

Abstract
Spatially modulated illumination fluorescence microscopy can in theory measure the sizes of objects with a diameter ranging between 10 and 200 nm and has allowed accurate size measurement of subresolution fluorescent beads (∼40–100 nm). Biological structures in this size range have so far been measured by electron microscopy. Here, we have labeled sites containing the active, hyperphosphorylated form of RNA polymerase II in the nucleus of HeLa cells by using the antibody H5. The spatially modulated illumination-microscope was compared with confocal laser scanning and electron microscopes and found to be suitable for measuring the size of cellular nanostructures in a biological setting. The hyperphosphorylated form of polymerase II was found in structures with a diameter of ∼70 nm, well below the 200-nm resolution limit of standard fluorescence microscopes.
INTRODUCTION
Gene transcription in eukaryotic nuclei is mediated by three distinct RNA polymerases (pols; types I, II, and III). Each type transcribes specific sets of genes with pol II transcribing the largest and most varied group. The largest subunit of pol II contains an atypical C-terminal domain, which is composed of tandem repeats (52 in humans) of seven amino acids. Serine residues in positions 2 and 5 of the repeat become phosphorylated upon transcription initiation and elongation. Phosphorylation of Ser2 (pol IIo) correlates with transcription elongation and can be detected with monoclonal antibody (mAb) H5 (Bregman et al., 1995; Komarnitsky et al., 2000).
Sites of transcription by pol II have been detected after labeling nascent RNA with tagged nucleotides (Jackson et al., 1993; Wansink et al., 1993, 1996; Iborra et al., 1996; Pombo et al., 1999b), and more recently using mAb H5 (Grande et al., 1997; Zeng et al., 1997; Pombo et al., 1999b). In a HeLa (subtetraploid) cell, there are ∼8000 discrete sites of pol II transcription distributed throughout the nucleoplasm, with diameters of ∼40–80 nm, as determined by electron microscopy (EM; Iborra et al., 1996; Jackson et al., 1998; Pombo et al., 1999b, 2000). Individual sites can only be seen in thin sections due to the complexity of staining patterns. Sucrose-embedded, Tokuyasu cryosections (100–150 nm in thickness) have proved particularly useful because they can be used for both light microscopy and EM measurements (Tokuyasu, 1973; Pombo et al., 1999a).
Fluorescence microscopy has been a significant force in modern biological studies. With the advent of green fluorescent protein (GFP) and the increased potential for the study of live cells, much work is currently underway to improve the resolution of fluorescence-based microscopy systems. The resolution limits of conventional light microscopes can be exceeded using various methods, including gathering light from more than one angle, using nonuniform excitation light, and using nonlinear absorption and emission of more than one photon (Gustafsson, 1999). Already, a fluorescence-based microscopy method has been used to measure the size of GFP-labeled mitochondria in live yeast cells with ∼100-nm resolution (Egner et al., 2002). Spatially modulated illumination (SMI)-microscopy, which uses nonuniform excitation to improve resolution, can, in theory, measure the sizes of objects with diameters ranging between 10 and 200 nm, depending on the excitation wavelength and signal-to-noise ratio (Failla et al., 2002a). It has allowed the accurate size measurement of fluorescent beads ranging from ∼40 to 100 nm (Failla et al., 2002b), but its applicability for biological samples has not been tested.
The SMI-microscope can be represented as an interferometer setup where two laser beams (colimated and vertically polarized) are focused into the back focal plane of two opposing objective lenses (for a detailed description of the SMI-microscope, see Schneider et al., 1999; Albrecht et al., 2002; also see Supporting Information, Figure 4). This configuration produces a periodic excitation along the axial direction, which gives rise to a similarly periodic emission profile from a fluorescent object placed between the two objectives (Figure 1A). The object causes variation of the emission profile (Figure 1A, iii, shaded area) in a size-dependent manner (Bailey et al., 1993). The ratio between the maximum of this variation and the maximum emission intensity gives the “modulation contrast” (R), which, for a range of sizes, is directly proportional to the object size, depending on the excitation wavelength (e.g., 30–120 nm for 488-nm excitation; Figure 1B). Previous SMI-microscopy size measurements have been obtained using computer-simulated objects or fluorescent beads of known size (Failla et al., 2002a,b). Here, we show that SMI-microscopy is suitable for measuring the size of sites containing the active form of pol II (pol IIO) in thin cryosections from human (HeLa) cells.
Figure 1.

Measuring the size of fluorescent objects with SMI-microscopy. (A) The SMI-microscope uses two collimated lasers to produce a periodic excitation profile. (A) z-stack images of a 50-nm bead, collected at 20-nm intervals, show variation of emitted light, with the focal plane of the bead corresponding to image 92. i, images 88–96. ii, images corresponding to the brightest peaks and troughs throughout the stack, from image 63 to 121. (Note that the lateral resolution of the SMI-microscope is that of a wide-field microscope.) iii, emission profile representing the intensity values at coordinates located at the center of the bead for each image in the z-stack. The shape of the profile varies with the size of the object and the modulation contrast (R) is proportional to the size of the object in a wavelength-dependent manner. au, arbitrary units. (B) Calibration curve, computer simulated by virtual microscopy, for beads of different sizes, shows the relationship between the modulation contrast (R) and object size for 488-nm excitation wavelength. Experimental values for beads of sizes 40-, 50-, and 100-nm fit this curve. The diameter of structures can be obtained by interpolation of experimental values of R into this calibration curve.
MATERIALS AND METHODS
Cell Culture, Fixation, and Cryosectioning
HeLa cells were cultured, fixed in 4 and then 8% paraformaldehyde in 250 mM HEPES, and frozen for cryosectioning as described previously (Pombo et al., 1999a,b). Cryosections (∼140 nm in thickness; deduced from interference color) were cut from the frozen blocks by using an UltraCut UCT 52 ultracryomicrotome (Leica, Milton Keynes, United Kingdom), captured in drops of 2.1 M sucrose in phosphate-buffered saline (PBS), and transferred to coverslips (for confocal laser scanning microscopy [CLSM] and SMI-microscopy) or metal grids (copper/palladium or nickel, coated with Formvar and glow-discharged; for EM).
Immunolabeling
Cryosections were immunolabeled as described previously (Pombo et al., 1999a,b), except Triton X-100 treatment was extended to 10 min. Pol IIO sites were indirectly immunolabeled (2 h) by using mouse mAb H5 against the hyperphosphorylated C-terminal domain of the largest subunit of pol II (phosphorylated at Ser2 of the heptad repeat; diluted 1/2000 in PBS+; Babco/Covance, Richmond, CA). For CLSM and SMI-microscopy, sections were incubated (1 h) with Alexa Fluor 488-conjugated antibodies raised against mouse IgG (1/1500; Molecular Probes, Eugene, OR) or, for a three-layer detection protocol, incubated (1 h) with antibodies raised in rabbit against mouse Ig (1/200; Valeant Pharmaceuticals, Costa Mesa, CA) and (1 h) with Alexa Fluor 488-conjugated antibodies raised in goat against rabbit IgG (1/4000; Molecular Probes). For EM, sections were incubated in H5 (as described above) and then (1 h) with antibodies raised in rabbit against mouse Ig (as described above) and (4 h) with 5-nm gold-conjugated antibodies raised in goat against rabbit IgG (1/50; British Biocell, Cardiff, United Kingdom). Sections were washed (1 h; 5×) in PBS+ between and after the final antibody incubations, except for washes after the 5-nm gold conjugates, which were >2 h. For CLSM and SMI-microscopy, sections were washed in 0.1% Tween 20 in PBS before nucleic acids were counterstained (45 min) in 1 μM TOTO-3 iodide (Molecular Probes) in PBS-Tween, washed successively in PBS-Tween and PBS (5×), and mounted in VectaShield (Vector Laboratories, Peterborough, United Kingdom). For EM, grids were washed (>2 h; 5×) in PBS+ after final antibody incubation, rinsed (5×) in PBS, fixed (10 min) in 0.5% glutaraldehyde in PBS, washed (10 min; 8×) in distilled water, and incubated (10 min; 4°C) in 0.3% uranyl acetate in 2% methylcellulose. Excess liquid was blotted and grids left to dry.
Control samples were prepared with omission of primary antibody or by pretreating samples with a saturating concentration of alkaline phosphatase (calf intestinal; 0.5 U/μl; 37°C; 16 h; New England BioLabs, Beverly, MA) to remove the phospho-epitopes recognized by H5.
Microscopy and Size Measurements
Fluorescent beads with diameters of 50 ± 8 nm were obtained from Duke Scientific (Palo Alto, CA).
For CLSM, images for H5 and TOTO-3 were collected sequentially on a Leica SP2 CLSM (100× objective, 1.3 numerical aperture, Milton Keynes, United Kingdom; immersion oil, n = 1.518, Leica, Wetzler, Germany), by using argon (488-nm) and HeNe (633-nm) lasers and pinhole equivalent to 1 Airy disk. Images (TIFF files) were transferred to Adobe Photoshop (Adobe Systems, Edinburgh, United Kingdom) and contrast stretched without further processing. For counting sites, a representative area of the nucleoplasm was selected in the TOTO-3 images, and all sites within the area were counted and the number per unit area was calculated. Enough cells were counted so that the percentage of the progressive mean was within 5% of the final mean value for at least the last 50% of measurements (Williams, 1977).
For EM, images were collected with a Tecnai 12 BioTwin transmission electron microscope (FEI Company, Hillsboro, OR) equipped with a Mega-View III charge-coupled device (CCD) camera (1376 × 1032 pixels, 12-bit images, pixel size 6.45 μm) running under analySIS software version 3.2 (Soft Imaging Systems, Munster, Germany). A cluster of gold particles was defined as one containing two or more particles lying within 40 nm of another (Iborra et al., 1996; Pombo et al., 1999b). The diameter (D) of a cluster was determined after measurement of the major (2a) and minor (2b) orthoganol axes where
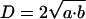 |
and the underlying objects were treated as spheres. Average diameters and the correction for the contribution of polar caps by using the sequential-subtraction method was carried out as described previously (Pombo et al., 1999a; also see Supporting Information, Part 3, Figure 5).
For SMI-microscopy, cryosections were visualized on an SMI-microscope (100× objective, 1.4 numerical aperture, Leica; immersion oil, n = 1.518, Carl Zeiss, Jena, Germany), with argon (488-nm) and krypton (647-nm) lasers, and a Quanticam CCD camera equipped with a four-color mosaic filter (pixel size 6.45 × 6.45 μm, final pixel size in the object space 107.2 × 107.2 nm; Phase, Lubeck, Germany); setup according to Albrecht et al. (2002) with some improvements to the detection light path (see Supporting Information, Part 1, Figure 4A). Coverslips were scanned using the CCD camera and a 647-nm laser to locate the cryosections. z-stacks of 160–180 images, step size 20 nm, were collected using the 488-nm laser and single binning in the mosaic filter and saved as KDF files. These files were opened in Image J (version 1.29; http://rsb.info.nih.gov/ij/index.html) by using a KDF reader plug-in (see Supporting Information, Part 2). For counting sites, the 160–180 images from each stack were merged in Image J, saved as a TIFF file, and opened in Adobe Photoshop before counting as for CLSM (see above). For size measurements of pol IIO sites (Figure 3A), areas containing ∼50 sites were chosen randomly, and all detected sites within these areas were analyzed. Emission profiles were obtained for all sites by using a Khoros Visual Programming Environment based on the Cantata Visual Programming Language (Khoros Software, version 2.2.0; Khoral, Albuquerque, NM) or Image J image files. Details of data collection and interpretation with SMI-microscopy software have been described previously (Failla et al., 2002a,b). For Image J, profiles were obtained by visual selection of a 5 × 5 pixel area centered on the maximum intensity of the site as for SMI-microscopy software (see Supporting Information, Part 2).
Figure 3.

Size of pol IIO sites obtained by SMI-microscopy and EM. (A) Pol IIO sites in cryosections (∼140 nm) were measured by SMI-microscopy (sites labeled as for Figure 2, A–C; 148 sites) and EM (sites labeled as for Figure 2D; 105 sites). The SMI-microscopy setup was that described in Albrecht et al. (2002). Sizes were grouped together into 10-nm ranges and frequencies expressed as a percentage of the total. Size measurements with EM (open bars) gave an average (weighted) diameter of 45 nm after correction for the contribution of polar caps (see text and Supporting Information, Part 3). Size measurements with SMI-microscopy (solid bars) gave an average (weighted) diameter of 74 nm (uncorrected as SMI-microscopy did not detect smaller/incomplete sites, see text and Supporting Information, Part 3). (B) Pol IIO sites were labeled for SMI-microscopy as described above (two-layer protocol) and with a three-layer protocol by using H5, rabbit anti-Ig antibodies and Alexa Fluor 488. The SMI-microscopy setup was improved by optimization of laser-camera alignment (Supporting Information, Part 1). Sizes were grouped as for A. Size measurements with the two-layer and three-layer protocols gave average (weighted) diameters of 82 nm (open bars) and 81 nm (solid bars), respectively.
For Figure 2, A and B, individual images or a z-projection image of the whole stack were saved as JPEG files and opened in Adobe Photoshop and contrast stretched without further processing.
Figure 2.

Measuring the size of pol IIO discrete sites. Pol IIO occur as sites throughout the nucleoplasm in sites SMI-microscopy (A and B), CLSM (C), and EM (D) images. HeLa cells were fixed, embedded in sucrose, and cryosectioned (∼140 nm in thickness). Pol IIO was indirectly immunolabeled using mAb H5 and Alexa Fluor 488 (A–C) or 5-nm gold particles (D); sections were counter-stained with TOTO-3 (insets in A and C) or uranyl acetate (D), and images were collected with an SMI-microscope (A and B), CLSM (C), or EM (D). (A) An x,y image from the center of a z-stack of 160 images collected with the SMI-microscope. Due to the modulation of the incident light (see text), some sites cannot be seen in this image. (B) In a projected image of the 160 z-stack images, all sites detected by SMI-microscopy can be seen. The inset shows an enlarged view of several sites. (C) CLSM detects a greater number of sites, which are absent from nucleoli (arrowhead; see text). (D) On the EM, sites occur as clusters (arrowheads) of gold particles, and lone particles (arrow) represent nonspecific background; the inset shows an enlarged view of one cluster. EM detects the highest number of sites due to increased resolution. (E) Emission profile produced by the site circled in B (inset) giving a diameter of 51 nm.
Sizes were determined analytically using the following standard algorithm: 1) determine site coordinate, 2) extract stacks with dimensions 9 × 9 × (160–180) centered on the coordinate, 3) extract emission profile, 4) process emission profile, 5) determine fit function iteratively, and 6) determine size from fit function parameters. Eight methods were used to determine the sizes of pol IIO sites from SMI-microscopy stacks of images (see Supporting Information, Part 2, Table 1).
Sizes were determined graphically from Khoros Cantata or Image J generated profiles by measurement of M1 and M2 (Figure 1A) and calculation of the modulation contrast (R), which was interpolated into the calibration curve for excitation wavelength 488 nm (Figure 1B; Failla et al., 2002a) by using MatLab (version 6.5; Mathworks, Natick, MA) to extract the size values. To take into account sectioning effects that can underestimate the sizes of structures, we used the sequential-subtraction method (Weibel, 1979; Pombo et al., 1999a) modified for axial measurements as explained in Supporting Information (Part 3, Figure 6).
RESULTS
Imaging pol IIO Sites with SMI-Microscopy, CLSM, and EM
We first tested whether sites containing active pol II could be detected by SMI-microscopy. Thin (∼140-nm) cryosections of HeLa cells were immunolabeled with mAb H5 that detects Ser2-phosphorylated pol IIO (Bregman et al., 1995), and images were collected by SMI-microscopy and CLSM (Figure 2, A–C). A two-layer antibody detection system was used with H5 (an IgM) and Alexa Fluor 488-conjugated anti-Ig antibodies, because this allows detection of all sites with CLSM (Pombo et al., 1999a). (Note that on the CLSM at least 20% of sites seen by EM are not resolved from others that lie within 200 nm; Pombo et al., 1999a.) With CLSM, a single optical section (z thickness >500 nm) at the focal plane contains all sites in an ∼140-nm-thick cryosection (Figure 2C). Pol IIO is found in discrete nucleoplasmic sites, particularly in euchromatin (Figure 2C, inset shows nucleic acid staining by using TOTO-3), and absent from nucleoli (arrowheads; also see Grande et al., 1997). Labeling is specific, because pretreatment with alkaline phosphatase to remove phospho-epitopes, or omitting H5, abolishes >97% of the signal (our unpublished data).
The SMI-microscope produces a stack of 160–180 images, collected at 20-nm intervals through the z-axis. Figure 2A shows a single image from a z-stack, and Figure 2B shows a projection of the whole stack. Due to the periodic excitation and consequent periodic emission of the microscope (Figure 1A), all sites can only be seen in a z-projection of the image stack. By using the SMI-microscopy setup as described in Albrecht et al. (2002), one-half the number of sites per unit area was detected compared with CLSM, with average 1.1 ± 0.2 and 2.7 ± 0.4 sites/μm2, respectively (n = 10 and 12 cells, compare Figure 2, B and C). Although a lower density of sites is expected for SMI-microscopy due to the poorer lateral resolution in comparison with CLSM, we improved the laser-camera alignment procedure of the SMI-microscope (see Supporting Information, Part 1, Figure S4) to increase light collection efficiency and give improved detection, and we obtained an average of 2.4 ± 0.5 sites/μm2 (n = 10 cells; our unpublished data). (Note that the SMI-microscope has the properties of a wide-field microscope in the lateral dimensions and that the detector pixel sizes for SMI-microscopy and CLSM were 0.11 and 0.06 μm in the object plane, respectively.) We also analyzed H5-labeled sites with EM (Figure 2D), which provides better resolution than CLSM, and the same sensitivity when using a three-layer antibody detection protocol (Pombo et al., 1999a,b). H5, rabbit anti-Ig antibodies and 5-nm goat anti-rabbit Ig were used in sequence before sections were counterstained with uranyl acetate. H5 sites are marked by clusters of 5-nm gold particles found throughout the nucleoplasm; clusters (Figure 2D, arrowheads) are defined as groups of two or more particles that are separated by ≤40 nm (Iborra et al., 1996). Lone particles (Figure 2D, arrow; lying >40 nm apart) represent background labeling (<4%), because they are present in equal numbers in controls preincubated with alkaline phosphatase to remove the phospho-epitope recognized by H5.
Size Measurements with SMI-Microscopy
We next measured the sizes of pol IIO sites labeled with H5 and Alexa Fluor 488, as described above, with SMI-microscopy (Figure 2, A and B). Emission profiles were obtained from z-stacks of 160–180 images (20-nm intervals). Profiles were extracted from the KDF image files collected by the SMI-microscope by using a workspace assembled by the authors, based on Khoros Cantata software or by using Image J software (see MATERIALS AND METHODS). Under optimal imaging conditions (i.e., without bleaching of the signal), these profiles had the same general appearance as those previously obtained from fluorescent beads and from profiles of 50-nm objects generated using virtual microscopy (compare profiles in Figures 1A, iii, and 2E; see also Albrecht et al., 2002; Failla et al., 2002a,b). (Note that the profile in Figure 2E shows an asymmetry which is not apparent in profiles from beads [Figure 1A, iii], possibly due to the irregular shape of the underlying structure and/or the unevenness of antibody labeling.] Size information from the profiles was obtained graphically from the plots (as in Figure 1A, iii, and B) or analytically by calculation of a fit function (see MATERIALS AND METHODS; Failla et al., 2002b). Algorithms to extract size parameters, after analytical determination of a fit function to the experimental profiles, have been described using computer-simulated objects (Failla et al., 2002a) or fluorescent beads (Failla et al., 2002b). Biological samples, however, are often less intense and therefore more sensitive to background noise, inhomogeneous illumination, and other aberrations; profiles from beads seemed smoother and more symmetrical than those from pol IIO sites (our unpublished data). We therefore investigated different procedures for analysis of biological data, by using the previously described algorithms as a starting point (see Supporting Information, Part 2, Table 1). Method variables included 1) the x,y coordinates of sites being deduced visually or from the maximum intensity from a 5 × 5 pixel mask around the sites; 2) the use of all 160–180 z-planes or removal of seemingly uninformative ones from the edges of the profile; and 3) the use of raw or background-subtracted profiles. We found that average diameters obtained by the eight methods varied by <5 nm (n = 69 sites). (Note that the comparison of individual site measurements gave statistically significant differences, with mean differences for the eight methods between 5.4 and 9.7 nm [range 0–40 nm; p < 0.001, repeated measures analysis of variance]; however, we could not associate this variability with any specific property of objects [e.g., dimmer sites were not more prone to variability]) Thereafter, we favored the method that uses 1) visual determination of the center of coordinates, 2) all informative images, and 3) background subtraction (see details and discussion in Supporting Information, Part 2, Method 3, Table 1).
Analytical determination of sizes from emission profiles requires a sophisticated evaluation tool but, in principle, it is possible to extract size data graphically by measuring values M1 and M2 and interpolation of the value R in the calibration curve (see MATERIALS AND METHODS; Figure 1A, iii, and B). First, we compared measurements obtained using graphical and analytical (number 3) methods from Khoros Cantata generated profiles of 50-nm fluorescent beads. We found no significant difference between the two methods (average diameters 47.2 ± 5.1 and 47.9 ± 5.0 nm for graphical and analytical methods, respectively; n = 48 beads). These two methods also gave no significant difference in average diameters of pol IIO sites (65.8 ± 16.3 and 65.9 ± 15.7 nm; n = 67 sites from 5 cells) or between measurements of individual sites (p = 0.92, paired t-test).
Emission profiles also can be generated without the need for the SMI-microscope software by using the program Image J from the KDF image stacks. Profiles for the 67 pol IIO sites (as described above) were generated using Image J, and M1 and M2 values were measured graphically, after subtracting a background level chosen visually by joining the baselines at the edges of profiles. Again there was no significant difference between these and values determined using either of the above-mentioned graphical or analytical methods (average diameter 65.4 ± 11.3 nm) or between measurements of individual sites (p = 0.71 and 0.74 when comparing with the graphical and analytical methods, respectively; paired t-test). These comparisons show that any of these methods can be used to determine the average size of sites; however, the analytical method is probably the most accurate because it is able to take into account variations in excitation wavelength, which can occur due to local fluctuation of the refractive index and may affect individual profiles.
The Size of pol IIO Sites
We then measured the size of pol IIO sites by using analytical method 3 and image profiles extracted with Khoros Cantata, after labeling pol IIO with H5 and Alexa Fluor 488. A random area was chosen from each of five cells before emission profiles were obtained for all sites within the selected areas (n = 148 sites). By using the SMI-microscopy setup described in Albrecht et al. (2002), diameters ranged from 8 to 124 nm (Figure 3A, solid bars) and were skewed toward the larger sizes.
Due to sectioning effects across each site, the size histogram is expected to contain a proportion of variously sized incomplete sites (i.e., sites that were cut during sectioning) and need to be taken into account to avoid underestimation of sizes (Weibel, 1979). This analysis also gives an indication of the sensitivity of the imaging or labeling method; if the expected frequency of incomplete sites is absent (i.e., negative frequencies are obtained), it indicates that the contrast or imaging methods are not sensitive enough to detect smaller or dimmer structures (Weibel, 1980). We had previously developed an appropriate modification of the sequential-subtraction method to correct for the contribution of polar caps upon EM (lateral) measurements of sites contained in cryosections (transparent media, because gold probes access and are visualized throughout the thickness of section; Pombo et al., 1999a; see Supporting Information, Figure 5). Here, we present a modification for the axial measurements performed with SMI-microscopy (see Supporting Information, Part 3, Figure 6). Starting with the largest sites measured, the expected frequency of incomplete sites is estimated and subtracted from the smaller size categories; the next size categories are then treated in the same way. During this correction, it became apparent that a number of smaller and/or incomplete sites were missing from the profiles (see Supporting Information, Figure 7A), suggesting that these were not detected. This result was seen with both SMI-microscopy setups tested; although most sites were detected with the improved setup, 50% did not give good modulation profiles and could not be measured. This is consistent with the fact that, when using a 488-nm laser, it is not possible to accurately measure objects that have diameters of <20 nm (Figure 1B; also see Failla et al., 2002a). Because incomplete sites were not detected, we estimated the average diameter without correction and obtained an average (weighted) diameter of 74 nm (range 8–124 nm; Figure 3A, solid bars). (The average (weighted) diameter after correction for incomplete sites was 81 nm (range 60–124 nm; see Supporting Information, Figure 7A).
We further tested whether the performance of the SMI-microscope could be improved using a three-layer detection protocol with H5, rabbit anti-mouse Ig antibodies and Alexa Fluor 488-conjugated anti-rabbit Ig antibodies. We used the improved SMI-microscopy setup to compare the sizes obtained with two- and three-layer detection systems. We found an average of 2.4 ± 0.5 and 2.9 ± 0.4 sites/μm2 (n = 10 and 11 cells) with two-layer and three-layer protocols, respectively (in a similar CLSM experiment the values were 2.7 ± 0.6 and 2.7 ± 0.3 sites/μm2, respectively; n = 16 and 12 cells; our unpublished data). Although most sites were detected with the improved setup, only 50% of the two-layer profiles could be analyzed compared with 89% of the three-layer profiles. The extra layer allows measurement of many more smaller/incomplete sites, possibly due to increased photon counts and therefore signal-to-noise ratio. Alternatively it is possible that an extra layer of antibodies physically increases the size of sites (at most by ∼2 × 9 = 18 nm; IgGs measure 9 nm). However, incomplete sites, expected due to sectioning, were still missing in the three-layer size histogram, suggesting an inherent difficulty in measuring sizes <50 nm (see Supporting Information, Figure 7B). Although the three-layer protocol gave rise to some larger sites (two-layer range 11–129 nm, three-layer range 8–170; Figure 3B), we detected no significant difference in the average (weighted) diameters (81.5 nm, n = 154 sites from 11 cells, and 80.2 nm, n = 338 sites from 15 cells, two- and three-layer, respectively). (Note that because the sections are only ∼140 nm in thickness, the largest sites measured with the three-layer protocol [150, 168, and 169 nm] cannot represent true sizes.)
Size Measurements with EM
We next determined the size of pol IIO sites on the EM in sections labeled in parallel with those used for SMI-microscopy. Previous estimates of the size of transcription sites obtained after labeling nascent RNA with modified nucleotides (e.g., Br-UTP) ranged from 40 to 82 nm (Iborra et al., 1996; Jackson et al., 1998; Pombo et al., 1999b). Here, we have labeled sites containing the active form of pol II by using an antibody (H5), measured their size in electron micrographs, and determined the average diameter by using stereological procedures (see MATERIALS AND METHODS and Supporting Information, Part 3, Figure S5). Diameters of clusters of gold particles ranged from 8 to 85 nm (n = 105 sites from 11 cells). After correction for the contribution of polar caps by using the sequential-subtraction method (see MATERIALS AND METHODS and Supporting Information; Part 3, Figure S5), the average (weighted) diameter of pol IIO sites was 44.6 nm (Figure 3A, open bars), in agreement with previous estimates.
The diameters obtained by SMI-microscopy are therefore in the same order of magnitude as those obtained by EM; however, they are larger, possibly due to the difficult detection of sites smaller than ∼50 nm and the inability of the current setup to measure objects <20 nm. It is equally possible that the use of immunogold probes causes a reduction in the size measured by EM. Immunogold probes are prepared by coating colloidal gold particles with Igs, which produces a number of binding sites on each gold particle. The multiple Igs surrounding the gold particle may cross-link the different antibodies present in the immunolabeling complex, giving smaller sites. Hence, the importance of future developments in fluorescence based methods that will allow direct measurements by using smaller probes and GFP-labeled compartments in living cells.
DISCUSSION
SMI-microscopy is a novel method of far field microscopy that can measure the size of fluorescent beads of ∼40–100 nm (Failla et al., 2002b). Its potential use for the measurement of subresolution structures within cells is an exciting prospect, especially in live-cell imaging, because the resolution of current CLSMs is limited to >200 and >500 nm in the x,y- and z-axes, respectively. SMI-microscopy has previously been used to measure sizes of virtual objects and fluorescent beads below this resolution limit, i.e., from 40 to 100 nm. Here, we show the first results for size determination of biological nanostructures. Sites containing pol IIO are likely markers for sites of transcription and were chosen because they are within the range of sizes appropriate for SMI-microscopy evaluation. Initial attempts to obtain SMI-microscopy emission profiles from sites labeled in whole cells were unsuccessful with the present prototype, probably due to the complexity of staining patterns in whole cells and problems of “out-of-focus” flare from sites above and below the focal plane. (Note that a maximum of ∼4000 transcription sites have been detected using whole-cell imaging, in comparison with ∼8000 on thin sections; Wansink et al., 1996; Fay et al., 1997; Pombo et al., 1999b.) The use of thin sections simplifies staining patterns and signal collected comes only from sites that lie within the section (with no flare from sites above or below as in images from whole cells); here, we show that sites in sections ∼140 nm in thickness produce informative emission profiles with SMI-microscopy.
The use of SMI-microscopy to analyze biological structures rather than fluorescent beads posed a new set of challenges; they are usually dimmer, occur with variable sizes and intensities, and are present within the biological specimen at variable distances from the glass coverslip. We therefore optimized the analytical methods used to extract size information from the profiles for use with biological structures. We tested a number of different algorithms (see Supporting Information, Part 2, Table 1); although individual size measurements varied significantly between some methods, there was no significant difference between the average sizes obtained. Experimental observations (see above) led us to choose a method that allows distinction between two sites lying close together, uses information from all z-planes, and uses subtraction of background to simplify size evaluation.
Theory suggests that the size of an object could be deduced by simple measurement of M1 and M2 from SMI-microscopy emission profiles and extrapolation into a curve of R versus object size (Figure 1; Failla et al., 2002a,b). We tested this hypothesis by comparing measurements of 50-nm beads by using the analytical method and graphical measurements of R; there was no significant difference in the average diameter or between individual measurements of fluorescent beads or sites containing pol IIO. Although the determination of R is trivial, the analytical method is likely to be the most accurate because it accounts for any changes in the modulation distance due to local fluctuations of the refractive index.
At present the only way to measure structures as small as pol IIO sites is to use EM, but to achieve maximum sensitivity, the probes used consist of three layers of antibodies, leading to a potential over- or underestimation of sizes (Iborra and Cook, 1998; Pombo et al., 1999b). SMI-microscopy may increase accuracy of size measurements, because it should allow the use of fewer antibody layers and possibly even directly labeled molecules, including single molecules tagged with GFP, provided that more sensitive detectors are incorporated into the system. Here, we compared the size of pol IIO sites labeled with three- and two-antibody layers for EM and SMI-microscopy, respectively. SMI-microscopy was successful in measuring these sites giving results in the same order of magnitude as EM (Figure 3A). Diameters from SMI-microscopy measurements were consistently larger than those obtained by EM, in part due to the inability of SMI-microscopy to measure sites <20 nm in its current setup. However, it also should be noted that the density of sites seen by EM is greater than that of SMI-microscopy and CLSM (our unpublished data), implying that some sites are missed or are not resolved by CLSM or SMI-microscopy. Due to sectioning effects, the size distribution is expected to contain a proportion of incomplete sites that result from cutting through sites (see Supporting Information, Figures 5 and 6). With SMI-microscopy, we see negative values in the size distribution after correcting for the contribution of these incomplete sites (see Supporting Information, Figure 7A), implying that the setup was not sensitive enough to detect or to measure all sites. The equivalent correction for EM size data does not give negative values, implying that EM can detect and measure all sites (see Supporting Information, Figure 7C).
Fewer sites were detected per unit area with SMI-microscopy compared with CLSM (Figure 2, B and C), and although adjustments to the alignment procedure and detection light path improved detection of sites, only ∼50% could be measured, suggesting that some sites are too small to be measured. It is possible that the proximity of larger/brighter sites may interfere with the properties of profiles from small sites; the use of a smaller pixel size would provide more resolution and improve this limitation. (Note the CCD camera in the current SMI-microscope setup gives images with a final pixel size of 107 nm and that this is not sufficient to consistently achieve 200-nm resolution.) Increasing photon counts by addition of another antibody layer allowed measurement of some of these small sites without having a significant effect on the average size of sites; however, sites with >140 nm were detected, indicating that an extra layer of antibodies may lead to an increase in the size of sites (Figure 3B). We predict that further improvements to the camera setup, to allow maximum collection of photons, will allow detection of more sites; this will prove important for detection of directly labeled molecules. Also, to measure sites below ∼20 nm, a shorter wavelength of excitation is required (Failla et al., 2002a). Despite these limitations the fact that SMI-microscopy can measure structures within cells close to EM values is highly encouraging. With the continuing development and fine tuning of SMI systems, it is likely that this novel form of light microscopy will prove invaluable in the quest for understanding of cellular substructures, for example, by measuring the size of GFP-labeled structures inside living cells.
Supplementary Material
Acknowledgments
We thank Antonio Cavallo for help with the Khoros Cantata workspace configuration, Alexander Kroll for help with image collection, and Mike Hollinshead (Henry Wellcome Imaging Facility, Imperial College London) for help with EM. Financial support for this study was provided by Medical Research Council (United Kingdom) (to S.M and A.P.), the Royal Society (United Kingdom) (to A.P.), and the Bundesminister für Bildung und Forschung and the Deutsche Forschungsgemeinschaft (Germany) (to V.F., U.S., and C.C.). European Molecular Biology Organization provided a Short Term Fellowship (to S.M.), the Deutsches Krebsforschungszentrum (DFG Project CR-60/16-3) handled traveling and accommodation expenses (for S.M. and A.P.).
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E04–01–0045. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E04–01–0045.
Abbreviations used: CLSM, confocal laser scanning microscopy; GFP, green fluorescent protein; mAb, monoclonal antibody; pol, RNA polymerase; SMI, spatially modulated illumination.
Online version of this article contains supporting material. Online version is available at www.molbiolcell.org.
References
- Albrecht, B., Failla, A.V., Schweitzer, A., and Cremer, C. (2002). Spatially modulated illumination microscopy allows axial distance resolution in the nanometer range. Appl. Opt. 41, 80–87. [DOI] [PubMed] [Google Scholar]
- Bailey, B., Farkas, D.L., Taylor, D.L., and Lanni, F. (1993). Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation. Nature 366, 44–48. [DOI] [PubMed] [Google Scholar]
- Bregman, D.B., Du, L., Van Der Zee, S., and Warren, S.L. (1995). Transcription-dependent redistribution of the large subunit of RNA polymerase II to discrete nuclear domains. J. Cell Biol. 129, 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egner, A., Jakobs, S., and Hell, S.W. (2002). Fast 100-nm resolution three-dimensional microscope reveals structural plasticity of mitochondria in live yeast. Proc. Natl. Acad. Sci. USA 99, 3370–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failla, A.V., Cavallo, A., and Cremer, C. (2002a). Subwavelength size determination by spatially modulated illumination virtual microscopy. Appl. Opt. 41, 6651–6659. [DOI] [PubMed] [Google Scholar]
- Failla, A.V., Spöri, U., Albrecht, B., Kroll, A., and Cremer, C. (2002b). Nanosizing of fluorescent objects by spatially modulated illumination microscopy. Appl. Opt. 41, 7275–7283. [DOI] [PubMed] [Google Scholar]
- Fay, F.S., Taneja, K.L., Shenoy, S., Lifshitz, L., and Singer, R.H. (1997). Quantitative digital analysis of diffuse and concentrated nuclear distributions of nascent transcripts, SC35 and poly(A). Exp. Cell Res. 231, 27–37. [DOI] [PubMed] [Google Scholar]
- Grande, M.A., van der Kraan, I., de Jong, L., and van Driel, R. (1997). Nuclear distribution of transcription factors in relation to sites of transcription and RNA polymerase II. J. Cell Sci. 110, 1781–1791. [DOI] [PubMed] [Google Scholar]
- Gustafsson, M.G. (1999). Extended resolution fluorescence microscopy. Curr. Opin. Struct. Biol. 9, 627–634. [DOI] [PubMed] [Google Scholar]
- Iborra, F.J., and Cook, P.R. (1998). The size of sites containing SR proteins in human nuclei: problems associated with characterizing small structures by immunogold labelling. J. Histochem. Cytochem. 46, 985–992. [DOI] [PubMed] [Google Scholar]
- Iborra, F.J., Pombo, A., Jackson, D.A., and Cook, P.R. (1996). Active RNA polymerases are localized within discrete transcription “factories” in human nuclei. J. Cell Sci. 109, 1427–1436. [DOI] [PubMed] [Google Scholar]
- Jackson, D.A., Hassan, A.B., Errington, R.J., and Cook, P.R. (1993). Visualization of focal sites of transcription within human nuclei. EMBO J. 12, 1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, D.A., Iborra, F.J., Manders, E.M., and Cook, P.R. (1998). Numbers and organization of RNA polymerases, nascent transcripts, and transcription units in HeLa nuclei. Mol. Biol. Cell 9, 1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarnitsky, P., Cho, E.J., and Buratowski, S. (2000). Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo, A., Hollinshead, M., and Cook, P.R. (1999a). Bridging the resolution gap: Imaging the same transcription factories in cryosections by light and electron microscopy. J. Histochem. Cytochem. 47, 471–480. [DOI] [PubMed] [Google Scholar]
- Pombo, A., Jackson, D.A., Hollinshead, M., Wang, Z., Roeder, R.G., and Cook, P.R. (1999b). Regional specialization in human nuclei: visualization of discrete sites of transcription by RNA polymerase III. EMBO J. 18, 2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo, A., Jackson, D.A., Iborra, F., Hollinshead, M., Kimura, H., Sugaya, K., and Cook, P.R. (2000). Transcription factories. Proceedings of the 12th European Congress on elution microscopy, Vol I, Biological Sciences, B461–B464. [Google Scholar]
- Schneider, B., Upmann, I., Kirsten, I., Bradl, J., Hausmann, M., and Cremer, C. (1999). A dual-laser, spatially modulated illumination fluorescence microscope. Microsc. Anal. 57, 5–7. [Google Scholar]
- Tokuyasu, K.T. (1973). A technique for ultracryotomy of cell suspensions and tissues. J. Cell Biol. 57, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansink, D.G., Schul, W., van der Kraan, I., van Steensel, B., van Driel, R., and de Jong, L. (1993). Fluorescent labeling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J. Cell Biol. 122, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansink, D.G., Sibon, O.C., Cremers, F.F., van Driel, R., and de Jong, L. (1996). Ultrastructural localization of active genes in nuclei of A431 cells. J. Cell Biochem. 62, 10–18. [DOI] [PubMed] [Google Scholar]
- Weibel, E.R. (1979). Stereological Methods, Vol. 1: Practical Methods for Biological Morphometry, London: Academic Press, 162–203. [Google Scholar]
- Weibel, E.R. (1980). Stereological Methods, Vol. 2: Theoretical Foundations, London: Academic Press, 175–214. [Google Scholar]
- Williams, M.A. (1977). Quantitative methods in biology. In: Practical Methods in Electron Microscopy, Vol. 6, ed. A.M. Glauert, Amsterdam: Elsevier North-Holland Biomedical Press, 5–84. [Google Scholar]
- Zeng, C., Kim, E., Warren, S.L., and Berget, S.M. (1997). Dynamic relocation of transcription and splicing factors dependent upon transcriptional activity. EMBO J. 16, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.