

Abstract
Antiangiogenic therapies like bevacizumab offer promise for cancer treatment, but acquired resistance, which often includes an aggressive mesenchymal phenotype, can limit the use of these agents. Upregulation of β1 integrin (ITGB1) occurs in some bevacizumab-resistant glioblastomas (BRG) whereby, mediating tumor–microenvironment interactions, we hypothesized that it may mediate a mesenchymal-type resistance to antiangiogenic therapy. Immunostaining analyses of β1 integrin and its downstream effector kinase FAK revealed upregulation in 75% and 86% of BRGs, respectively, compared with pretreatment paired specimens. Furthermore, flow cytometry revealed eight-fold more β1 integrin in primary BRG cells compared with cells from bevacizumab-naïve glioblastomas (BNG). Fluorescence recovery after photobleaching of cells engineered to express a β1-GFP fusion protein indicated that the mobile β1 integrin fraction was doubled, and half-life of β1 integrin turnover in focal adhesions was reduced markedly in BRG cells compared with bevacizumab-responsive glioblastoma multiforme cells. Hypoxia, which was increased with acquisition of bevacizumab resistance, was associated with increased β1 integrin expression in cultured BNG cells. BRGs displayed an aggressive mesenchymal-like phenotype in vitro. We found that growth of BRG xenograft tumors was attenuated by the β1 antibody, OS2966, allowing a 20-fold dose reduction of bevacizumab per cycle in this model. Intracranial delivery of OS2966 through osmotic pumps over 28 days increased tumor cell apoptosis, decreased tumor cell invasiveness, and blunted the mesenchymal morphology of tumor cells. We concluded that β1 integrin upregulation in BRGs likely reflects an onset of hypoxia caused by antiangiogenic therapy, and that β1 inhibition is well tolerated in vivo as a tractable strategy to disrupt resistance to this therapy.
Introduction
Glioblastoma multiforme is the most common malignant brain tumor in adults, with a median survival close to 1 year (1). To date, only 3 drugs have been approved by the U.S. Food and Drug Administration for glioblastoma treatment, including, most recently, bevacizumab (Avastin; Genentech) in 2009 (2). Despite improvements in clinical metrics with bevacizumab including cognitive benefits and reduction of steroid use (3), 40% of treated patients developed acquired resistance in phase II trials (4). Thus, as has been the case with other cancers treated with antiangiogenic therapy, the promise of antiangiogenesis in glioblastoma remains unfulfilled in part due to acquired resistance.
Integrins are cell-adhesion molecules that mechanosense the microenvironment and elicit extracellular-matrix (ECM)–induced signaling in both normal and pathologic states such as inflammation and cancer. Importantly, integrins lie at the interface of the cell and microenvironment, playing a key role in tumor progression and regulating growth and survival pathways. Upregulation of integrins has been associated with epithelial malignancies (5), particularly during invasion, metastasis, and angiogenesis (6, 7). There is growing evidence for the role of aberrantly expressed integrins in glioblastoma pathophysiology (8). β3 and β5 integrins have been implicated in angiogenesis, and several approaches targeting these molecules are under investigation in the clinic (9). β1 integrins, which coordinate much broader functional activities such as inflammation, proliferation, adhesion, and invasion, have recently been implicated in therapeutic resistance in multiple solid cancer models (10–13) and hematopoietic malignancies (14, 15). Importantly, this β1 integrin-mediated resistance is thought to occur at the level of the tumor cells themselves. β1 integrin also has important functions during tumor vascularization such as VEGF-dependent (16) and VEGF-independent angiogenesis by promoting endothelial cell migration (17). Notably, as shown in glioblastoma models (18–20), we found that bevacizumab causes U87MG, a commonly studied glioblastoma cell line, to grow more invasively (Supplementary Fig. S1A), and orthotopic bevacizumab-resistant glioblastoma (BRG) xenografts infiltrate the brain via vessel co-option, which has been shown to require β1 integrins (21), whereas bevacizumab-naïve glioblastoma (BNG) xenografts remain well circumscribed (Supplementary Fig. S1B; ref. 22). This evidence, when taken together, led us to investigate the novel hypothesis that β1 integrin drives resistance to antiangiogenic therapy by promoting multiple mechanisms at the interface of tumor cells and the microenvironment.
Materials and Methods
Collection and analysis of human clinical specimens
Human specimens were obtained from the University of California, San Francisco (UCSF; San Francisco, CA) Brain Tumor Tissue Bank (22). Fluorescence immunohistochemistry of human paraffin-embedded tissues for β1 integrin (ab52971; Abcam), CA9 (Novus Biologicals), FAKY397 (ab4803; Abcam), GFAP (ab4674; Abcam), and laminin (ab14055; Abcam) was conducted with either standard indirect or tyramide signal amplification (PerkinElmer) as described (21) following standard citrate buffer (Abcam) antigen retrieval.
Cells and cell lines
U87MG, MDA-MB-231, and SW-1080 cell lines were obtained from and authenticated by American Type Culture Collection and passed in less than 6 months. Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, nonessential amino acids (NEAA), and antibiotics. The bevacizumab-resistant primary glioblastoma cells, SF7796 and SF8106, and the bevacizumab-naïve primary glioblastoma cells, SF7996 and SF8244, were obtained and grown as described (22). Bevacizumab-resistant (BRG1, BRG2, and BRG3) and bevacizumab-naïve (N1, N2, and N3) cell lines were derived from fresh clinical resection specimens at UCSF as described (22) and were propagated in 50% to 50% DMEM/F-12 with 10% FBS, NEAA, and antibiotics. Primary SF8106 and SF7796 glioma cultures were transfected with the pFB-Neo Retroviral Vector (Agilent) containing a cDNA expressing the β1-GFP fusion protein (gift of Martin Humphries; University of Manchester, UK), followed by selection in 5 μg/mL geneticin (Gibco), as described (23). Experimental hypoxia was induced by growth in 1% oxygen as described (24).
Short hairpin RNA-mediated integrin knockdown
U87MG, BRG2, and BRG3 cells were infected with short hairpin RNA (shRNA) lentiviral particles (Santa Cruz Biotechnology) targeting β3 integrin (sc-63292-V), β1 integrin (sc-35674-V), or vector control (shCTRL, sc-108084) per manufacturer’s protocol. After 48 hours, subcultured cells were selected in 1 μg/mL puromycin for 1 week.
Flow cytometry
Cells were trypsinized in 0.25% trypsin with EDTA, counted, resuspended in PBS at 106 cells/mL, and kept on ice. Cell surface β1 integrin was labeled with anti-β1 integrin antibodies OS2966 (OncoSynergy, Inc.), CD29 (Clone TS-2/16, BioLegend), or CD29 (Clone K20, BD). Cell surface c-MET was detected with Clone 95106 monoclonal antibody (mAb; R&D Systems). Flow cytometry was conducted with a BD FACS Calibur II machine, and data were analyzed using FlowJo software (Tree Star, Inc.).
Western blot analysis
Western blots were conducted as described previously (25), using primary antibody to β1 integrin (Biolegend; Abcam) along with αV, α5, α4, and β3 antibodies (Cell Signaling).
Fluorescence recovery after photobleaching
SF8106/B1-GFP and SF7996/B1-GFP cells were seeded at 10,000 cells per well on chambered coverglass (Fisher), which had been coated overnight with 10 μg/mL fibronectin (Sigma-Aldrich). Individual cells were analyzed on a Zeiss LSM 510 NLO Meta confocal microscope. Target-bleach regions were normalized to background observational bleaching. Images were collected every 30 seconds until fluorescence recovery plateau. Fractional fluorescence recovery and half-time of recovery were calculated as described (26).
Dynamic in vitro assays and timelapse microscopy
Adhesion assays were conducted as described (21) on plates coated with 10 μg/mL fibronectin (Sigma). Scratch assays were conducted as described (27) on plates coated with 10 μg/mL fibronectin. Cell spreading assays were conducted by plating 105 cells on a 30-mm dish coated with 10 μg/mL fibronectin. Time-lapse phase-contrast microscopy was conducted as described (21) with a Zeiss Observer Z1 inverted microscope with an AxioCam MRm and accompanying AxioVision 4.8 software. Briefly, 30-mm dishes were placed upon a heated stage adapter (Stable Z with heated lid, Bioptechs), and images were captured every 90 to 180 seconds for up to 24 hours. Media were covered with sterile embryo-tested mineral oil (Sigma).
Cell adhesion was quantified as described (21). Cell spreading was quantitated as cell area (measured hourly) and percentage of spread cells per high-power field. For cell area, all whole cells in a ×40 field were measured manually in ImageJ (NIH) at time zero and every hour for 5 hours. Means were calculated at each timepoint and compared. The percentage of spread cells per ×40 field was similarly calculated hourly (not including time zero) for 12 hours. Cell migration and dendricity factor for cell shape were measured as described (22, 28).
Cell proliferation
Bromodeoxyuridine (BrdU) immunofluorescence was conducted by incubating cells in 10 μmol/L BrdU (Sigma) for 1 hour in complete media followed by fixation in 1% paraformaldehyde. Cells were permeated in 0.1% Triton-X100 (Sigma) in PBS on ice for 5 minutes followed by incubation in biotinylated sheep anti-BrdU antibody (ab2284, Abcam) overnight at 4°C. Following PBS washes, cells were incubated with streptavidin-Alexa555 (Invitrogen) and coverslipped with VectaShield mounting medium with DAPI (VectorLabs). BrdU-positive cells per high-power field were counted in three 40× fields per well, 3 wells per experiment with repeats.
Tumor spheroid assays
Aggregation assays were conducted by suspending tumor cells in PBS on ice for 15 minutes. To visualize β1 integrin, cells were incubated with Alexa 647 conjugated anti-CD29 (clone TS-2/16) for 15 minutes, spun down, and rinsed. Cell growth in acidified media was conducted as described (29). Spheroidal growth assays were conducted in 6-well ultra-low adhesion plates (CoStar) with standard conditions for growth of neuro-spheres (29) for 3 to 5 days. Spheroid dissolution assays were conducted by plating spheroids on culture dishes coated with 10 μg/mL of bovine serum albumin (BSA) control, fibronectin, laminin, or collagen type IV (all ECM from Sigma) for 5 to 7 days.
Xenograft studies
Animal procedures were approved by the UCSF Institutional Animal Care and Use Committee (IACUC). A total of 105 to 106 cells were injected s.c. or 105 cells were injected stereotactically into the right caudate (orthotopic model) of 5- to 8-week-old female athymic mice. Mice were randomized into treatment groups after tumors were established (mean s.c. volume = 40–60 mm3 or 10 days after intracranial implantation). Bevacizumab and human immunoglobulin G (IgG) control (Sigma) were administered at 10 mg/kg intra-peritoneally (i.p.) twice weekly. Inhibitory anti-β1 integrin mAb OS2966 or control IgG was administered at 1, 5, or 10 mg/kg i.p. twice weekly. For orthotopic studies, OS2966 or IgG (160 μg total) was delivered intratumorally over 28 days via rigid cannula connected to Model 1004 osmotic pumps (Alzet) implanted s.c. between the scapulae. Subcutaneous tumors were measured with calipers 1 to 2 times weekly, with volume calculated as described (24). Treatment continued until mice had to be sacrificed per IACUC criteria (e.g., 2.1-cm maximal dimension, cutaneous ulceration, or symptoms) or study end.
Tissue preparation and immunohistochemistry
Mice were sacrificed and tissue collected and prepared as described (21). Cryostat sections (20–30 μm in size) were collected and mounted directly on slides and were processed for immunohistochemistry as described (21). For TUNEL staining, the TMR red In Situ Cell Death Detection Kit (Roche) was used. Immunohistochemistry was conducted using antibodies against β1 (Abcam), laminin (Abcam), GFAP (Millipore), and GLUT-1 (Millipore). Collagen IV immunostaining was described previously (25).
Statistical analyses
Student t test was used for parametric comparisons between paired variables. The Mann–Whitney U test was used for nonparametric pairwise comparisons. Multivariate analyses were done with ANOVA with post hoc Dunnett (most analyses) or Bonferroni tests (e.g., xenograft studies) as appropriate. P < 0.05 was deemed statistically significant.
Results
Upregulation of β1 integrin after acquired resistance to bevacizumab
We recently reported by microarray analysis and confirmatory PCR that transcription of β1 integrin was upregulated in glioblastomas exhibiting the nonenhancing infiltrative pattern of growth that is viewed as unique to bevacizumab resistance (22). To further verify this result, we conducted immunohistochemistry on BRGs and their paired pretreatment specimens and found increased β1 integrin in 9 of 12 cases (75%, Fig. 1A). This increased immunoreactivity was observed in both angiogenic blood vessels (Fig. 1A, arrows) and tumor cells (Fig. 1B). Consistent with bona fide integrin signaling, we found increased phosphorylated focal adhesion kinase staining in tumor cells in 6 of 7 BRG cases compared with pretreatment, with staining tending to be close to ECM (Fig. 1C; refs. 30, 31). Furthermore, β1 integrin was increased in primary cells from BRG clinical specimens compared with primary cultures established from BNG specimens by fluorescence-activated cell sorting (P < 0.05; Fig. 1D). To rule out contributions from patient variability among clinically derived samples that could affect integrin expression, we experimentally derived a bevacizumab-resistant xenograft from U87MG (U87-BEV; ref. 25). Screening of integrin expression revealed that U87-BEV showed significantly higher expression of β1 integrin, along with αV and α5 integrins, 2 potential β1 heterodimer partners, which interact to promote fibronectin binding (32), compared with U87-IGG, a control line treated only with IgG (Supplementary Fig. S2A and S2B).
Figure 1.

β1 integrin expression and bevacizumab resistance in glioblastoma. A, immunofluorescent staining for β1 integrin (green); collagen IV, an endothelial marker (red); and 4′, 6-diamidino-2-phenylindole nuclear counterstain in clinical specimens from 2 patients before and after development of bevacizumab resistance and a control patient at first and second surgery. Scale bar, 100 μm. B, high-powered view from another BRG shows β1 integrin immunoreactivity in glioma cells including glial fibrillary acidic protein-positive cells (arrows). Scale bar, 50 μm. C, phosphorylated focal adhesion kinase (FAKY397, green) is enriched in post-bevacizumab tissues and correlated with ECM consistent with increased integrin activity. Scale bar, 120 μm. D, dissociated cells from resistant glioblastoma (GBM) have significantly higher β1 integrin on flow cytometry than primary tumor cells (*, P < 0.05, t test). E, FRAP conducted on cells expressing β1-GFP fusion protein revealed more rapid β1 integrin turnover in focal adhesions and a higher integrin mobile fraction in bevacizumab-resistant SF8106 cells than in bevacizumab-responsive SF7996 cells. Experiment was repeated 4 times, with distinct curves each time (P < 0.001, t test). F, BRG cells are more adherent to classical ECM proteins than several glioblastoma cell lines in addition to the highly adherent MDA-MB-231 breast carcinoma (BSA, negative control; PLL, positive control; FN, fibronectin; C4, collagen type IV; LN, laminin). Data are mean ± SD. See also Supplementary Fig. S2.
Because the percentage of integrin that is mobile and the turnover of this mobile integrin in focal adhesions sustains integrin signaling, we engineered BRG and BNG cells to express a β1-GFP fusion protein and used fluorescence recovery after photobleaching (FRAP) to measure integrin turnover in focal adhesions (26). We found 2-fold faster β1 integrin turnover in focal adhesions and double the mobile fraction of β1 integrin in bevacizumab-resistant SF8106 cells than in bevacizumab-responsive SF7996 cells (Fig. 1E and Supplementary Fig. S2C). Thus, compared with BNG cells, bevacizumab-resistant cells exhibited more mobile β1 integrin, reflective of reduced cellular membrane barriers to β1 integrin mobility in these cells and more rapid turnover of β1 integrin in focal adhesions.
To verify the functional relevance of increased β1 integrin in glioblastoma cells, we conducted cell adhesion assays and found significantly greater adhesion to ECM in BRGs compared with classic glioblastoma lines and even the highly adhesive MDA-MB-231 human breast adenocarcinoma, which served as a positive control (Fig. 1F). Thus, the β1 integrin subunit is functionally upregulated in glioblastoma after acquired bevacizumab resistance.
Hypoxia associated with high β1 integrin expression in glioblastoma cells
Hypoxia is a common cellular stress, particularly in the setting of fast-growing cancers and variably during the course of antiangiogenic therapy. Hypoxia upregulates β1 integrin expression in the setting of wound healing via hypoxia-inducible factor-1α (HIF-1α)–stimulated β1 transcription (33). We showed nearly 80% more CA9 staining after bevacizumab resistance as compared with before, with areas of CA9 staining corresponding with high β1 integrin expression in patient glioblastoma specimens (Supplementary Fig. S3A). To verify that antiangiogenic therapy acutely increases β1 integrin expression in growing tumors in vivo, we stained glioblastoma tumors from mice taken within days of the last bevacizumab treatment. Striking β1 integrin immunoreactivity was observed in the treated tumors compared with controls, particularly in the hypoxic tumor core (Supplementary Fig. S3B). To confirm this mechanism, we cultured U87MG cells for 6 to 48 hours in 1% oxygen to simulate microenvironmental hypoxia. We observed a significant increase in β1 integrin expression in glioma cells in vitro (Supplementary Fig. S3C). This was a rapid and reversible cellular response, which we also showed in primary human glioblastoma cells and breast and colorectal carcinoma cells (Supplementary Fig. S3D). Therefore it is likely that hypoxia contributes to β1 integrin upregulation during bevacizumab therapy, thus promoting mechanisms of survival and evasion.
β1 integrin inhibition attenuates mesenchymal phenotype and function of BRG cells
To further establish a functional role for β1 integrin in the malignant phenotype of bevacizumab glioblastoma cells, we established several clones of the BRG3 line expressing shRNA-targeting β1 integrin (shBETA1) and shRNA-targeting control sequences (shCTRL; Supplementary Fig. S4A–S4D). An epithelioid morphologic shift, including polygonal shape and tendency for growth in sheets, was observed in the shBETA1 clones compared with shCTRL cells (Supplementary Fig. S5A). Indeed, individual cell size as measured by cell area was significantly increased in the shBETA1 clones compared with shCTRL (Supplementary Fig. S5B). In addition, dendricity factor, a unitless measure of cell shape, confirmed mesenchymal morphology in the shCTRL cells compared with more epithelioid morphology in the shBETA1 clones (Supplementary Fig. S5C). To verify functional impairment of mesenchymal phenotype, we examined the knockdown cells in various static and dynamic in vitro assays. As hypothesized, BRG3-shBETA1 clones showed significantly decreased capacity for mesenchymal functions, including adhesion (Fig. 2A), cell spreading (Fig. 2B and C and Supplementary Movie S1), and migration (Fig. 2D and E and Supplementary Movie S2) on ECM compared with BRG3-shCTRL cells. Furthermore, bevacizumab-naïve cells showed epithelial-like migration with broad lamellipodia (Supplementary Movie S3) in contrast with the saltatory mesenchymal-type migration seen in the BRG3 line. In addition, in vitro proliferation (Fig. 2F) was significantly attenuated in the BRG3-shBETA1 clones compared with BRG3-shCTRL cells. This effect of β1 integrin knockdown in the BRG3 line on cell growth in vitro was determined to be cytostatic, rather than cytotoxic (Supplementary Fig. S5D). Similar results on adhesion, migration, and proliferation were found when the BRG3 line was treated with the inhibitory β1 integrin mAb, OS2966 (Supplementary Fig. S6A and S6B and Supplementary Movie S4). Thus, β1 integrin is important for multiple tumor cell functions implicated in antiangiogenic therapy evasion, including persistent proliferation and mesenchymal phenotype.
Figure 2.

β1 integrin knockdown in resistant glioblastoma cells inhibits mesenchymal function. β1 integrin knockdown clones are impaired in cell adhesion (A; *, P < 0.01, ANOVA with post hoc Dunnett test), dynamic cell spreading (B; *, P < 0.05, **, P < 0.01, ANOVA with post hoc Dunnett test), and percentage of spread cells (C; *, P < 0.01, Mann–Whitney test) on fibronectin. D and E, dynamic time-lapse analysis in a scratch wound assay shows impaired migration in knockdown clones versus control (*, P < 0.05, ANOVA with post hoc Dunnett test). β1 integrin knockdowns show decreased proliferation as shown by BrdU immunofluorescence (F; *, P < 0.01, t test). Scale bar, 120 μm. Data are mean ± SD. See also Supplementary Figs. S3 to S5.
Spheroidal growth is impaired by β1 integrin inhibition
Microenvironmental hypoxia (34) and acidification (29) are 2 suspected consequences of antiangiogenic therapy, and both activate HIF-1α and HIF-2α expression. Both hypoxia-associated factors have also been linked to malignant stem-like phenotypes in glioblastoma cells including in vitro spheroid formation. Indeed, freshly isolated bevacizumab-resistant U87-BEV cells are more highly prone to spheroid formation in vitro compared with the control U87-IGG line under normal culture conditions (Supplementary Fig. S7A). Implantation of glioblastoma spheroids has been shown to be associated with decreased survival in orthotopic xenograft studies compared with control cells (29). Importantly, we observed that acutely dissociated BRG1 and BRG3 glioblastoma cells in suspension show the highest β1 integrin expression at cell–cell contacts (Fig. 3A). Therefore, to investigate a potential role for β1 integrins in glioblastoma spheroid formation, we cultured U87MG cells under acidic stress. Spheroid formation in acidic media was attenuated in U87-shBETA1 (Supplementary Fig. S7B) cells compared with control U87-shCONTROL cells (Fig. 3B). Similar results were obtained with parental U87MG cells treated with an inhibitory β1 integrin antibody (Supplementary Fig. S7C). To more directly study spheroid formation, we grew glioblastoma cells in suspension in nonadherent plates. U87 cells with stable knockdown of β1 integrin (U87-shBETA1) showed significantly smaller spheroid diameters than control U87-shCONTROL cells (Fig. 3C). The same result was found in knockdown clones from the clinically derived BRG3 line (Fig. 3D). Furthermore, confirming the role of β1 integrins in spheroidal maintenance, we showed dissolution of glioblastoma spheroids when plated on classic β1 integrin ECM substrates, particularly fibronectin (Fig. 3E). Thus, the β1 integrin subunit is intimately involved in cell–cell interactions and malignant spheroidal growth in glioma cells in vitro, a phenotype that has been associated with increased malignancy in vivo.
Figure 3.
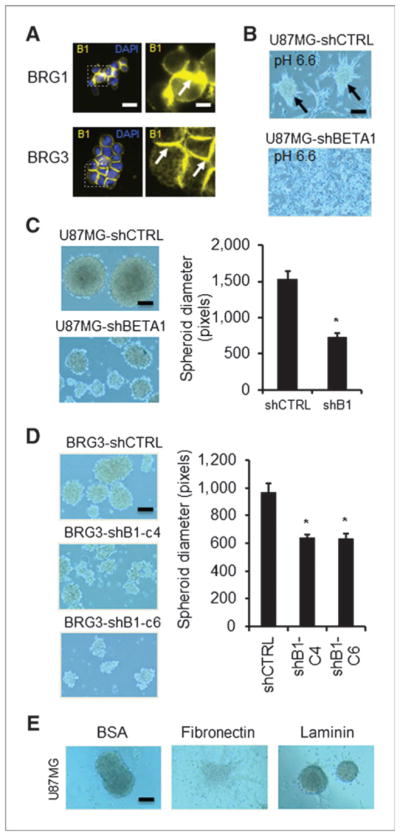
β1 knockdown in glioma cells inhibits spheroidal growth. A, immunofluorescence shows acutely enriched β1 immunoreactivity at cell–cell contacts in 2 BRG lines in an in vitro aggregation assay. Scale bars, 30 and 10 μm, respectively. B, β1 knockdown in U87MG attenuates spheroid formation (arrows) in acidic media. Scale bar, 120 μm. Formal spheroidal growth assay confirms impairment of spheroid diameter with β1 knockdown in both U87MG (C; *, P < 0.05, t test; scale bar, 120 μm) and BRG3 clones (D; *, P < 0.05, ANOVA with post-hoc Dunnett test; scale bar, 120 μm). E, U87MG spheroids unravel when plated on fibronectin, but not BSA control substrate consistent with a β1-mediated mechanism. Scale bar, 120 μm. Data are mean ± SD. See also Supplementary Fig. S6.
β1 integrin inhibition prevents BRG growth in vivo
To test the role of β1 integrin in glioma growth and bevacizumab resistance in vivo, we used several glioma cell models in athymic mice. First, we showed attenuation of tumor growth in the BRG2 and BRG3 lines after stable knockdown of β1 integrin (polyclonal, approximately 70% knockdown). In both cases, tumor growth was significantly impaired in the shBETA1 cells compared with shCTRL cells (Fig. 4A and B). We then inoculated mice with clones of BRG3 that had more than 90% knockdown of β1 integrin. Consistent with a gene–dosage effect, we showed complete prevention of in vivo tumor growth for 6 months (Fig. 4C). Thus, β1 integrin inhibition overcomes growth of both BNG and BRG cells in vivo.
Figure 4.

β1 integrin inhibition in BRG cells attenuates growth in vivo. Growth of polyclonal β1 integrin knockdowns in a subcutaneous xenograft model was attenuated in both BRG2 (A; *, P < 0.05, ANOVA with post hoc Bonferroni test) and BRG3 (B; *, P < 0.05, ANOVA with post hoc Bonferroni test) lines. C, more than 90% knockdown of β1 integrin in BRG3 clones prevents in vivo growth for up to 6 months. D and E, inhibitory anti-integrin antibody, OS2966, similarly inhibits growth of BRG3 cells in vivo at 1 or 5 mg/kg i.p. biweekly compared with control IgG (*, P < 0.05, ANOVA with post hoc Bonferroni test). F, 5 mg/kg of OS2966 induces more apoptosis in subcutaneous tumors than IgG. Scale bar, 100 μm. Data are mean ± SEM. See also Supplementary Fig. S7A.
To provide proof-of-concept for a clinically relevant treatment modality, we tested a therapeutic antibody approach in a glioma xenograft model. Mice inoculated with the BRG3 cell line were systemically treated with inhibitory β1 integrin monoclonal antibodies (OS2966) at 2 doses. Both groups of treated mice had significantly smaller tumors than mice treated with 5 mg/kg of control IgG (Fig. 4D and E). Strikingly, 56% (5/9) of mice with BRG3 tumors treated with 5 mg/kg OS2966 showed complete tumor regression after 8 weeks of treatment. We observed apoptosis on histology in the treated group but not in the IgG control (Fig. 4F). Finally, to verify the above findings in an orthotopic model, we stereotactically implanted BRG3 cells into the striatum of mice. Ten days after inoculation, we implanted cannulae through the same burr hole for chronic intratumoral delivery of either OS2966 or IgG control via s.c. osmotic pumps that provided antibody delivery for 28 days. Successful tumoral delivery of antibodies was verified with immunofluorescence detection of OS2966 in horizontal brain sections (Supplementary Fig. S8). BRG3 cells in the control condition formed highly invasive tumors, and 55.2% ± 11% of invading cells were associated with the neurovasculature according to GLUT-1 immunoreactivity for endothelial cells (Fig. 5A). OS2966 treatment resulted in complete regression of tumors (3/3 mice) as supported by massive induction of tumor cell apoptosis on TUNEL staining (Fig. 5B). Finally, OS2966 treatment inhibited striatal invasion as shown by number of invading cells per section (Fig. 5C) and invasion distance (Fig. 5D). Accordingly, OS2966 treatment seemed to directly affect invasive morphology of tumor cells (Fig. 5E). Thus, blockade of β1 integrin function with inhibitory monoclonal antibodies attenuates growth of a recurrent bevacizumab-resistant glioma line in vivo.
Figure 5.

OS2966 inhibits growth and invasion of orthotopically implanted BRG3 cells. A, low-power epifluorescent microscopy shows parenchymal invasion of BRG3 cells in IgG-treated control mice. Many instances of this invasion (>50%) occurred perivascularly consistent with vessel cooption (arrows). Scale bar, 120 μm. B, induction of apoptosis after OS2966 versus IgG control mAb treatment (*, P < 0.05, t test). Scale bar, 120 μm. Invasion of BRG3 cells in OS2966-treated mice was significantly attenuated as shown by number of invasive cells (C; *, P < 0.05, t test), invasion distance (D; *, P < 0.05, t test), and was supported by altered morphology of single invading cells including length and number of cellular processes (E). Scale bar, 50 μm. Data are mean ± SD. See also Supplementary Fig. S7.
β1 integrin inhibition potentiates bevacizumab treatment
Not all patients respond to bevacizumab, and significant morbidity is associated with its long-term use. Therefore, there is clinical need for a novel therapeutic approach to improve response rates, prevent resistance, and potentially reduce the necessary dose of bevacizumab. Toward this end, full-dose combination therapy of OS2966 with bevacizumab was attempted in the U87MG subcutaneous xenograft model in 2 separate investigations. However, both studies had to be halted before their endpoints due to rapid cutaneous tumor ulceration in 100% (5/5 and 10/10 mice, respectively) of the combination therapy animals by the third week of treatment. These findings are suggestive of synergy between OS2966 and bevacizumab. To explore this question further, we hypothesized that the addition of OS2966 alternating with low-dose bevacizumab would enhance efficacy in U87MG xenografts (Fig. 6A). Indeed, alternating OS2966 and bevacizumab at 1 mg/kg, each was equivalent to a biweekly standard dose of bevacizumab (10 mg/kg) in this setting (Fig. 6B). Furthermore, the percentage of animals with complete regression of tumors was nearly equivalent, with 44% (4/9) of mice in the alternating low-dose group compared with 56% (5/9) of mice in the standard dose group. Thus, consistent with its multiple roles in promoting therapeutic resistance, β1 integrin is potentially a promising adjunct target in combination with antiangiogenic therapies, including bevacizumab.
Figure 6.

OS2966 potentiates efficacy of bevacizumab in a glioma model. A, study design of experimental groups with standard bevacizumab therapy at 10 mg/kg biweekly (group I, BEV) versus alternating bevacizumab and OS2966 at 1 mg/kg (group II, low-dose combo). B, inhibition of tumor growth with low-dose combo (group II) was equivalent to high-dose bevacizumab (group I) compared with IgG controls in subcutaneous U87MG xenografts (*, P < 0.05, ANOVA with post hoc Bonferroni test). Shown are mean ± SEM.
Discussion
Our data show for the first time that resistance to anti-angiogenic therapy is associated with increased expression of β1 integrin as well as increased mobility of β1 integrin and increased turnover of β1 integrin in focal adhesions. We also showed that resistance to antiangiogenic therapy can be overcome by targeting tumor cell interactions with the glioma microenvironment via β1 integrin inhibition. In contrast to approaches that target a specific integrin–ligand interaction (e.g., natalizumab, which targets α4 integrin, blocking the ability of α4β1 or α4β7 to bind VCAM-1), a single drug targeting β1 would inhibit the ability of tumor cells to bind a spectrum of ECM ligands like fibronectin, collagen IV, and laminin, each of which we found to be upregulated in BRGs (22). This approach could potentially be applied more broadly in several medical indications. Inhibition of β1 integrin overcomes resistance to antiangiogenesis therapy via multiple potential mechanisms: (i) preventing vessel cooption (and/or growth/invasion upon any classical ECM substrate; ref. 21); (ii) reducing viability of tumor cells after insults (35); (ii) directly inhibiting tumor cell proliferation; (iii) directly inhibiting the vascularization process (36); and (iv) inhibiting the aggressive mesenchymal phenotype, including spheroidal growth. In addition, β1 integrin inhibition alternating with bevacizumab can reduce the necessary dose of antiangiogenic agent, thus potentially reducing drug-related morbidity.
It is tempting to speculate that because breast and other cancer cells also respond to insults with an increase in β1 integrin expression (e.g., Supplementary Fig. S3D), this approach may be applicable for the treatment of a wide variety of high-grade and/or treatment-refractory adenocarcinomas. This hypothesis warrants further study. Indeed, β1 integrin has proved to be a viable oncological target in animal models of solid and blood-borne cancers for multiple processes, including (i) prevention and growth of metastasis (37); (ii) overcoming resistance to radiation (35) and chemotherapy (38–42); and (iii) inhibition of local carcinoma spread. In addition, β1 integrin upregulation has been shown to be prognostic for breast (38, 43), lung (44), and pancreatic (37) cancers.
A limitation of the current study is the inability to directly study the effect of antiangiogenesis mechanistically in vitro. Bevacizumab acts at the endothelium, causing ischemia and other tissue-level processes, which cannot be accurately modeled with isolated tumor cells or cocultures. Therefore, the spectrum of adaptations to antiangiogenic therapy cannot currently be reliably assessed in vitro. We have addressed this limitation with a multimodal approach using clinical specimens and their cellular derivatives in surrogate in vitro assays (e.g., hypoxia, acidified media) and relevant preclinical murine models. Of note, these murine models were generated by treating tumors with bevacizumab, which targets human, not murine VEGF, and would therefore target only tumor cells rather than stromal cells. However, we have found that bevacizumab is equally effective in xenograft treatment as an agent-targeting mouse and human VEGF (personal communication, C. David James, Department of Neurological Surgery, University of California, San Francisco, CA; 2008).
The orthotopic model with bevacizumab-resistant BRG3 cells revealed several important findings. First, we confirmed that parenchymal invasion of bevacizumab-resistant glioma cells occurs primarily via vessel cooption. Second, OS2966 delivered via osmotic pump nearly completely abolished invasion of BRG3 cells. In addition, inhibition of β1 integrin in these studies resulted in tumor cell apoptosis and disrupted tumor mass formation. Because of the finite life of the osmotic pumps, survival studies were not possible; however, the results provide solid proof-of-concept for intracranial delivery of OS2966 as a treatment for BRG via modalities like convection-enhanced delivery, which would enable more high-flow intratumoral infusion of OS2966 (45). Although local delivery of β1 integrin-targeted therapy would be appealing by increasing local concentrations of the therapeutic agent and minimizing systemic exposure, it should be noted that an antibody targeting β1 integrin was nontoxic when systemically delivered in our studies and other studies (35).
Although we have shown that alternating low doses of β1 integrin inhibition with bevacizumab can reduce the necessary weekly dose of bevacizumab by 20 times and can inhibit growth of bevacizumab-resistant glioma cells, the question remains whether alternate or combination therapy can attenuate development of resistance to bevacizumab over longer treatment periods. To define the full potential of our approach in the clinic will require exploring various dosing schedules and combinations in vivo. Still, our data suggest that β1 integrin inhibition as monotherapy for BRG or combined with bevacizumab to reduce the risk of resistance are promising therapeutic strategies.
Supplementary Material
Acknowledgments
The authors thank Rick Horwitz for helpful discussion.
Grant Support
This work was supported by funding from the James S. McDonnell Foundation, the American Cancer Society (ACS), and NIH 5K02NS64167-2 (M.K. Aghi); the Howard Hughes Medical Institute (A. Jahangiri); and NIH grant 1R01CA124891 and ACS RSG-07-1110-01-CCE (C.C. Park).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
W. Shawn Carbonell is employed as a cofounder, President, and CEO, and has ownership interest (including patents) in OncoSynergy, Inc. C.C. Park has ownership interest (including patents) in Oncosynergy Ltd. M.K. Aghi is employed as a scientific advisory board member/shareholder, has ownership interest (including patents), and is a consultant/advisory board member of Oncosynergy. No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: W.S. Carbonell, M. DeLay, A. Jahangiri, C.C. Park, M.K. Aghi
Development of methodology: W.S. Carbonell, M. DeLay, A. Jahangiri, M.K. Aghi
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): W.S. Carbonell, M. DeLay, A. Jahangiri, M.K. Aghi
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): W.S. Carbonell, M. DeLay, M.K. Aghi
Writing, review, and/or revision of the manuscript: W.S. Carbonell, M. DeLay, A. Jahangiri, C.C. Park, M.K. Aghi
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M.K. Aghi
Study supervision: W.S. Carbonell, M.K. Aghi
References
- 1.Stupp R, Dietrich PY, Ostermann Kraljevic S, Pica A, Maillard I, Maeder P, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20:1375–82. doi: 10.1200/JCO.2002.20.5.1375. [DOI] [PubMed] [Google Scholar]
- 2.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–5. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kienast Y, Winkler F. Therapy and prophylaxis of brain metastases. Expert Rev Anticancer Ther. 2010;10:1763–77. doi: 10.1586/era.10.165. [DOI] [PubMed] [Google Scholar]
- 4.Vredenburgh JJ, Desjardins A, Herndon JE, II, Dowell JM, Reardon DA, Quinn JA, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–9. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 5.Gotzmann J, Mikula M, Eger A, Schulte-Hermann R, Foisner R, Beug H, et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. 2004;566:9–20. doi: 10.1016/s1383-5742(03)00033-4. [DOI] [PubMed] [Google Scholar]
- 6.Foubert P, Varner JA. Integrins in tumor angiogenesis and lymphangiogenesis. Methods Mol Biol. 2012;757:471–86. doi: 10.1007/978-1-61779-166-6_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tchaicha JH, Reyes SB, Shin J, Hossain MG, Lang FF, McCarty JH. Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by β8 integrin. Cancer Res. 2011;71:6371–81. doi: 10.1158/0008-5472.CAN-11-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reardon DA, Nabors LB, Stupp R, Mikkelsen T. Cilengitide: an integrin-targeting arginine-glycine-aspartic acid peptide with promising activity for glioblastoma multiforme. Expert Opin Investig Drugs. 2008;17:1225–35. doi: 10.1517/13543784.17.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker GC. Integrins: molecular targets in cancer therapy. Curr Oncol Rep. 2006;8:96–103. doi: 10.1007/s11912-006-0043-3. [DOI] [PubMed] [Google Scholar]
- 10.Cordes N, Park CC. beta1 integrin as a molecular therapeutic target. Int J Radiat Biol. 2007;83:753–60. doi: 10.1080/09553000701639694. [DOI] [PubMed] [Google Scholar]
- 11.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. 1999;5:662–8. doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- 13.Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9:665–74. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 14.Hazlehurst LA, Landowski TH, Dalton WS. Role of the tumor micro-environment in mediating de novo resistance to drugs and physiological mediators of cell death. Oncogene. 2003;22:7396–402. doi: 10.1038/sj.onc.1206943. [DOI] [PubMed] [Google Scholar]
- 15.Matsunaga T, Fukai F, Miura S, Nakane Y, Owaki T, Kodama H, et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia. 2008;22:353–60. doi: 10.1038/sj.leu.2405017. [DOI] [PubMed] [Google Scholar]
- 16.Leenders WP, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, et al. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res. 2004;10:6222–30. doi: 10.1158/1078-0432.CCR-04-0823. [DOI] [PubMed] [Google Scholar]
- 17.Azam F, Mehta S, Harris AL. Mechanisms of resistance to antiangiogenesis therapy. Eur J Cancer. 2010;46:1323–32. doi: 10.1016/j.ejca.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 18.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12:233–42. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janouskova H, Maglott A, Leger DY, Bossert C, Noulet F, Guerin E, et al. Integrin α5β1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high grade glioma. Cancer Res. 2012;72:3463–70. doi: 10.1158/0008-5472.CAN-11-4199. [DOI] [PubMed] [Google Scholar]
- 21.Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE. 2009;4:e5857. doi: 10.1371/journal.pone.0005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Lay M, Jahangiri A, Carbonell WS, Hu YL, Tsao S, Tom MW, et al. Microarray analysis verifies two distinct phenotypes of glioblastomas resistant to anti-angiogenic therapy. Clin Cancer Res. 2012;18:2930–42. doi: 10.1158/1078-0432.CCR-11-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons M, Messent AJ, Humphries JD, Deakin NO, Humphries MJ. Quantification of integrin receptor agonism by fluorescence lifetime imaging. J Cell Sci. 2008;121:265–71. doi: 10.1242/jcs.018440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu YL, Delay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72:1773–83. doi: 10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of anti-angiogenic therapy resistance. Clin Cancer Res. 2013 Mar 21; doi: 10.1158/1078-0432.CCR-12-1281. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wehrle-Haller B. Analysis of integrin dynamics by fluorescence recovery after photobleaching. Methods Mol Biol. 2007;370:173–202. doi: 10.1007/978-1-59745-353-0_13. [DOI] [PubMed] [Google Scholar]
- 27.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–33. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 28.Carbonell WS, Murase S, Horwitz AF, Mandell JW. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: an in situ time-lapse confocal imaging study. J Neurosci. 2005;25:7040–7. doi: 10.1523/JNEUROSCI.5171-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011;18:829–40. doi: 10.1038/cdd.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ljubimova JY, Fugita M, Khazenzon NM, Das A, Pikul BB, Newman D, et al. Association between laminin-8 and glial tumor grade, recurrence, and patient survival. Cancer. 2004;101:604–12. doi: 10.1002/cncr.20397. [DOI] [PubMed] [Google Scholar]
- 31.Ljubimova JY, Fujita M, Khazenzon NM, Ljubimov AV, Black KL. Changes in laminin isoforms associated with brain tumor invasion and angiogenesis. Front Biosci. 2006;11:81–8. doi: 10.2741/1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall JF, Rutherford DC, McCartney AC, Mitjans F, Goodman SL, Hart IR. Alpha v beta 1 is a receptor for vitronectin and fibrinogen, and acts with alpha 5 beta 1 to mediate spreading on fibronectin. J Cell Sci. 1995;108 (Pt 3):1227–38. doi: 10.1242/jcs.108.3.1227. [DOI] [PubMed] [Google Scholar]
- 33.Clarke JL, Ennis MM, Yung WK, Chang SM, Wen PY, Cloughesy TF, et al. Is surgery at progression a prognostic marker for improved 6-month progression-free survival or overall survival for patients with recurrent glioblastoma? Neuro Oncol. 2011;13:1118–24. doi: 10.1093/neuonc/nor110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koh MY, Lemos R, Jr, Liu X, Powis G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011;71:4015–27. doi: 10.1158/0008-5472.CAN-10-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park CC, Zhang HJ, Yao ES, Park CJ, Bissell MJ. Beta1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Res. 2008;68:4398–405. doi: 10.1158/0008-5472.CAN-07-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S, Bell K, Mousa SA, Varner JA. Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am J Pathol. 2000;156:1345–62. doi: 10.1016/s0002-9440(10)65005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grzesiak JJ, Tran Cao HS, Burton DW, Kaushal S, Vargas F, Clopton P, et al. Knockdown of the β(1) integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. Int J Cancer. 2011;129:2905–15. doi: 10.1002/ijc.25942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang C, Park CC, Hilsenbeck SG, Ward R, Rimawi MF, Wang YC, et al. β1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011;13:R84. doi: 10.1186/bcr2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesniak D, Xu Y, Deschenes J, Lai R, Thoms J, Murray D, et al. Beta1-integrin circumvents the antiproliferative effects of trastuzumab in human epidermal growth factor receptor-2-positive breast cancer. Cancer Res. 2009;69:8620–8. doi: 10.1158/0008-5472.CAN-09-1591. [DOI] [PubMed] [Google Scholar]
- 40.Damiano JS, Dalton WS. Integrin-mediated drug resistance in multiple myeloma. Leuk Lymphoma. 2000;38:71–81. doi: 10.3109/10428190009060320. [DOI] [PubMed] [Google Scholar]
- 41.Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR) Oncogene. 2000;19:4319–27. doi: 10.1038/sj.onc.1203782. [DOI] [PubMed] [Google Scholar]
- 42.Aoudjit F, Vuori K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene. 2001;20:4995–5004. doi: 10.1038/sj.onc.1204554. [DOI] [PubMed] [Google Scholar]
- 43.Pontiggia O, Sampayo R, Raffo D, Motter A, Xu R, Bissell MJ, et al. The tumor microenvironment modulates tamoxifen resistance in breast cancer: a role for soluble stromal factors and fibronectin through β1 integrin. Breast Cancer Res Treat. 2012;133:459–71. doi: 10.1007/s10549-011-1766-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oshita F, Kameda Y, Hamanaka N, Saito H, Yamada K, Noda K, et al. High expression of integrin beta1 and p53 is a greater poor prognostic factor than clinical stage in small-cell lung cancer. Am J Clin Oncol. 2004;27:215–9. doi: 10.1097/01.coc.0000054894.64867.80. [DOI] [PubMed] [Google Scholar]
- 45.Lieberman DM, Laske DW, Morrison PF, Bankiewicz KS, Oldfield EH. Convection-enhanced distribution of large molecules in gray matter during interstitial drug infusion. J Neurosurg. 1995;82:1021–9. doi: 10.3171/jns.1995.82.6.1021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.