

Abstract
Controversy has plagued tumor virology since the first tumor viruses were described over 100 years ago. Methods to establish cancer causation, such as Koch’s postulates, work poorly or not at all for these viruses. Kaposi’s sarcoma herpesvirus (KSHV/HHV8) and Merkel cell polyomavirus (MCV) were both found using nucleic acid identification methods but they represent opposite poles in the patterns for tumor virus epidemiology. KSHV is uncommon and has specific risk factors that contribute to infection and subsequent cancers. MCV and Merkel cell carcinoma (MCC), in contrast, is an example in which mutations to our normal viral flora contribute to cancer. Given the near-ubiquity of human MCV infection, establishing cancer causality relies on molecular evidence that does not fit comfortably within traditional infectious disease epidemiological models. These two viruses reveal some of the challenges and opportunities for inferring viral cancer causation in the age of molecular biology.
Introduction
Seven known human tumor viruses cause about 1 in every 6 cancers worldwide[1, 2]. Beyond the large public health impact, this is remarkable because there are so few of these viruses: of the thousands of viruses causing infection, only a minute proportion have been established to cause cancer (Table 1) and even then most people infected with a cancer virus never develop tumors. This review focuses on the two most recently described tumor viruses, Kaposi’s sarcoma herpesvirus (KSHV) and Merkel cell polyomavirus (MCV), which were discovered in 1994 and 2008, respectively. They reveal new opportunities, as well as new limits, for discovering infectious cancer causes in the age of molecular biology.
Table 1.
Human Tumor Viruses
| Year | Virus | Abbreviation | Notable cancers |
|---|---|---|---|
| 1964 | Epstein-Barr virus | EBV | Burkitt’s lymphoma, Nasopharyngeal carcinoma, Hodgkin disease, Gastric carcinoma |
| 1965 | Hepatitis B virus | HBV | Hepatocellular carcinoma |
| 1980 | Human T- lymphotropic virus-I | HTLV-1 | Adult T cell leukemia |
| 1983 | High-risk human papillomavirus | HPV | Cervical cancer, Head and neck cancer |
| 1989 | Hepatitis C virus | HCV | Hepatocellular carcinoma |
| 1994 | Kaposi’s sarcoma- associated herpesvirus | KSHV | Kaposi’s sarcoma, Primary effusion lymphoma, Multicentric Castleman’s disease |
| 2008 | Merkel cell polyomavirus | MCV | Merkel cell carcinoma |
Causality, Cancer and Molecular Virology
Controversies surround tumor viruses, largely on the fundamental question of whether or not they cause cancer. Causality itself is a topic that generates arguments not only among scientists but also among philosophers, statisticians, computer scientists and others. One tends to suppose that there exists well-defined criteria that must be met for an agent to be called a tumor virus. Either the agent meets these requirements or it does not. Instead, adjudicating causality is a normative process that no one person can successfully determine. Similar to a famous description for innovation, causality “only exists when the correctly credentialed hivemind agrees that it exists”[3]. But determining cancer virus causality is not an empty intellectual exercise because it has profound consequences that can be measured in lives prematurely lost when diagnostics, medicines and vaccines are not developed or employed.
EBV was discovered in 1964[4], yet declared to be a legitimate human carcinogen only in 1997 by the International Agency for Cancer Research[5]. During these 32 years, ~3.7 million persons developed EBV-induced cancers (based on unadjusted 2008 estimates[1]). More recently the successes of human papillomavirus (HPV) and hepatitis B virus (HBV) control show that targeting the fundamental viral cause for a cancer can massively alter the burden of infectious cancers. The debate over AIDS and HIV provides an even more stark case for the practical importance of causal inference. Over 300,000 preventable HIV infections occurred in South Africa between 2000 and 2005 as a result of a government policy withholding distribution of antiretroviral prophylaxis for pregnant women on the basis that HIV is not the cause of AIDS[6]. This policy was supported by fringe science that did not take into account any modern sense of viral causality[7–9].
The reasons why viruses have until relatively recently been neglected as causes for cancer are complex[10]. Viral cancers—like all diseases—are multifactorial and only rare examples exist of a clear 1-to-1 correspondence between virus infection and neoplasia. Most persons who are exposed to a tumor virus never develop disease, although this should hardly be surprising since asymptomatic infection is a feature for almost all pathogens. Further, for every bona fide human cancer virus that has been found, there have been dozens of false leads and dead-ends that have littered the scientific literature with conflicting, confusing and contentious descriptions of virus-cancer links. Evidence that herpes simplex virus (HSV) 2 is the likely cause of cervical cancer led to a large body of evidence[11, 12], the interpretation of which was clarified only after years of research following the discoveries of HPV type 16 and 18 by zur Hausen’s group[11, 13, 14]. Since both HPV and HSV are sexually transmitted, confounding and overlapping epidemiologies for these two viruses is not surprising in retrospect. A more recent and remarkable example was discovery of a simple endogenous murine retrovirus, XMRV, which had cryptically jumped from the mouse genome into human prostate cancer cell lines during mouse xenograft studies[15]. The virus was discovered over a decade later, long after the mouse passaging experiments had been forgotten, and therefore was reasonably suspected to be a novel human cancer virus--though the discovering authors were appropriately cautious in ascribing any causative etiology[16]. Each valid tumor virus requires years of confirmatory research to begin to unravel its association with cancers and so the failure to recognize true virus-cancer associations by the general public is understandable.
Classifying Causation
For more detailed discussions of causal inference, see here[17–22]. Attempts to define causality date back at least to the era of Galileo Galilei (1564–1642) and (controversially) even have been ascribed to him [23]: An agent causes disease when it is both necessary and sufficient for the disease to occur. This has been called a complete causal effect[17].
Beyond the most simplistic examples, however, this is not a useful definition, especially not for infectious cancers. All infectious diseases emerge from a complex interplay of multiple factors (immunity, host genetics, age of infection, etc.), so that no infection alone is sufficient to cause cancer. Various causal factors (e.g., immunodeficiency and viral infection) may be overlapping and synergistic with each other so that the attributable risk from a viral infection may not be obvious[17]. Further, depending on how cancers are classified, not all types of a cancer would necessarily be caused by a single agent. A good example of this is hepatocellular carcinoma, which can arise after exposure to HBV, hepatitis C virus (HCV) or chemical mutagens[24]. And so, the necessary and sufficient definition becomes meaningless after even the most trivial scientific description of viral tumors.
Skepticism about causal inference reached a high point a century later when the empiricist David Hume (Figure 1) outlined the difficulty, perhaps even impossibility, of describing causality through external criteria such as necessity and sufficiency (Figure 2). Hume instead used a counterfactual argument to define causality: we can say object A causes object B when B follows A and when A does not occur, then B does not occur [11, 18, 25].
Figure 1.

Major theorists in causal inference.
Figure 2.

A) Example of a circuit in which Switch 1 (but not Switch 2) is “causing” a light to turn on, although neither switch is necessary (From [122]). B) Example for EBV causality for Burkitt lymphoma using this reasoning. In this example, EBV is not necessary. Since other factors (“switches”) also needed, such as cMyc rearrangements or immune suppression, it is not sufficient either.
Koch’s Postulates
This was hardly helpful to 19th Century microbiologists. To establish scientific rigor for the new field of microbiology, Robert Koch famously formulated a set of postulates for microbial causes of disease in a series of lectures to the International Congress in Berlin in 1890[22]. These postulates have since tenaciously taken hold in microbiology because they are easy to remember and, at first glance, seem to be universally true. Koch’s eponymous postulates were partially articulated by Koch’s mentor, Jakob Henle in 1840[26], and later expanded and codified by Koch so they are also often referred to as the Henle-Koch Postulates[27]:
The parasite occurs in every case of the disease in question and under circumstances which can account for the pathological changes and clinical course of the disease.
It occurs in no other disease as a fortuitous and nonpathogenic parasite.
After being fully isolated from the body and repeatedly grown in pure culture, it can induce the disease anew.
The simplicity and clarity of this proposition is so persuasive that these postulates are usually included in the canon of a sound undergraduate science education. But, even Koch was aware of their limitations[8] during his disputes with Petenkoffer over whether Vibrio cholerae, which Koch had discovered[28], caused epidemic cholera disease (an argument leading Pettenkofer to swill pure cultures of the bacteria and then develop diarrhea but not classic cholera disease[29, 30]). Even for diseases that have an intuitively obvious cause, rigorous application of Henle-Koch’s postulates leave open possibilities for doubt. A skeptic could argue, for example, that Neisseria meningitidis is not the cause spinal meningitis since there are many infectious causes for meningitis and ~10% of healthy adults are asymptomatic N. meningititdis carriers. Even animal models for disease can be disputed as not fully recapitulating human spinal meningitis. Yet, detection of gram-negative diplococci in cerebrospinal fluid leaves no reasonable room to doubt whether or not to give antibiotics to a symptomatic patient.
The principal problem with Henle-Koch’s postulates is not so much that they are wrong but that they have little value since they are inapplicable to the majority of pathogens. When negative, the results say little about whether a candidate pathogen causes disease or not. For viruses, these issues become even more complex[31]. As obligate cellular parasites, viruses cannot be formally evaluated in pure culture. Under the most assiduous gradient isolation conditions, the purity of a virus culture can still be questioned. Viral cancers, which generally are non-permissive for viral replication[32], may have no virions to isolate and are therefore ineligible for the Henle-Koch rules. HIV denialists make use of these ambiguities, which in turn require complex--and ultimately unsatisfying—rebuttals by epidemiologists[33] trying to fulfill ersatz Henle-Koch’s postulates. Ironically, Werner Henle, Jakob’s grandson, and Werner’s wife, Gertrude, contributed to establishing EBV as a cancer-causing virus by experimental lymphocyte immortalization studies[27] rather than by attempting to fulfill his grandfather’s postulates. Henle-Koch’s postulates are a brilliant example of precision in scientific thinking but they hold little practical value for 21st Century tumor virology since they cannot prove nor disprove most candidate tumor viruses to cause cancers. Rivers attempted to address these failings in 1937 using immunologic evidence without the requirement for purified virus culture[34]. More recently, modifications of the Henle-Koch postulates have tried to take into account molecular biology advances[21, 35].
Hill’s Epidemiologic Criteria for Causality
Intriguingly parallel to the theoretical vs. empirical descriptions by Galileo and Hume, in the 1960s, Austin Bradford Hill provided an empirical counterbalance to Koch’s theoretical disease causation paradigm. Hill and Richard Doll pioneered the application of various epidemiologic study designs to the question of whether cigarette smoking causes lung cancer. This contentious topic required a deeply analytical approach that could withstand attacks by vested tobacco industry interests as well as academic skeptics. In an influential address to the British Royal Society of Medicine in 1965[19], Hill outlined nine criteria to address causal inference (Table 2).
Table 2.
Hill’s Criteria for Causation in Tumor Virology
| 1. STRENGTH OF ASSOCIATION | A strong association between a virus and a cancer is most consistent with causality unless confounded by a some other exposure, which is likely to be obvious. A weak association, however, does not give evidence against causality. Example: High-risk HPV is more strongly associated with cervical cancer than herpes simplex 2, which is confounded by sexual exposure. |
| 2. CONSISTENCY | Consistent findings observed by different persons, in different places, circumstances and times. |
| 3. SPECIFICITY | A viral exposure is limited to specific types cancer and not others. This was considered a weak criterion since there are well-established examples in which multiple types are disease are caused by one type of exposure or infection. The more specific the association between a virus and a cancer, the higher the probability of a causal relationship. |
| 4. TEMPORALITY | Exposure to the virus must occur prior to the onset of the cancer, in contrast to a “passenger infection”. |
| 5. BIOLOGICAL GRADIENT | Originally described as a dose-response effect to show that increased smoking results in increased lung cancer rates. This is less obvious for a biological exposure such as a virus but has been interpreted as the virus is more likely to be found at the site of the tumor than at nontumor sites among infected persons. |
| 6. PLAUSIBILITY | Hill noted that plausibility for a disease mechanism is limited by current medical knowledge. Even implausible mechanisms may gain acceptance as greater understanding of the underlying biology becomes known. |
| 7. COHERENCE | A virus-cancer association should not seriously conflict with known facts on the cancer’s natural history and biology. Supportive data, such a CIN histopathology in cervical cancer, that are coherent with a proposed agent,, increase the likelihood for a causal relationship. |
| 8. EXPERIMENT | This is generally the most difficult of the Hill criteria to achieve but it supposes the ability to change either exposure or continued infection in a randomized clinical trial and measure a change in cancer outcome. Examples of this include vaccination programs for HPV and HBV. |
| 9. ANALOGY | Are related viruses clearly established to cause cancers in animals or humans? |
In comparison to the Henle-Koch’s postulates, Hill’s criteria were shaded more towards practical measures that help inform public health professionals about potential interventions. The criterion for consistency, for example, implies that in the real world, chance variation or isolated erroneous reports might contribute to a spurious association (or fail to detect a real association). But taken as a whole, the evidence for a causal association should be reproducible and consistent in different settings, in studies performed by different investigators. Hill was also careful to state that none of these criteria, except the correct temporal relationship, were absolute and he emphasized the need to rationally weigh all of the evidence to come to a best judgment[19, 20]. As an early advocate for randomized clinical trials, he also included experimental epidemiology into his criteria and the need for counterfactual reasoning[20].
Concerns regarding the universality of Hill’s criteria emerged during advances in molecular pathobiology. Even though they are an improvement over Henle-Koch’s postulates, Hill’s criteria also have unspoken biases about pathogenic relationships. Hill proposed that a dose-response or biological gradient is consistent with causality. And so, the greater the exposure to a causal factor, the more likely for disease to occur. This is a sensible idea but one can readily construct a counter-example: if an autoimmune disease is caused by a candidate pathogen, it would be reasonable to hypothesize that those persons with more robust immune responses are also more likely to both 1) clear the infection and 2) to suffer disease. An epidemiologic study then, would find the paradoxical result that cases are significantly less likely to harbor the pathogen than comparison populations. While the agent would actually cause the autoimmune disorder, an epidemiologist would find its biologic gradient to be the opposite of that predicted from Hill’s criteria. This is not a trivial point: over the past 150 years most of the straightforward microbial disease associations have been readily and reproducibly found. There exist chronic diseases, such as cancers and autoimmune disorders, in which infection is suspected to play a role but no progress has been made in detecting the etiologic agents. As with MCV, new pathogens that violate existing paradigms may lead to a more profound understanding of basic pathobiology and its reflection in the epidemiology of human disease.
Enter KSHV and KS
In the 1970s, the herpesvirus cytomegalovirus (CMV) was first proposed as the likely cause for KS, based mainly on antibody reactivity studies[36]. Careful electron microscopy soon revealed scattered herpes-like capsids in an African KS tumor[37]. KS had first been described by Moriz Kaposi in 1872 as an aggressive tumor in five patients that quickly led to death[38]. It is a complex tumor containing neoplastic endothelial cells, immune infiltrates and well-formed, non-neoplastic neo-angiogenic structures (Figure 3A). Subsequent studies, revealed KS to be more indolent tumor occurring generally in elderly men, particularly those with Mediterranean and Ashkanazi Jewish ethnicities. KS was assumed to be uncommon until it was found to be one of the most frequent tumors reported in Eastern African countries; in some cancer registries, the third most common cancer in adults[39]. Another surprise occurred with the emergence of KS as a transplantation-associated tumor during high-dose immunosuppressive treatment[40], suggesting that the tumor is controlled by immune surveillance. By this time, it was also recognized that KS clusters with secondary lymphoid malignancies[41], including lymphomas and Castleman’s disease[42].
Figure 3. A) Kaposi’s sarcoma.
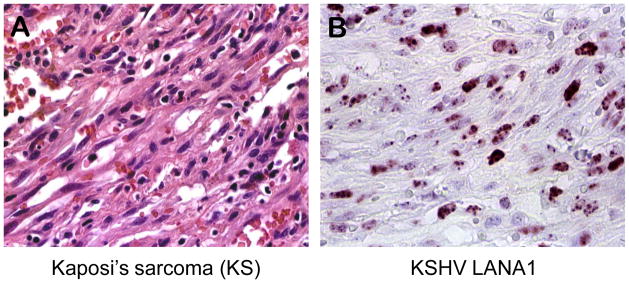
Micrograph of a Kaposi’s sarcoma lesion showing spindle tumor cells of endothelial/lymphatic origin. The tumor cells are associated with infiltrating lymphoid cells and abnormal microvasculature, some still containing red blood cells. (hematoxylin and eosin staining). B) KSHV LANA1. Micrograph of Kaposi’s sarcoma tissue section immunostained with an antibody targeting the latency associated nuclear antigen 1 (LANA1). KS spindle tumor cells show nuclear distribution of LANA1 expression.
The emergence of KS as the prominent manifestation of AIDS in the early 1980s re-energized efforts to finds its cause. The link to CMV was disputed[43] and over a dozen other agents (including HPV[44] and mycoplasma[45]) were proposed and discarded as causes for this cancer. One widely-held hypothesis was that HIV itself was responsible for the tumorigenic changes in endothelial cells leading to KS[46], either through induction of inflammatory cytokines or by expression of the tat transactivator protein[47]. This was based in part on the failure to find any other organism consistent with the etiology of KS.
Careful epidemiologic examination, however, provided clear clues on the causal agent for KS. In 1990, Beral and coauthors published a provocative paper[48] entitled “Kaposi’s sarcoma among persons with AIDS: A sexually transmitted infection” that rigorously analyzed AIDS-KS statistics. Its key conclusions included 1) HIV is not the direct cause of KS (HIV-positive hemophilics have low rates of KS while over 100 years of research had shown that KS occurs among persons without HIV), 2) the KS agent has low general prevalence in Europe and North America but may be common in geographic regions such as sub-Saharan Africa, 3) the agent is sexually transmitted among gay and bisexual men in the United States and epidemic clustering can be found, and 4) unlike HIV, the agent has a low risk for parenteral transmission. Subsequently, each of these predictions would be found to be correct.
We used a DNA subtractive hybridization and PCR enrichment technique, developed by the Lisitsyns and Wigler called representational difference analysis[49], to isolate the KS agent DNA from an AIDS-KS tumor. Analysis of these DNA fragments revealed a previously undescribed human herpesvirus (KSHV or HHV8), localized to AIDS-KS tumors and absent from most nonKS tissues from HIV-infected and uninfected persons[50]. That this was a new herpesvirus was confirmed through in vitro isolation and antibody detection[51, 52], and subsequently, by electron microscopy[53] and whole genome sequencing[54].
Immediately thereafter, laboratories sought to find whether KSHV fulfills Hill’s criteria for causality of KS[55]. Molecular biology provided the tools to search for this virus even though less than 1% of its genome had been initially uncovered. KSHV was quickly confirmed to be present in nearly all AIDS-KS (perhaps all KS after taking into account diagnostic and specimen handling issues) as well as nonAIDS KS, e.g., [56–58], meeting criteria of strength and consistency of association. Identification of the virus in primary effusion lymphomas [59], as well as most Castleman’s disease tumors[60], provided the opportunity to develop a serologic test based for antibodies to the KSHV LANA protein[52] (Figure 3B). This led quickly to a series of studies[61–64] showing the virus to not be ubiquitous but it instead follows the patterns predicted by Beral et al. [48]: KSHV is generally uncommon in developed countries but highly prevalent in parts of Africa, it is readily transmitted through homosexual but not heterosexual activity (the precise risk behavior responsible for this difference is still unclear) and it, unlike HIV, is not readily transmitted from contaminated blood products. Detection of the virus genome[65, 66] and then KSHV antibodies[67, 68] demonstrated that KSHV infection precedes KS disease, fulfilling the key Hill temporal requirement. Two years after discovery, the virus was sequenced and found to contain a number of plausible oncogenes[54], particularly oncogenic proteins that aid in immune evasion[69, 70]. By 1999, even the most difficult-to-achieve Hill criterion, experimental epidemiologic evidence, had been inadvertently fulfilled through a randomized clinical trial of AIDS patients given an antiherpesvirus drug that was highly protective against new occurrence of KS (though ineffective against established KS tumors)[71].
In hindsight, the overwhelming weight of scientific evidence within two years concretely showed that KSHV to be the infectious cause for KS. This is not to say that there was no controversy over this relationship[72]. Both PCR contamination[73, 74] and cross-reactive capsid serologies[75] erred in the direction of falsely detecting KSHV infection in healthy control populations, and gave results incompatible with KSHV being the agent causing KS. Similar false-positive KSHV detections would prompt spurious associations for KSHV with other cancers, leading to the incorrect conclusion that KSHV is not specific for KS. Clarifying the true prevalence and disease associations for KSHV required dozens of research studies[55]. Ironically, despite years of controversy, the patterns of KSHV infection are now generally accepted to be near-identical to those initially reported in the very first studies[50,52,62,64].
Why did Hill’s criteria work so well for KSHV and KS? This is likely due to all clinical forms of KS requiring KSHV infection, which is a property shared by few viral cancers. African and New Guinean Burkitt’s lymphoma, for example, are nearly universally EBV-infected but only half of sporadic US tumors contain EBV genome. All cervical cancers, but not all head-neck cancers, are driven by high-risk HPV. Even for KSHV, approximately half of Castleman’s tumors are negative for KSHV and have a poorly-defined etiology. Given convergent tumor viral evolution to target relatively few tumor suppressor pathways, it should not be surprising that noninfectious carcinogens might mimic tumor virus infection. Epidemiologists have to intellectually contend with this if they seek to understand and prevent these cancers.
A second helpful property of KSHV is that it is not ubiquitous (in North America, Asia and Europe) since a high prevalence would pose little extra risk for KS through sexual KSHV transmission. The virus is much more common in many African and indigenous South American populations[76], consistent with casual, nonsexual transmission playing a role in these settings. KSHV coevolved with humans and follows patterns of human migration[77]. At some point, it was nearly lost from most populations outside of Africa suggesting that KSHV is rather poorly-adapted to the humans compared to other human herpesviruses--and therefore might be an exceptionally good vaccine candidate. Within historical times, however, it is likely there was an undocumented epidemic spread of KSHV into settled Old World populations, resulting in the current pattern of high focal prevalence in East Africa diminishing into other African and Eastern Mediterranean areas, and extending into Silk Road Uyger and Hazakh Chinese populations[78, 79]. Two additional technical, but important, aspects to KSHV and KS causation studies are notable. The first is that initial epidemiologic studies were performed using strictly blinded and randomized testing. Under these conditions, any random testing errors would bias towards the null hypothesis--that there is no association between KSHV infection and KS. This is the most important, easiest and least expensive (but often overlooked) control in virus-cancer association studies. Secondly, ongoing cohort HIV studies provided an irreplaceable resource for addressing issues related to KSHV infection and AIDS-KS.
AIDS-KS incidence has reduced by 85% during the era of effective antiretroviral therapy in developed countries[80]. Unfortunately, no KSHV-specific diagnostic tests or therapies have been developed outside of the research setting and this cancer remains a major unaddressed public health problem in many developing countries, particularly in Africa.
Epidemiology Fails Merkel Cell Polyomavirus
MCV was found as a viral infection of Merkel cell carcinomas in 2008[81]. Like KS, Merkel cell carcinoma (MCC) is a cancer sensitive to immune surveillance and occurs more frequently in AIDS [82] and transplant populations[83, 84]. It is one of the most aggressive skin cancers (Figure 4A and B) and has few effective therapeutic options once disseminated[85]. We sought to develop a new approach to finding tumor viruses based on computational (rather than physical) nucleic acid subtraction. To achieve this, Feng et al. developed digital transcriptome subtraction (DTS) relying on first identifying a high-fidelity (Hi-Fi) sequence dataset and then eliminating human sequence alignments[86]. The remaining candidate sequence pool (<0.5%) can then be examined with relaxed alignment filters or by direct experimentation.
Figure 4.
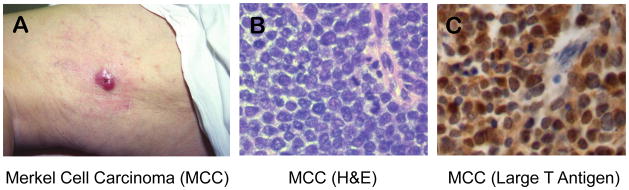
A) Dermatologic, clinical appearance of Merkel cell carcinoma as a raised, red nodule on skin (courtesy of Dr. K. Busam). B) Histologic appearance of Merkel cell carcinoma showing sheets of small, round, blue tumor cells with high mitotic activity (arrowheads). C) MCV Large T antigen expression in Merkel cell carcinoma showing predominantly nuclear localization of the viral oncoprotein.
MCV was the first human pathogen (January 2008) found by unbiased sequencing although a number of other groups simultaneously pursued similar approaches for other non-neoplastic diseases[87]. DTS uses polyadenylated mRNA (Refseq) sequencing to achieve a “genome simplification” as described by Wigler et al.[49] that improves detection of novel sequences. Assuming that a viral gene product is directly driving tumor cell proliferation, at least one viral mRNA copy should be present in each tumor cell, which is a level that can be readily achieved by direct cDNA sequencing. This approach also reduces contaminating, incidental nonpolyadenylated bacterial sequences and so it is useful in examining non-sterile site tumors. Applying DTS to MCC tumors revealed one with a unique gene fragment similar to but not identical to known polyomavirus sequences[81]. The remaining viral genome was isolated from tumors by rapid amplification of cDNA ends (RACE) and PCR cloning.
MCV is clonally-integrated into the tumor genome[81] so that daughter cells within a tumor have identical virus integration sites even though the individual integration sites differ between MCC patients. The MCV large T (LT) antigen oncoprotein for viruses isolated from tumors also possesses truncation mutations that eliminate full-length expression of LT protein[88]. Since the C-terminal helicase domain of LT is essential for MCV replication, and integration is not compatible with replication of the normally episomal, double-stranded DNA polyomavirus, tumor-derived MCV is replication incompetent. Initially, only 8 of 10 MCC tumors were found positive for MCV and the virus could be detected at low levels in various nonMCC control tissues[81]. Infection appears to be asymptomatic but probably is life-long.
Establishing causality for MCV and MCC is problematic from the perspective of Hill’s criteria. Most studies (but not all[89]) agree that there are at least two forms of MCC, one that is MCV infected at ~1 virus copy per cell or higher (75–80%) and the remainder which are not MCV-infected except as a coincidental skin infection[90]. Patient cohort studies suggest that MCV-positive MCC are generally less virulent and have better prognoses than the MCV-negative variety[91, 92]. Prospective studies provide evidence for a weak but consistent correlation between MCV infection and MCC[93].
Patients with MCV-positive MCC have significantly higher antibody titers against MCV capsid antigens than MCV-negative MCC patients or control patients[94, 95], although this data seems at first to be contradictory since MCV capsid antigens are not expressed in tumor tissues (H. Feng, pers. comm). Thus, Hill’s criterion for strength of association between MCV and all forms of MCC is in question. Worse yet, widespread human MCV infection[94–99] implies that MCV cannot be a specific causal factor for a rare cancer like MCC—much like earlier criticisms of EBV cancer causality. Epidemiologic evidence, which worked well for KS and KSHV, fails to convincingly establish or refute a relationship between MCC and MCV.
MCV, Over the Hill?
Molecular biology came to the rescue of the MCV-MCC hypothesis in ways scarcely imaginable to A.B. Hill when he articulated his criteria in 1965. Clonality of MCV in tumors provides unambiguous proof that the virus is present in MCV-positive tumors prior to the beginning of tumor cell proliferation. This molecular confirmation for the correct temporal sequence relies on technologies and a body of scientific knowledge that were largely unavailable to Hill. Furthermore, since MCV is mutated and integrated in MCC, measurement of total MCV burden does not reflect the tumor-causing form of the virus.
Rolling circle amplification, which only detects the free episomal form of the virus, can be used to detect wild-type virus in nonMCC tissues[97, 100]. It is not exposure to whole, intact MCV that leads to MCC, instead, the association is between a well-defined mutated form of MCV and MCC. When this is taken into account, a very highly specific association between mutated MCV and MCC is reproducibly found[88] [101] and Hill’s criteria begin to be fulfilled for the mutant virus. MCV is also present at high copy number in MCV-positive MCC, a necessary precondition if MCV is directly driving cell proliferation in the tumor. Tumor surveys reveal high level MCV T antigen expression present only in MCC tumor specimens[90] (Figure 4C). Non-quantitative PCR frequently detects low level viral genome in non-cancerous tissues, consistent with MCV being a component of our healthy viral flora[102], and cannot be used to reliably distinguish cancer-associated from cancer-unassociated virus. Thus, specificity cannot be addressed by a dichotomous present/absent test for the virus but also should take into consideration high copy number vs. low copy number infections. Further, subtle pathologic features and miRNA ontology studies distinguish MCV-positive from MCV-negative MCC[103–105] arguing that there really are two or more biological forms of MCC, only one of which is MCV-infected, adding to the strength of association. As with spinal meningitis, clinically similar forms of MCC may have different etiologies, one of which appears to be caused by infection with a mutated version of a common floral virus.
Finally, experimental studies confirm the molecular relationship between virus and cancer. If MCV-positive MCC tumor cells require the presence of foreign viral oncoproteins for cell survival and proliferation, causality would be assured. This was tested by siRNA knockdown studies of both large T (LT)[106, 107] and small T (sT)[108, 109] antigen oncoproteins in MCC tumor cell lines. Knockdown of both LT and sT cause MCV-positive tumor cells to undergo necroptotic cell death while knockdown of sT alone arrests MCV-positive tumor cell proliferation. MCV-negative cells are not affected by these knockdowns. MCV oncoprotein expression is therefore essential for MCV-positive tumor cells. Laboratory support for this comes from studies of MCV sT, which transforms rodents cells, acts to deregulate cap-dependent protein translation[109] and to target the Fbw7 tumor suppressor[110]. Fbw7 inhibition by MCV sT has the curious function of amplifying expression of both MCV LT as well as a variety of cellular oncoproteins.
This provides a new model for viral carcinogenesis. MCV is part of our viral skin flora but is usually clinically silent. Sequentially increased risk occurs under a precise set of circumstances in which an individual is infected (common), and then loses immune surveillance against MCV proteins (uncommon), and finally undergoes a precise set of mutations to the virus itself (rare) [92, 111]. Humans are genetically complex chimeras in which floral viruses, including MCV, might undergo mutations—as opposed to mutations in human somatic cells—and contribute to emergence of some cancers.
Future Tumor Virus Cancer Causation
Not all cancers are caused by viruses so what rules should we use to determine whether or not a virus causes cancer? Cancers are genetically heterogenous[112] and small fractions of common cancers might be triggered by viruses. Is it possible to rigorously establish a virus as the cause of 5% of a particular cancer? Rigid application of either Henle-Koch postulates or Hill’s criteria is unlikely to be useful, yet establishing even a small etiologic fraction contributed by viruses might have important public health consequences.
Molecular viral pathogenesis is rapidly changing with completion of human genome annotation and development of cheap, accurate high-throughput sequencing methods. It is becoming ever easier to distinguish “self” from “non-self” nucleic acid sequences and these techniques are revealing that we typically carry a vast viral as well as bacterial flora. MCV is just one of 9 new human polyomaviruses discovered since 2007[113]. Dozens of previously-undiscovered papillomavirus genotypes have been recently described. Also, evidence increases for nonviral host or environmental factors: whether cMYC rearrangements in EBV-related Burkitt’s lymphoma [114, 115] or aflatoxin B1 in HBV-related hepatocellular carcinoma[116] play causative roles in virus-caused tumors. Even the definition of an infectious disease has become strained by discovery of endogenous viruses passed through Mendelian inheritance[117], and whole cancer cells being “infectious”[118]. Any causation rules have to be broad enough to take into account these and other biologic processes described in the future.
Counterfactual reasoning [20, 119] is one attractive approach that does not require precise knowledge of the underlying pathobiology. If we eliminate a particular virus, using a vaccine or drug, and the cancer rate decreases, then we can say the virus causes the cancer even though we don’t know how this occurs. Unfortunately, this is an impractical ideal for most human tumor viruses. There are no antivirals or vaccines to eliminate most candidate viruses, and the development of new ones generally will not be attempted until a causal virus-cancer relationship has already been established. Though not a true counterfactual measure, knockdown of viral oncogenes in the laboratory[107–109, 120, 121] can provide useful surrogate support for a causal relationship.
Causality implies a law-like formalism[122] that does not operate well in cancer biology except at the most superficial levels. A probablistic approach might best fit our needs for making public health decisions on whether certain viruses do or do not cause specific human cancers. Probabilistic causation provides a practical yet ultimately rigorous method for determining causation without relying on criteria or postulates. Rather than testing for the dichotomous event that virus A causes cancer B, a probabilistic approach states the cancer B is more probable in the presence of virus A. While “correlation does not imply causation”, if the correlation is strong, reproducible and predictive, then it approaches having the same value as a causative conclusion. While we still do not know a precise mechanism that leads EBV to cause Burkitt’s lymphoma, there is little doubt that it does so in at least those cases with tumor cells that are uniformly infected with the virus. Bayesian inference, a mathematical method of estimating new probabilities based on new evidence[123], for a causal association might be appropriate for virus-cancer relationships that can be tested quantitatively.
Moreover, there may be particular cases in which biological data is so compelling that any explanation other than causality is highly unlikely (“singular or token” causality[122]). Among 219 squamous cell lung carcinomas examined in The Cancer Genome Anatomy Project, one showed integrated HPV-16 at the DAX-1 gene with high level expression of the HPV E6 and E7 oncogenes[124]. If confirmed, these data strongly imply that in at least one case, HPV can cause lung cancer. Of course, if found once, it is likely to be found again at some low frequency (“generic or type-level” causality).
Even under circumstances where no mechanism is known, a probabilistic approach to causation allows investigators to gather new evidence until the role of the agent in the cancer becomes mechanistically clear. As in the case of herpes simplex vs. HPV in cervical cancer, presence of the agent may ultimately be found to be a confounded correlation or a very complex case in which multiple distinct but related genotypes of a virus are mechanistically involved in carcinogenesis.
Despite huge efforts to sequence tumor cell genomes, precise molecular causes are known for only a portion of human tumors. This leaves open the possibility that there are additional pathways, perhaps including viruses, which contribute to human carcinogenesis. KSHV and MCV are useful models for exploring how we can determine whether or not new candidate viral carcinogens induce cancers and, equally important, develop new diagnostic therapeutic and preventive measures based on this knowledge.
Acknowledgments
The authors thank Dan Normolle and Robin Weiss for helpful discussions and Miles Davis for help with the manuscript. This manuscript was supported by American Cancer Society Research Professorships and by grants from NIH NCI (CA136806, CA136363 and CA170354).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Patrick S. Moore, Email: psm9@pitt.edu.
Yuan Chang, Email: yc70@pitt.edu.
References
- 1.de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–15. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 2.de Martel C, Franceschi S. Infections and cancer: established associations and new hypotheses. Crit Rev Oncol Hematol. 2009;70:183–94. doi: 10.1016/j.critrevonc.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 3.Frank T. Getting to Eureka. Harper’s Magazine. 2013:7–9. [Google Scholar]
- 4.Epstein MA, Achong BG, Barr YM. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet. 1964;15:702–3. doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- 5.IARC. Epstein-Barr virus and Kaposi’s sarcoma herpesvirus/human herpesvirus 8. Lyon: World Health Organization; 1997. [Google Scholar]
- 6.Chigwedere P, Seage GR, 3rd, Gruskin S, Lee TH, Essex M. Estimating the lost benefits of antiretroviral drug use in South Africa. J Acquir Immune Defic Syndr. 2008;49:410–5. doi: 10.1097/qai.0b013e31818a6cd5. [DOI] [PubMed] [Google Scholar]
- 7.Duesberg PH, Mandrioli D, McCormack A, Nicholson JM, Rasnick D, Fiala C, et al. AIDS since 1984: no evidence for a new, viral epidemic--not even in Africa. Ital J Anat Embryol. 2011;116:73–92. [PubMed] [Google Scholar]
- 8.Weiss RA, Jaffe HW. Duesberg, HIV and AIDS. Nature. 1990;345:659–60. doi: 10.1038/345659a0. [DOI] [PubMed] [Google Scholar]
- 9.Duesberg P. HIV is not the cause of AIDS. Science. 1988;241:514–7. doi: 10.1126/science.3399880. [DOI] [PubMed] [Google Scholar]
- 10.Ernberg I, Klein G. Effects on apoptosis, cell cycle and transformation, and comparative aspects of EBV with other DNA tumor viruses. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al., editors. Human Herpesviruses: Biology, Therapy and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007. pp. 514–39. [PubMed] [Google Scholar]
- 11.Rawls WE, Tompkins WA, Figueroa ME, Melnick JL. Herpesvirus type 2: association with carcinoma of the cervix. Science. 1968;161:1255–6. doi: 10.1126/science.161.3847.1255. [DOI] [PubMed] [Google Scholar]
- 12.McDougall JK, Crum CP, Fenoglio CM, Goldstein LC, Galloway DA. Herpesvirus-specific RNA and protein in carcinoma of the uterine cervix. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:3853–7. doi: 10.1073/pnas.79.12.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durst M, Gissmann L, Ikenberg H, zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A. 1983;80:3812–5. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boshart M, Gissmann L, Ikenberg H, Kleinheinz A, Scheurlen W, zur Hausen H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. Embo J. 1984;3:1151–7. doi: 10.1002/j.1460-2075.1984.tb01944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paprotka T, Delviks-Frankenberry KA, Cingoz O, Martinez A, Kung HJ, Tepper CG, et al. Recombinant origin of the retrovirus XMRV. Science. 2011;333:97–101. doi: 10.1126/science.1205292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urisman A, Molinaro RJ, Fischer N, Plummer SJ, Casey G, Klein EA, et al. Identification of a Novel Gammaretrovirus in Prostate Tumors of Patients Homozygous for R462Q RNASEL Variant. PLoS Pathog. 2006;2:e25. doi: 10.1371/journal.ppat.0020025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Rothman KJ, Greenland S. Causation and causal inference in epidemiology. Am J Public Health. 2005;95 (Suppl 1):S144–50. doi: 10.2105/AJPH.2004.059204. [DOI] [PubMed] [Google Scholar]
- 18.Greenland S, Robins JM, Pearl J. Confounding and collapsibility in causal inference. Statistical Science. 1999;14:29–48. [Google Scholar]
- 19.Hill AB. Environment and disease: association or causation? Proc Roy Soc Med. 1965;58:295–300. doi: 10.1177/003591576505800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofler M. The Bradford Hill considerations on causality: a counterfactual perspective. Emerg Themes Epidemiol. 2005;2:11. doi: 10.1186/1742-7622-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falkow S. Molecular Koch’s postulates applied to bacterial pathogenicity--a personal recollection 15 years later. Nat Rev Microbiol. 2004;2:67–72. doi: 10.1038/nrmicro799. [DOI] [PubMed] [Google Scholar]
- 22.Evans AS. Causation and disease: the Henle-Koch postulates revisited. The Yale journal of biology and medicine. 1976;49:175–95. [PMC free article] [PubMed] [Google Scholar]
- 23.Bunge M. Causality: The Place of the Causal Principle in Modern Science. Cleveland and New York: Meridian Books; 1959. [Google Scholar]
- 24.Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45:529–38. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Menzies P. Counterfactual theories of causation. 2009. Stanford Encyclopedia of Philosophy; Fall. 2009. [Google Scholar]
- 26.Henle J, Rosen G. Jacob Henle: On miasmata and contagia. Baltimore: The Johns Hopkins press; 1938. [Google Scholar]
- 27.Evans AS. Causation and disease: A chronological journey. Am J Epidemiol. 1978;108:249–58. doi: 10.1093/oxfordjournals.aje.a112617. [DOI] [PubMed] [Google Scholar]
- 28.Koch R. An Address on Cholera and its Bacillus. Br Med J. 1884;2:453–9. doi: 10.1136/bmj.2.1236.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wildner M, Hofman A. Re: “Epidemiologic interactions, complexity, and the lonesome death of Max von Pettenkofer”. Am J Epidemiol. 2008;168:119–20. doi: 10.1093/aje/kwn128. author reply 20–1. [DOI] [PubMed] [Google Scholar]
- 30.Morabia A. Epidemiologic interactions, complexity, and the lonesome death of Max von Pettenkofer. Am J Epidemiol. 2007;166:1233–8. doi: 10.1093/aje/kwm279. [DOI] [PubMed] [Google Scholar]
- 31.Huebner RJ. Criteria for etiologic association of prevalent viruses with prevalent diseases; the virologist’s dilemma. Ann N Y Acad Sci. 1957;67:430–8. doi: 10.1111/j.1749-6632.1957.tb46066.x. [DOI] [PubMed] [Google Scholar]
- 32.Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10:878–89. doi: 10.1038/nrc2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darby SC, Ewart DW, Giangrande PL, Dolin PJ, Spooner RJ, Rizza CR. Mortality before and after HIV infection in the complete UK population of haemophiliacs. UK Haemophilia Centre Directors’ Organisation. Nature. 1995;377:79–82. doi: 10.1038/377079a0. [DOI] [PubMed] [Google Scholar]
- 34.Rivers TM. Viruses and Koch’s Postulates. J Bacteriol. 1937;33:1–12. doi: 10.1128/jb.33.1.1-12.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fredericks DN, Relman DA. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clin Microbiol Rev. 1996;9:18–33. doi: 10.1128/cmr.9.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giraldo G, Kourilsky FM, Henle W, Mike V, Huraux JM, Andersen HK, et al. Antibody patterns to herpesviruses in Kaposi’s sarcoma: serological association of European Kaposi’s sarcoma with cytomegalovirus. Int J Cancer. 1975;15:839–48. doi: 10.1002/ijc.2910150516. [DOI] [PubMed] [Google Scholar]
- 37.Walter PR, Philippe E, Nguemby-Mbina C, Chamlian A. Kaposi’s sarcoma: Presence of herpes-type particles in a tumor specimen. Human Pathol. 1984;15:1145–46. doi: 10.1016/s0046-8177(84)80309-3. [DOI] [PubMed] [Google Scholar]
- 38.Kaposi M. Idiopathic multiple pigmented sarcoma of the skin. Arch Dermatol Syphil. 1872;4:265–73. English translation in CA: A Cancer Journal for Clinicians, New York 1982, 32, 342–347. [Google Scholar]
- 39.Cook-Mozaffari P, Newton R, Beral V, Burkitt DP. The geographical distribution of Kaposi’s sarcoma and of lymphomas in Africa before the AIDS epidemic. Br J Cancer. 1998;78:1521–8. doi: 10.1038/bjc.1998.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Penn I. Kaposi’s sarcoma in organ transplant recipients: Report of 20 cases. Transplantation. 1979;27:8–11. doi: 10.1097/00007890-197901000-00003. [DOI] [PubMed] [Google Scholar]
- 41.Safai B, Miké V, Giraldo G, Beth E, Good RA. Association of Kaposi’s sarcoma with second primary malignancies. Cancer. 1980;45:1472–9. doi: 10.1002/1097-0142(19800315)45:6<1472::aid-cncr2820450629>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 42.Ulbright TM, Santa Cruz DJ. Kaposi’s sarcoma: relationship with hematologic, lymphoid, and thymic neoplasia. Cancer. 1981;47:963–73. doi: 10.1002/1097-0142(19810301)47:5<963::aid-cncr2820470524>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 43.Ambinder RF, Newman C, Hayward GS, Biggar R, Melbye M, Kestens L, et al. Lack of association of cytomegalovirus with endemic African Kaposi’s sarcoma. J Infect Dis. 1985;156:133. doi: 10.1093/infdis/156.1.193. [DOI] [PubMed] [Google Scholar]
- 44.Huang YQ, Li JJ, Rush MG, Poiesz BJ, Nicolaides A, Jacobson M, et al. HPV-16-related DNA sequences in Kaposi’s sarcoma. Lancet. 1992;339:515–8. doi: 10.1016/0140-6736(92)90338-4. [DOI] [PubMed] [Google Scholar]
- 45.Wang RY-H, Shih JW-K, Weiss SH, Grandinetti T, Pierce PF, Lange M, et al. Mycoplasma penetrans infection in homosexual men with AIDS: High seroprevalence and association with Kaposi’s sarcoma. Clin Infect Dis. 1993;17:724–9. doi: 10.1093/clinids/17.4.724. [DOI] [PubMed] [Google Scholar]
- 46.Palca J. Kaposi’s sarcoma gives on key fronts. Science. 1992;255:1352–4. doi: 10.1126/science.1311867. [DOI] [PubMed] [Google Scholar]
- 47.Ensoli B, Barillari G, Gallo RC. Cytokines and growth factors in the pathogenesis of AIDS-associated Kaposi’s sarcoma. Immunological Reviews. 1992;127:147–55. doi: 10.1111/j.1600-065x.1992.tb01412.x. [DOI] [PubMed] [Google Scholar]
- 48.Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123–8. doi: 10.1016/0140-6736(90)90001-l. [DOI] [PubMed] [Google Scholar]
- 49.Lisitsyn N, Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–51. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- 50.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;265:1865–69. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 51.Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi’s sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood. 1995;86:2708–14. [PubMed] [Google Scholar]
- 52.Moore PS, Gao SJ, Dominguez G, Cesarman E, Lungu O, Knowles DM, et al. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcomae. J Virol. 1996;70:549–58. doi: 10.1128/jvi.70.1.549-558.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, et al. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996;2:342–6. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 54.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8) Proc Natl Acad Sci U S A. 1996;93:14862–7. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarid R, Olsen SJ, Moore PS. Kaposi’s sarcoma-associated herpesvirus: epidemiology, virology, and molecular biology. Adv Virus Res. 1999;52:139–232. doi: 10.1016/s0065-3527(08)60299-7. [DOI] [PubMed] [Google Scholar]
- 56.Boshoff C, Whitby D, Hatziionnou T, Fisher C, van der Walt J, Hatzakis A, et al. Kaposi’s sarcoma-associated herpesvirus in HIV-negative Kaposi’s sarcoma. Lancet. 1995;345:1043–44. doi: 10.1016/s0140-6736(95)90780-7. [DOI] [PubMed] [Google Scholar]
- 57.Su I-J, Hsu Y-S, Chang Y-C, Wang I-W. Herpesvirus-like DNA sequence in Kaposi’s sarcoma from AIDS and non-AIDS patients in Taiwan. Lancet. 1995;345:722–23. [PubMed] [Google Scholar]
- 58.Moore PS, Chang Y. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma lesions from persons with and without HIV infection. New Eng J Med. 1995;332:1181–5. doi: 10.1056/NEJM199505043321801. [DOI] [PubMed] [Google Scholar]
- 59.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–91. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 60.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86:1276–80. [PubMed] [Google Scholar]
- 61.Miller G, Rigsby MO, Heston L, Grogan E, Sun R, Metroka C, et al. Antibodies to butyrate-inducible antigens of Kaposi’s sarcoma-associated herpesvirus in patients with HIV-1 infection. New England Journal of Medicine. 1996;334:1292–7. doi: 10.1056/NEJM199605163342003. [DOI] [PubMed] [Google Scholar]
- 62.Gao SJ, Kingsley L, Li M, Zheng W, Parravicini C, Ziegler J, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat Med. 1996;2:925–8. doi: 10.1038/nm0896-925. [DOI] [PubMed] [Google Scholar]
- 63.Martin JN, Ganem DE, Osmond DH, Page-Shafer KA, Macrae D, Kedes DH. Sexual transmission and the natural history of human herpesvirus 8 infection. N Engl J Med. 1998;338:948–54. doi: 10.1056/NEJM199804023381403. [DOI] [PubMed] [Google Scholar]
- 64.Kedes DH, Operskalski E, Busch M, Kohn R, Flood J, Ganem D. The seroepidemiology of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nat Med. 1996;2:918–24. doi: 10.1038/nm0896-918. [DOI] [PubMed] [Google Scholar]
- 65.Whitby D, Howard MR, Tenant-Flowers M, Brink NS, Copas A, Boshoff C, et al. Detection of Kaposi’s sarcoma-associated herpesvirus (KSHV) in peripheral blood of HIV-infected individuals predicts progression to Kaposi’s sarcoma. Lancet. 1995;364:799–802. doi: 10.1016/s0140-6736(95)91619-9. [DOI] [PubMed] [Google Scholar]
- 66.Moore PS, Kingsley LA, Holmberg SD, Spira T, Gupta P, Hoover DR, et al. Kaposi’s sarcoma-associated herpesvirus infection prior to onset of Kaposi’s sarcoma. AIDS. 1996;10:175–80. doi: 10.1097/00002030-199602000-00007. [DOI] [PubMed] [Google Scholar]
- 67.Gao S-J, Kingsley L, Hoover DR, Spira TJ, Rinaldo CR, Saah A, et al. Seroconversion to antibodies against Kaposi’s sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi’s sarcoma. New Eng J Med. 1996;335:233–41. doi: 10.1056/NEJM199607253350403. [DOI] [PubMed] [Google Scholar]
- 68.Renwick N, Halaby T, Weverling GJ, Dukers NH, Simpson GR, Coutinho RA, et al. Seroconversion for human herpesvirus 8 during HIV infection is highly predictive of Kaposi’s sarcoma. AIDS. 1998;12:2481–8. doi: 10.1097/00002030-199818000-00018. [DOI] [PubMed] [Google Scholar]
- 69.Moore PS, Chang Y. Antiviral activity of tumor-suppressor pathways: clues from molecular piracy by KSHV. Trends Genet. 1998;14:144–50. doi: 10.1016/s0168-9525(98)01408-5. [DOI] [PubMed] [Google Scholar]
- 70.Moore PS, Chang Y. Kaposi’s sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu Rev Microbiol. 2003;57:609–39. doi: 10.1146/annurev.micro.57.030502.090824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N Engl J Med. 1999;340:1063–70. doi: 10.1056/NEJM199904083401402. [DOI] [PubMed] [Google Scholar]
- 72.Cohen J. Controversy: Is KS really caused by new herpesvirus? Science. 1994;268:1847–8. doi: 10.1126/science.7604255. [DOI] [PubMed] [Google Scholar]
- 73.Rady PL, Yen A, Rollefson JL, Orengo I, Bruce S, Hughes TK, et al. Herpesvirus-like DNA sequences in non-Kaposi’s sarcoma skin lesions of transplant patients. Lancet. 1995;345:1339–40. doi: 10.1016/s0140-6736(95)92538-4. [DOI] [PubMed] [Google Scholar]
- 74.Lin J-C, Lin S-C, Mar E-C, Pellett PE, Stamey FR, Stewart JA, et al. Is Kaposi’s sarcoma-associated herpesvirus detectable in semen of HIV-infected homosexual men? Lancet. 1995;346:1601–2. doi: 10.1016/s0140-6736(95)91931-7. [DOI] [PubMed] [Google Scholar]
- 75.Lennette ET, Blackbourn DJ, Levy JA. Antibodies to human herpesvirus type 8 in the general population and in Kaposi’s sarcoma patients. Lancet. 1996;348:858–61. doi: 10.1016/S0140-6736(96)03240-0. [DOI] [PubMed] [Google Scholar]
- 76.Boshoff C, Weiss RA. Epidemiology and pathogenesis of Kaposi’s sarcoma-associated herpesvirus. Philos Trans R Soc Lond B Biol Sci. 2001;356:517–34. doi: 10.1098/rstb.2000.0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hayward GS. KSHV strains: the origins and global spread of the virus. Semin Cancer Biol. 1999;9:187–99. doi: 10.1006/scbi.1998.0116. [DOI] [PubMed] [Google Scholar]
- 78.Fu B, Sun F, Li B, Yang L, Zeng Y, Sun X, et al. Seroprevalence of Kaposi’s sarcoma-associated herpesvirus and risk factors in Xinjiang, China. J Med Virol. 2009;81:1422–31. doi: 10.1002/jmv.21550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang X, He B, Zhang Z, Liu T, Wang H, Li X, et al. Human herpesvirus-8 in northwestern China: epidemiology and characterization among blood donors. Virol J. 2010;7:62. doi: 10.1186/1743-422X-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Engels EA, Pfeiffer RM, Goedert JJ, Virgo P, McNeel TS, Scoppa SM, et al. Trends in cancer risk among people with AIDS in the United States 1980–2002. AIDS. 2006;20:1645–54. doi: 10.1097/01.aids.0000238411.75324.59. [DOI] [PubMed] [Google Scholar]
- 81.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet. 2002;359:497–8. doi: 10.1016/S0140-6736(02)07668-7. [DOI] [PubMed] [Google Scholar]
- 83.Koljonen V, Kukko H, Tukiainen E, Bohling T, Sankila R, Pukkala E, et al. Incidence of Merkel cell carcinoma in renal transplant recipients. Nephrol Dial Transplant. 2009 doi: 10.1093/ndt/gfp334. [DOI] [PubMed] [Google Scholar]
- 84.Na R, Grulich AE, Meagher NS, McCaughan GW, Keogh AM, Vajdic CM. Comparison of de novo cancer incidence in Australian liver, heart and lung transplant recipients. Am J Transplant. 2013;13:174–83. doi: 10.1111/j.1600-6143.2012.04302.x. [DOI] [PubMed] [Google Scholar]
- 85.Lemos B, Nghiem P. Merkel cell carcinoma: more deaths but still no pathway to blame. J Invest Dermatol. 2007;127:2100–3. doi: 10.1038/sj.jid.5700925. [DOI] [PubMed] [Google Scholar]
- 86.Feng H, Taylor JL, Benos PV, Newton R, Waddell K, Lucas SB, et al. Human transcriptome subtraction by using short sequence tags to search for tumor viruses in conjunctival carcinoma. J Virol. 2007;81:11332–40. doi: 10.1128/JVI.00875-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.MacConaill L, Meyerson M. Adding pathogens by genomic subtraction. Nat Genet. 2008;40:380–2. doi: 10.1038/ng0408-380. [DOI] [PubMed] [Google Scholar]
- 88.Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105:16272–7. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rodig SJ, Cheng J, Wardzala J, DoRosario A, Scanlon JJ, Laga AC, et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J Clin Invest. 2012;122:4645–53. doi: 10.1172/JCI64116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shuda M, Arora R, Kwun HJ, Feng H, Sarid R, Fernandez-Figueras MT, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer. 2009;125:1243–9. doi: 10.1002/ijc.24510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst. 2009;101:938–45. doi: 10.1093/jnci/djp139. [DOI] [PubMed] [Google Scholar]
- 92.Chang Y, Moore PS. Merkel cell carcinoma: a virus-induced human cancer. Annual review of pathology. 2012;7:123–44. doi: 10.1146/annurev-pathol-011110-130227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Faust H, Andersson K, Ekstrom J, Hortlund M, Robsahm TE, Dillner J. Prospective study of merkel cell polyomavirus and risk of merkel cell carcinoma. Int J Cancer. 2013 doi: 10.1002/ijc.28419. [DOI] [PubMed] [Google Scholar]
- 94.Tolstov YL, Pastrana DV, Feng H, Becker JC, Jenkins FJ, Moschos S, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer. 2009;125:1250–6. doi: 10.1002/ijc.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pastrana DV, Tolstov YL, Becker JC, Moore PS, Chang Y, Buck CB. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009;5:e1000578. doi: 10.1371/journal.ppat.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Viscidi RP, Rollison DE, Sondak VK, Silver B, Messina JL, Giuliano AR, et al. Age-Specific Seroprevalence of Merkel Cell Polyomavirus, BK Virus, and JC Virus. Clin Vaccine Immunol. 2011;18:1737–43. doi: 10.1128/CVI.05175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Faust H, Pastrana DV, Buck CB, Dillner J, Ekstrom J. Antibodies to merkel cell polyomavirus correlate to presence of viral DNA in the skin. J Infect Dis. 2011;203:1096–100. doi: 10.1093/infdis/jiq173. [DOI] [PubMed] [Google Scholar]
- 98.Chen T, Hedman L, Mattila PS, Jartti T, Ruuskanen O, Soderlund-Venermo M, et al. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J Clin Virol. 2011;50:125–9. doi: 10.1016/j.jcv.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 99.Touze A, Gaitan J, Arnold F, Cazal R, Fleury MJ, Combelas N, et al. Generation of Merkel cell polyomavirus (MCV)-like particles and their application to detection of MCV antibodies. J Clin Microbiol. 2010;48:1767–70. doi: 10.1128/JCM.01691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schowalter RM, Pastrana DV, Pumphrey KA, Moyer AL, Buck CB. Merkel Cell Polyomavirus and Two Previously Unknown Polyomaviruses Are Chronically Shed from Human Skin. Cell Host Microbe. 2010;7:509–15. doi: 10.1016/j.chom.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Laude HC, Jonchere B, Maubec E, Carlotti A, Marinho E, Couturaud B, et al. Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010:6. doi: 10.1371/journal.ppat.1001076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tolstov YL, Knauer A, Chen JG, Kensler TW, Kingsley LA, Moore PS, et al. Asymptomatic primary Merkel cell polyomavirus infection among adults. Emerging infectious diseases. 2011;17:1371–80. doi: 10.3201/eid1708.110079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Higaki-Mori H, Kuwamoto S, Iwasaki T, Kato M, Murakami I, Nagata K, et al. Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum Pathol. 2012 doi: 10.1016/j.humpath.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 104.Renwick N, Cekan P, Masry PA, McGeary SE, Miller JB, Hafner M, et al. Multicolor microRNA FISH effectively differentiates tumor types. J Clin Invest. 2013 doi: 10.1172/JCI68760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xie H, Lee L, Caramuta S, Hoog A, Browaldh N, Bjornhagen V, et al. microRNA Expression Patterns Related to Merkel Cell Polyomavirus Infection in Human Merkel Cell Carcinoma. J Invest Dermatol. 2013 doi: 10.1038/jid.2013.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Houben R, Adam C, Baeurle A, Hesbacher S, Grimm J, Angermeyer S, et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int J Cancer. 2012;130:847–56. doi: 10.1002/ijc.26076. [DOI] [PubMed] [Google Scholar]
- 107.Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84:7064–72. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shuda M, Chang Y, Moore PS. Merkel Cell Polyomavirus Positive Merkel Cell Carcinoma Requires Viral Small T Antigen For Cell Proliferation. Journal of Investigative Dermatology. 2013 doi: 10.1038/jid.2013.483. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest. 2011;121:3623–34. doi: 10.1172/JCI46323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kwun HJ, Shuda M, Feng H, Camacho CJ, Moore PS, Chang Y. Merkel Cell Polyomavirus Small T Antigen Controls Viral Replication and Oncoprotein Expression by Targeting the Cellular Ubiquitin Ligase SCF(Fbw7) Cell Host & Microbe. 2013;14:125–35. doi: 10.1016/j.chom.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arora R, Chang Y, Moore PS. MCV and Merkel cell carcinoma: a molecular success story. Curr Opin Virol. 2012;2:489–98. doi: 10.1016/j.coviro.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.DeCaprio JA, Garcea RL. A cornucopia of human polyomaviruses. Nat Rev Microbiol. 2013;11:264–76. doi: 10.1038/nrmicro2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kelly GL, Rickinson AB. Burkitt lymphoma: revisiting the pathogenesis of a virus-associated malignancy. Hematology Am Soc Hematol Educ Program. 2007:277–84. doi: 10.1182/asheducation-2007.1.277. [DOI] [PubMed] [Google Scholar]
- 115.Shah KM, Young LS. Epstein-Barr virus and carcinogenesis: beyond Burkitt’s lymphoma. Clin Microbiol Infect. 2009;15:982–8. doi: 10.1111/j.1469-0691.2009.03033.x. [DOI] [PubMed] [Google Scholar]
- 116.Liu Y, Chang CC, Marsh GM, Wu F. Population attributable risk of aflatoxin-related liver cancer: systematic review and meta-analysis. Eur J Cancer. 2012;48:2125–36. doi: 10.1016/j.ejca.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Weiss RA. The discovery of endogenous retroviruses. Retrovirology. 2006;3:67. doi: 10.1186/1742-4690-3-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murgia C, Pritchard JK, Kim SY, Fassati A, Weiss RA. Clonal origin and evolution of a transmissible cancer. Cell. 2006;126:477–87. doi: 10.1016/j.cell.2006.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Phillips CV, Goodman KJ. Causal criteria and counterfactuals; nothing more (or less) than scientific common sense. Emerg Themes Epidemiol. 2006;3:5. doi: 10.1186/1742-7622-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–8. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood. 2005;105:2510–8. doi: 10.1182/blood-2004-08-3052. [DOI] [PubMed] [Google Scholar]
- 122.Pearl J. Causality: Models, Reasoning and Inference. 2. New York, New York: Cambridge University Press; 2010. [Google Scholar]
- 123.Efron B. Mathematics. Bayes’ theorem in the 21st century. Science. 2013;340:1177–8. doi: 10.1126/science.1236536. [DOI] [PubMed] [Google Scholar]
- 124.Khoury JD, Tannir NM, Williams MD, Chen Y, Yao H, Zhang J, et al. Landscape of DNA virus associations across human malignant cancers: analysis of 3,775 cases using RNA-Seq. J Virol. 2013;87:8916–26. doi: 10.1128/JVI.00340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]