, D., Bogyo, M., Yu, J., Mothes, W., Jones, T.R., Rapoport, T.A., and Ploegh, H.L. (1996a). Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384, 432–438. [DOI] [PubMed] [Google Scholar]
- Wuestehube, L.J., Duden, R., Eun, A., Hamamoto, S., Korn, P., Ram, R., and Schekman, R. (1996). New mutants of Saccharomyces cerevisiae affected in the transport of proteins from the endoplasmic reticulum to the Golgi complex. Genetics 142, 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y., Wong, S.H., Zhang, T., Subramaniam, V.N., and Hong, W. (1997). GS15, a 15-kilodalton Golgi soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) homologous to rbet1. J. Biol. Chem. 272, 20162–20166. [DOI] [PubMed] [Google Scholar]
- Xu, Y., Martin, S., James, D.E., and Hong, W. (2002). GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol. Biol. Cell 13, 3493–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori, T., Yamamoto, A., Moriyama, Y., Futai, M., and Tashiro, Y. (1991). Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266, 17707–17712. [PubMed] [Google Scholar]
- Yuan, L., Barriocanal, J.G., Bonifacino, J.S., and Sandoval, I.V. (1987). Two integral membrane proteins located in the cis-middle and trans-part of the Golgi system acquire sialylated N-linked carbohydrates and display different turnovers and sensitivity to cAMP-dependent phosphorylation. J. Cell Biol. 105, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaal, K.J.M., et al. (1999). Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell 99, 589–601. [DOI] [PubMed] [Google Scholar]
- Zerfaoui, M., Fukuda, M., Langlet, C., Mathieu, S., Suzuki, M., Lombardo, D., and El-Battari, A. (2002). The cytosolic and transmembrane domains of the beta 1, 6 N-acetylglucosaminyltransferase (C2GnT) function as a cis to medial/Golgi-targeting determinant. Glycobiology 12, 15–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.