

Summary
Allogeneic hematopoietic cell transplantation (allo-HCT) is increasingly being performed to treat patients with hematologic malignancies. However, separating the beneficial graft-versus-tumor (GVT) or graft-versus-leukemia effects from graft-versus-host disease (GVHD) has been difficult and remains a significant challenge toward improving therapeutic efficacy and reducing toxicity of allo-HCT. GVHD is induced by donor T cells that also mediate potent anti-tumor responses. However, despite the largely shared effector mechanisms, extensive animal studies have demonstrated the potential of dissociating the GVT effect from GVHD. Also in many clinical cases, long-term remission was achieved following allo-HCT, without significant GVHD. A better mechanistic understanding of the immunopathophysiology of GVHD and GVT effects may potentially help to improve allo-HCT as well as maximize the benefit of GVT effects while minimizing GVHD. In this article, we review the role of IFN-γ in regulation of alloresponses following allo-HCT, with a focus on the mechanisms of how this cytokine may separate GVHD from GVT effects.
Keywords: allogeneic hematopoietic cell transplantation, DLI, GVHD, GVT, IFN-γ, leukemia
Overview of IFN-γ in immune regulation
Interferon-γ (IFN-γ), the only type II IFN, can be produced by CD4+ T-helper type 1 (Th1) cells, activated CD8+ T cells, natural killer (NK) cells, natural killer T (NKT) cells, B cells, and antigen-presenting cells (APCs) (1). IFN-γ exhibits pleiotropic activities on a wide range of cell types and plays a central role in host defense by influencing both innate and adaptive immunity. During innate immune responses, IFN-γ plays an important role in eliminating intracellular pathogens via activation of macrophages and NK cells (2, 3). With regard to adaptive immune responses, IFN-γ is crucial for CD4+ Th1 cell differentiation and CD8+ T-cell proliferation during the primary and expansion phase, and for controlling the overexpansion of activated T cells in the contraction phase (4, 5).
IFN-γ receptor (IFN-γR) is composed of two heterodimeric chains: IFN-γR1 and IFN-γR2. IFN-γR1 or the α ligand-binding chain is ubiquitously expressed in almost every lymphoid and non-lymphoid cell type. IFN-γR2 or the β signal-transducing chain is highly expressed on myeloid and B cells, and variably expressed on T cells, depending on the activation status and subset commitment (6, 7). IFN-γ induces transcriptional activation of IFN-γ-inducible genes predominantly via the JAK/STAT1 (Janus kinase/signal transducer and activator of transcription 1) signaling pathway. IFN-γ-JAK-STAT1 activation may mediate proliferative or anti-proliferative/apoptotic effects depending on the levels of IFN-γR cell surface expression. In T cells, IFN-γ binding to IFN-γR complex on naive T cells leads to the activation of the JAK-STAT1 signaling pathway and subsequent upregulation of the downstream transcription factor T-bet prior to IL-12Rβ2 expression and IL-12-dependent STAT4 activation, which are required for T-cell differentiation into the Th1 subset (8–11). Activation of STAT1 negatively regulates STAT3 activated by IL-6, IL-17, IL-21, and IL-23, as well as STAT6 activated by IL-4 (12). Upon activation, T-bet suppresses the development of Th2 and Th17 cells by impairing the function of GATA-binding protein 3 (GATA3) and RAR-related orphan receptor γ (RORγt), allowing full maturation of Th1 cells (13–15). Once differentiated, Th1 effectors quickly downregulate surface expression of IFN-γR2 as a potential mechanism to prevent apoptosis induced by strong activation of STAT1 and continue to proliferate in the presence of high levels of IFN-γ (7, 16). Accordingly, IFN-γR2-transgenic mice show impaired Th1 responses (6) and forced expression of IFN-γR2 on human T cells makes them susceptible to IFN-γ-induced apoptosis. Activated antigen-specific CD8+ T cells also show downregulation or loss of IFN-γR2 cell surface expression (17). In contrast to IFN-γ-producing T cells, other T-cell subsets, B cells, and myeloid cells display constitutive IFN-γR2 expression and susceptibility to IFN-γ-STAT1-mediated apoptosis (6, 7, 18, 19).
Th1 cells may regain IFN-γR2 expression following T-cell receptor engagement in the absence of APCs or deprivation of IL-2, restoring the susceptibility to IFN-γ-STAT1-induced apoptosis (18, 20, 21). In addition to ligand-dependent loss of IFN-γR2 in activated T cells, T cells that are unable to produce and respond to IFN-γ can display ligand-independent internalization of IFN-γ R2 (7, 20). It has been implicated that anti-inflammatory signals prompt internalization of IFN-γR2, resulting in a T-cell population refractory to IFN-γR signaling. In contrast, exposure to inflammatory signals may induce IFN-γR2 expression on the surface of T cells, making these cells sensitive to IFN-γR-STAT1 signal-mediated apoptosis (22).
IFN-γ signaling plays a crucial and complex role in the regulation of immune responses. Recent studies have indicated that the immune regulatory effect of IFN-γ can also be mediated by its signaling in regulatory T cells (Tregs) (see discussion below). Moreover, IFN-γ signaling in non-hematopoietic cells plays a critical role in regulating T-cell responses in disease settings (23).
Role of IFN-γ in GVHD
Allogeneic hematopoietic cell transplantation (allo-HCT) or bone marrow transplantation (allo-BMT) is a potentially curative treatment for many hematologic malignancies (24, 25). In spite of reduced leukemic relapse rates resulting from graft-versus-tumor (GVT) effects following HLA-mismatched compared to HLA-identical transplants, the high incidence of intractable GVHD presents an enormous obstacle to HLA-mismatched allo-HCT. GVHD is induced mainly by donor T cells that recognize the mismatched host major histocompatibility complex (MHC) and minor histocompatibility antigens (miHA) and is characterized by damage in multiple organs/tissues, such as the gastrointestinal tract, liver, skin, mucosa, and lung.
Role of IFN-γ in GVHD in irradiated allo-HCT recipients
Acute GVHD-associated gut damage has been proposed to be mediated by Th1-type immune responses characterized by large amounts of IFN-γ and IL-2 production during T-cell activation (26). Endotoxin or lipopolysaccharide (LPS) from Gram-negative bacteria plays an important role in GVHD pathogenesis by stimulating inflammatory cytokine production of innate immune cells, particularly, in allogeneic recipients with damage in the gastrointestinal tract induced by the conditioning regimen (27). IFN-γ produced by activated donor T cells has been shown to exacerbate gut GVHD by promoting the apoptosis of intestinal epithelial crypt cells and acting synergistically with LPS to stimulate cytokine production (predominantly TNF-α) from innate immune cells in the intestine (28). However, IFN-γ was also found to mediate protection against gut GVHD in other studies (29–31) (see discussion below).
An increasing number of reports demonstrate that IFN-γ may play a protective role in recipients of allo-HCT. We have previously shown that IFN-γ mediates a protective effect against acute GVHD in lethally irradiated mice receiving a single injection of exogenous IL-12 on the day of allo-HCT (32–35). IL-12 is a potent immunostimulatory cytokine and inducer of Th1 cell activity as well as cytotoxic T-lymphocyte and NK cell function (36). Early reports using parent-to-non-irradiated F1 allo-HCT models showed that IL-12 stimulates the development of acute GVHD, but inhibits chronic GVHD by converting a Th2 response to a Th1 response (37, 38). Paradoxically, a single injection of IL-12 at the time of allo-HCT mediates marked protection against acute GVHD in lethally irradiated recipients (32, 33). In addition to resulting in a significant delay in GVHD-associated mortality, IL-12 treatment also reduced the extent of early weight loss and other clinical manifestations of acute GVHD. This protective effect of IL-12 against acute GVHD was demonstrated in different murine allo-HCT models, including multiple fully MHC plus miHA-mismatched (e.g. B10.A-to-C57BL/6, C3H-to-C57BL/6, and BALB/c-to-C57BL/6) and haploidentical C57BL/6 (H-2b)- or CBD2F1 (H-2kxd)-to-B6D2F1 (H-2bxd) strain combinations (34, 35, authors’ unpublished observation). In allo-HCT recipients treated with a single injection of IL-12, serum IFN-γ levels were markedly increased at days 2 and 3 post-HCT but became almost undetectable at day 4. In contrast, IFN-γ was undetectable in untreated controls at days 2 and 3 and reached a high level at day 4 (predominantly produced by alloreactive donor T cells) post-HCT (33). Furthermore, IL-12-mediated GVHD protection was completely eliminated following treatment with neutralizing anti-IFN-γ mAb R4-6A2 (33). In some experiments, IL-12 even accelerated GVHD when higher doses of R4-6A2 were administered. As R4-6A2 treatment only slightly delayed the onset of GVHD-associated mortality in non-IL-12-treated controls, we conclude that IFN-γ is required for IL-12-mediated GVHD protection but not for the induction of GVHD.
IFN-γ knockout (KO) mice provide a powerful tool for evaluating the role of IFN-γ in the pathogenesis of GVHD. Using this model, two independent groups have demonstrated for the first time that neither donor- nor host-derived IFN-γ is required for the development of lethal acute GVHD in mice receiving MHC-mismatched allo-HCT (39, 40). Indeed, IFN-γ can be protective for allo-HCT recipients. In a single MHC class II-mismatched murine allo-HCT model, IFN-γ significantly inhibits the induction of GVHD by donor CD4+ T cells (41). Co-injection of CD4+ T cells along with bone marrow cells from IFN-γ-deficient C57BL/6 mice into lethally irradiated B6.C-H2bm12 (H2-Ab1bm12; referred to as bm12) mice (disparate at class II) led to 100% mortality by 20 days. In contrast, bm12 mice receiving CD4+ T cells and bone marrow cells from wild-type C57BL/6 mice survived long term (41). IFN-γ also inhibited GVHD induced by donor CD8 T cells. Infusion of IFN-γ-deficient allogeneic bone marrow and CD4-depleted spleen cells induced severe lethal GVHD, while recipients of similar allo-HCT from wildtype donors developed only mild GVHD (30, 42).
We have used IFN-γ KO BALB/c and C57BL/6 mice to determine whether the IFN-γ that mediates the protective effect of IL-12 is donor or host derived. Treatment with IL-12 significantly prolonged the survival in both wildtype and IFN-γ KO C57BL/6 mice receiving allo-HCT from wildtype BALB/c mice. However, similar treatment did not protect against GVHD in wildtype C57BL/6 mice receiving allo-HCT from IFN-γ KO BALB/c donors, demonstrating that donor-derived IFN-γ is required for the protective effect of IL-12. Fas-mediated donor CD4+ T-cell apoptosis is one of the mechanisms involved in the inhibition of GVHD by IL-12 (34), suggesting that IFN-γ may play an important role in IL-12-induced Fas expression and apoptosis of host-reactive donor T cells during GVHD induction. IL-18, another potent inducer of IFN-γ, has also been reported to inhibit GVHD in lethally irradiated allo-HCT recipients (43, 44). Similar to the effect of IL-12 in inhibiting GVHD, the effect of IL-18 is also IFN-γ-dependent and requires expression of Fas on donor T cells (43).
In GVHD settings, donor CD4+ T cells differentiate into Th2 and Th17 phenotypes in the absence of IFN-γ, IFN-γR, or T-bet, and preferentially accumulate in lung and skin at least partially due to concomitant changes in the profile of tissue-homing chemokine receptor expression (23, 29, 45–48). Mice receiving allogeneic T cells lacking IFN-γ or T-bet showed accumulation of Th17 cells in the lung and the development of idiopathic pneumonia syndrome (IPS) (29, 45). However, deletion of the IL-17 gene in IFN-γ-deficient donor T cells did not significantly ameliorate injuries in the lung tissue (29), suggesting that the lung GVHD in recipients of IFN-γ-deficient allo-HCT is mediated predominantly by Th2-type alloresponses. In contrast, skin infiltration by donor T cells and the associated injuries were significantly reduced in mice receiving allogeneic T cells lacking both IFN-γ and IL-17 compared to mice receiving allogeneic T cells lacking only IFN-γ, indicating that Th17 cells play an important role in the induction of skin GVHD (29). Interestingly, although lack of IFN-γR signaling in T cells also leads to augmented Th2- and Th17-type responses, IFN-γR-deficient T cells are significantly less effective in inducing GVHD compared to IFN-γ-deficient and even wildtype T cells (29, 48, 49). Mice receiving IFN-γR-deficient allogeneic T cells experience significantly less GVHD-associated lung injury compared to mice receiving IFN-γ-deficient or wildtype allogeneic T cells (23, 29, 48). Of note, although both IFN-γ- and IFN-γR-deficient T cells default to a Th2 phenotype (e.g. producing IL-4), the latter produces IFN-γ (50). It has been suggested that the reduced GVHD in mice receiving IFN-γR-deficient allo-HCT is due to impaired CXCR3 expression and trafficking of donor T cells into GVHD target organs (49). However, IFN-γ produced by IFN-γR-deficient donor T cells is indeed protective, as in vivo neutralization of IFN-γ by anti-IFN-γ antibodies markedly exacerbated lung GVHD in recipients of IFN-γR-deficient allo-HCT (48). This study also suggests that the protective effect of donor-derived IFN-γ can also be mediated by its interaction with recipient cells. Both wildtype and IFN-γR-deficient allo-HCT significantly increased lung GVHD in chimeras with defective IFN-γ signaling compared to those with intact IFN-γ signaling in non-hematopoietic cells, regardless of whether or not IFN-γ signaling is intact in the recipient hematopoietic cells (29). This indicates that IFN-γ signaling in recipient non-hematopoietic cells, but not in hematopoietic cells, is critical for IFN-γ-mediated inhibition of lung GVHD.
Role of IFN-γ in GVHD in non-conditioned allo-HCT recipients
In a non-irradiated C57BL/6-to-B6D2F1 allo-HCT model, the GVH response is associated with a massive increase in IFN-γ production (51, 52). Administration of IFN-γ-deficient T cells or neutralization of IFN-γ in this model resulted in a delay in GVHD mortality that was associated with impaired elimination of recipient cells and chronic GVHD-like features including lymphoproliferation, autoantibody production, and a lupus-like renal disease (53–55). It has been shown that the Fas/FasL but not perforin pathway is required to eliminate host hematopoietic cells (56). Complete elimination of IFN-γ by injection of neutralizing antibody against IFN-γ in non-conditioned B6D2F1 mice receiving allo-HCT from IFN-γ-deficient C57BL/6 donors resulted in an enhanced expansion of donor CD8+ T cells with increased expression of the activation marker CD44. However, these T cells, due to impaired FasL expression, exhibit a significantly reduced capacity to eliminate host hematopoietic cells (57). Unlike FasL expression, perforin gene expression and perforin-mediated cytotoxicity are only marginally affected in the absence of IFN-γ (57). Of note, in the non-irradiated allo-HCT models discussed above, the recipients were transplanted with donor lymph node and spleen cells without bone marrow cells, so that the inoculum contains no or minimal numbers of hematopoietic stem cells (HSCs). Therefore, hematopoietic failure due to destruction of recipient hematopoietic cells is a likely cause of early mortality in these models and the delay in mortality by IFN-γ elimination could be due to impaired Fas/FasL cytotoxicity. As the recipients of allo-HCT from IFN-γ-deficient donors had greater weight loss and increased destruction of parenchymal GVHD target tissues than those receiving allo-HCT from wildtype donors, IFN-γ is likely to be protective against tissue GVHD in non-irradiated recipients.
Delayed administration of allogeneic donor lymphocyte infusion (DLI) without conditioning treatment in established mixed allogeneic hematopoietic chimeras has been shown to eliminate recipient hematopoietic cells [referred to as lymphohematopoietic GVH response (LGVHR)] without inducing severe GVHD (24, 58). The ability of DLI to mediate LGVHR without severe GVHD in established mixed chimeras is largely due to the lack of conditioning-induced tissue inflammation, an important checkpoint controlling the migration of GVH-reactive T cells into the epithelial GVHD target tissues (59). In this model, mixed chimeras can be prepared by injection of a mixture of T-cell-depleted donor and recipient bone marrow cells or by non-myeloablative conditioning and allo-BMT, followed 5–8 weeks later by administration of allogeneic donor spleen cells (as DLI) without conditioning. Allogeneic DLI from IFN-γ-deficient donors was significantly less effective compared to that from wildtype donors in eliminating recipient hematopoietic cells in mixed chimeras, indicating a critical role for DLI cell-produced IFN-γ in the induction of LGVHR (31). Interestingly, the reduced LGVHR was associated with significantly increased parenchymal tissue damage, loss of body weight, and mortality in chimeras receiving allogeneic DLI from IFN-γ-deficient donors.
Separation of LGVHR and the GVHR targeting parenchymal tissues by IFN-γ is not a specific phenomenon in non-conditioned allo-HCT. In a sublethally irradiated C57BL/6-to-B6D2F1 allo-HCT model (Fig. 1), mice receiving wildtype donor splenocytes alone died rapidly, whereas those receiving wildtype donor splenocytes plus bone marrow survived long-term (31). Recipients in both groups showed rapid elimination of host hematopoietic cells but minimal parenchymal tissue injury. However, mice receiving allo-HCT from IFN-γ-deficient donors died rapidly regardless of whether or not donor marrow cells were given, and they exhibited severe parenchymal injury but prolonged survival of host hematopoietic cells (31). Similar results were observed in a C57BL/6→bm12 combination, in which IFN-γ elimination significantly accelerated GVHD mortality in lethally irradiated recipients of allogeneic donor marrow and T cells, but reduced the death rate in sublethally irradiated mice receiving allogeneic T cells alone (41). Together, these studies suggest that IFN-γ inhibits GVHD and the associated parenchymal tissue damage, while promoting LGVHR in allo-HCT recipients. The role of IFN-γ in selectively eliminating recipient hematopoietic cells (i.e. LGVHR) can explain its effect of facilitating GVT responses (see discussion below).
Fig. 1. Interferon-γ (IFN-γ) promotes LGVHR while attenuating tissue graft-versus-host disease (GVHD).

Sublethally irradiated mice receiving allogeneic donor spleen cells (SPC) alone from wildtype (WT) donors succumb to hematopoietic failure with moderate tissue GVHD, while most mice receiving a similar number of WT SPCs plus bone marrow (BM) cells survive long-term. In contrast, administration of allogeneic SPCs from IFN-γ-deficient donors induces severe tissue injury, leading to death prior to complete elimination of host hematopoietic cells, and addition of donor BM cells cannot prevent death despite significantly improved hematopoiesis.
Possible mechanisms for inhibition of GVHD by IFN-γ
IFN-γ is a critical cytokine in immune regulation that is involved in T-cell priming, proliferation, contraction, effector function development, and memory generation. The wide range of IFN-γR expression enables IFN-γ to directly regulate the function of almost every lineage of immune cells, such as T cells, B cells, NK cells, APCs, and other myeloid immune cell populations. Furthermore, IFN-γ may also control T-cell responses within tissues by signaling through IFN-γ R expressed on parenchymal cells.
IFN-γ inhibits donor T-cell expansion by promoting apoptosis and suppressing proliferation
IFN-γ plays an important role in the maintenance of T-cell homeostasis. Although IFN-γ has been shown to promote T-cell expansion and memory formation (60), this cytokine also induces contraction of the effector T-cell pool. IFN-γ promotes Fas-mediated activation-induced cell death of CD4+ T cells (20) and restrains CD4+ T-cell activation in vitro by promoting caspase-8-dependent apoptosis through the transcriptional activation of STAT1, one of the major transcription factors induced by IFN-γR (4). The absence of IFN-γ leads to enhanced expansion and reduced death of antigen-specific CD8+ T cells in a Listeria infection model (61). The proapoptotic effect of IFN-γ has been implicated in the elimination of activated CD4+ and CD8+ T cells and contraction of the effector T-cell pool (62–64).
IFN-γ elimination is associated with a significant increase in donor T cells in allo-HCT settings (30, 42), underscoring the involvement of IFN-γ in controlling the expansion of alloreactive T cells. Previous studies revealed that IL-12 and IL-18 treatment promotes Fas-mediated apoptosis of recipient antigen-activated donor T cells via an IFN-γ-dependent mechanism (34, 39, 43). Significantly inhibited/delayed donor CD8+ T-cell apoptosis in recipients of allo-HCT from IFN-γ-deficient donors compared to those receiving allo-HCT from wildtype donors provides direct evidence that IFN-γ promotes apoptosis of alloreactive T cells in GVHD settings (30).
T-cell proliferation is governed by the ordered activation of cyclin-dependent kinases (CDKs). One of the CDK inhibitors p27Kip1 is an important negative regulator of both T-cell proliferation and memory T-cell generation (65–68). It has been shown that IFN-γ inhibits cell proliferation by preventing growth factor-induced downregulation of p27Kip1 (69). Moreover, IFN-γ has also been found to inhibit cell cycle progression of activated CD4+ T cells by stimulating nitric oxide production by macrophages (70). These observations indicate a role for IFN-γ in regulation of T-cell division, and raised the possibility that blockade of IFN-γ may result in over-proliferation of donor T cells in allogeneic recipients. Using a CD4+ cell-depleted allo-HCT model, we have shown that IFN-γ elimination significantly accelerates the cell division of alloreactive donor CD8+ T cells (30).
IFN-γ eliminates alloreactive T cells in GVHD target tissues by interacting with recipient non-hematopoietic cells
IFN-γ has been shown to regulate T-cell expansion, contraction, and differentiation in response to peptide vaccination via mechanisms independent of IFN-γR signaling in T cells (71–73). Studies using bone marrow chimeras with IFN-γR expression on only hematopoietic or non-hematopoietic cells demonstrated that following allo-HCT, the chimeras lacking IFN-γ signaling in lung parenchymal cells developed a significant increase in lung GVHD compared to mice expressing normal levels of IFN-γR in the lung (23). The programmed cell death-1 (PD-1)/programmed cell death-1 ligand 1 (PD-L1) pathway provides critical negative regulation in the process of T-cell activation (74). Blockade of PD-1 engagement accelerates GVHD mortality in an IFN-γ-dependent mechanism (75). Additional studies demonstrated that inhibition of lung GVHD by IFN-γ is largely attributed to IFN-γ-induced upregulation of PD-L1 expression in lung tissues (29), leading to the elimination (i.e. apoptosis) of lung-infiltrating alloreactive donor T cells (74) (Fig. 2). IFN-γ has also been shown to upregulate PD-L1 expression on hepatic stellate (76) and endothelial cells (77), and to ameliorate liver GVHD by inducing apoptosis of tissue-infiltrating T cells in allo-HCT recipients (29). However, the involvement of IFN-γ in mediating gut GVHD has been controversial. As discussed above, IFN-γ may augment gut GVHD by inducing intestinal epithelial crypt cell apoptosis and by stimulating production of inflammatory cytokines by innate immune cells in the intestine (28). However, an association was also reported between the lack of donor-derived IFN-γ and increased colon GVHD (30, 31) (Fig. 2). Furthermore, IFN-γ signaling in gut tissue cells was found to induce PD-L1 expression that protects against tissue damage by eliminating gut-infiltrating alloreactive T cells (29).
Fig. 2. Interferon-γ (IFN-γ) inhibits graft-versus-host disease (GVHD) through its signaling in parenchymal tissues cells.

IFN-γ signaling in non-hematopoietic cells within GVHD target tissues upregulates PD-L1 expression, leading to apoptosis of tissue-infiltrating alloreactive T cells and protection against tissue injury. Compared to other GVHD target organs, intestine is more sensitive to apoptosis induced by IFN-γ and IFN-γ-stimulated inflammation. However, IFN-γ elimination has been shown to increase intestinal injury in allo-HCT recipients, suggesting that the IFN-γ-mediated protective effect may outweigh its adverse effects. Dead cells are indicated by a black colored nucleus.
Role of IFN-γ in donor T-cell migration
Chemokine/chemokine receptor interactions and adhesion molecules play critical roles in GVHD pathogenesis by regulating tissue migration of alloreactive donor T cells (78). IFN-γ is critical in the induction of Th1-associated chemokines CXCL9 (MIG), CXCL10 (IP-10), and CXCL11 (I-TAC), as well as their receptor CXCR3 expression on activated T cells (79–81). Rapid upregulation of INF-γ induced chemokines CXCL9 and CXCL10 in the liver is involved in the initiation of hepatic GVHD following allo-HCT (82). Recent studies using IFN-γR- or CXCR3-deficient mice indicate that the recruitment of CXCR3+ CD4 T cells by CXCL9, CXCL10, and CXCL11 is important in the induction of GVHD (49, 83). However, the pattern of chemokine and chemokine receptor expression differs depending on the types of Th cell responses, and therefore, CXCR3 or CXCR3-binding chemokines may not play a significant role in GVHD induced by Th2 and Th17 cells. It has been shown that the recruitment of activated Th2 and Th17 cells into GVHD target organs (e.g. lung and skin) is mediated by the expression of CCR3, CCR4, and CCR6, and concomitant expression of the corresponding chemokine ligands in the GVHD target tissues (29).
Role of IFN-γ in Treg function
The role of IFN-γ in the generation and function of Tregs remains largely unexplored. In an in vitro culture system involving exposure of CD4+ T cells to allogeneic immature DCs, IFN-γ was found to preferentially induce apoptosis in the non-Treg population and promoted conversion of non-Tregs into Foxp3+ Tregs. This led to an enrichment of Tregs that could inhibit allograft rejection in a donor-specific manner (84). Using a collagen-induced arthritis (CIA) model, an accelerated onset and more severe form of CIA that is associated with a functional defect in CD4+CD25+ Tregs has been demonstrated in mice lacking IFN-γR (85). Similarly, in a spontaneous autoimmune encephalomyelitis (EAE) murine model, deficiency in STAT1 significantly accelerates disease development due to a reduced number and impaired function of CD4+CD25+ Tregs in these mice (86). These studies suggest that IFN-γ signaling in Tregs is critical for their development and/or function. However, it has also been reported that IFN-γ signaling has no effect on the suppressive function, but negatively regulates the expansion of Tregs (87, 88, authors’ unpublished observation). Tregs deficient in STAT1 (87, 88) or IFN-γR (our unpublished observation) exhibit increased expansion both in vitro and in vivo, and no defect in the immunosuppressive capacity compared to wildtype Tregs. Furthermore, administration of STAT1-deficient Tregs (expanded in vitro) suppressed GVHD in a similar manner as wildtype Tregs (87). Together, these studies indicate that the role of IFN-γ signaling in Treg function depends on the particular disease setting.
Regulation of Treg function by IFN-γ signaling is further complicated by the role of IFN-γ in Treg polarization. Similar to conventional T cells, natural Tregs (nTregs) can also be polarized into distinct effector Treg (eTreg) subsets that co-opt expression of different Th subset-specific transcription factors (e.g. T-bet for Th1-Tregs, GATA3 for Th2-Tregs, and RORγt for Th17-Tregs), and endowed with the properties optimized for suppression of T-cell responses elicited under the same inflammatory conditions (89). IFN-γ, as a major Th1-type cytokine, has been shown to promote the differentiation of T-bet+CXCR3+ eTregs that can effectively suppress conventional Th1 cell responses (90–92). Furthermore, Tregs could also be specialized in a site-specific manner. In mice challenged with intracellular pathogens, IL-27-dependent Th1-type eTregs are specialized to control Th1 responses in the primary sites of infection, whereas eTregs suppressing Th1 responses in the periphery are dependent on IFN-γ (90). These studies not only provide a possible explanation for why the role of IFN-γ in Treg function is different depending on the particular disease setting but also suggest the possibility of preventing GVHD by selectively promoting Tregs that are specialized to inhibit alloimmunity in GVHD target tissues.
In vivo studies using IFN-γ-deficient mice or neutralizing antibody against IFN-γ revealed a close association of IFN-γ elimination with impaired development and function of Tregs. IFN-γ-deficient mice exhibit significantly impaired generation and function of CD4+CD25+ Tregs, and blocking IFN-γ dramatically reduced the suppressive effect of alloantigen-specific Tregs (93). IFN-γ was also found to play a critical role in conversion of CD4+CD25− T cells to CD4+ Tregs, and the lack of this cytokine led to an increased susceptibility to EAE (94). Moreover, IFN-γ production by alloantigen-reactive CD4+CD25+ Tregs is critical for their ability to prevent allograft rejection or GVHD (88, 93, 95). These studies indicate that IFN-γ may potentially attenuate GVHD through its role in regulating Treg generation and function, and that a reduction in the number and/or function of Tregs might contribute to the exacerbation of GVHD in the absence of IFN-γ. However, it should be noted that IFN-γ can mediate GVHD protection independent of Tregs, at least in mice receiving CD4+ T-cell-depleted allo-HCT (30).
Role of IFN-γ in GVT effects
An important benefit of allo-HCT is that donor T cells mediate strong GVT or graft-versus-leukemia (GVL) effects. However, these effects must be achieved without severe GVHD. IFN-γ production is essential for tumor eradication by T cells (96). The role of IFN-γ in the induction of GVT effects following allo-HCT was initially revealed by studies in IL-12-treated allo-HCT recipients. Using an EL4 leukemia/lymphoma (H-2b) model, we have shown that a single injection of IL-12 led to simultaneous protection from GVHD- and leukemia-induced mortality in irradiated C57BL/6 mice receiving allo-HCT from A/J (H-2a) donors (97). IFN-γ is also required for the preservation of GVT effects by IL-12, as administration of anti-IFN-γ antibody significantly attenuated the anti-tumor activity of allogeneic T cells in IL-12-treated allo-HCT recipients (97). In murine allo-HCT models, the development of acute GVHD is largely CD4+ T cell-dependent in most fully MHC plus minor antigen-mismatched strain combinations. In these models, acute GVHD cannot be prevented by CD8+ T-cell depletion from the donor graft or induced by injection of substantial numbers of CD4+ T-cell-depleted donor spleen cells (97–100). However, donor CD8+ but not CD4+ T cells are required for the induction of GVT effects against EL4, an MHC class II-negative T-cell lymphoma (97, 98). Together, these studies indicate that IFN-γ is required for the optimal induction of CD8+ T-cell-mediated GVL effects and inhibition of CD4+ T-cell-induced GVHD in IL-12-treated allo-HCT recipients.
The role of IFN-γ in the induction of GVT effects was also demonstrated in IFN-γ-deficient allo-HCT models. Using a lethal total body irradiation (TBI)-conditioned CD4+ T-cell-depleted allo-HCT model, we showed that IFN-γ promotes GVT effects against EL4 cells while inhibiting GVHD (42). Similar results were also obtained in a renal cell carcinoma model, in which lethal TBI-conditioned BALB/c mice were transplanted with T-cell-depleted marrow cells and purified CD8+ T cells from wildtype or IFN-γ-deficient C57BL/6 donors. GVT effects against carcinoma cells were achieved in mice receiving wildtype donor cells without severe GVHD, but these effects were completely diminished in those receiving IFN-γ-deficient donor cells (101). In this model, significantly reduced GVT effects were also detected in mice receiving FasL-deficient donor cells, indicating a crucial role of FasL in the optimal induction of GVT effects.
IFN-γ also enhances GVT effects that involve both CD4+ and CD8+ T cells. In a sublethally irradiated mouse allo-HCT model (31), we have shown that donor cell-derived IFN-γ not only inhibits GVHD but also promotes the induction of GVL effects against P815 tumor cells (a mastocytoma cell line), which is attributed to both CD4+ and CD8+ T-cell-mediated anti-tumor responses (102, 103). In vivo and clinical studies related to allo-HCT have shown that delayed administration of DLI in established mixed chimeras induces GVT effects without severe GVHD (24). A20 is a spontaneously derived B-cell lymphoma cell line (generated from BALB/c mice) that has been widely used to assess GVT effects in mice. A20 tumor cells express both MHC class I and class II and are susceptible to anti-tumor alloresponses of both CD4+ and CD8+ T cells (104, 105). We have shown that administration of IFN-γ-deficient DLI in mixed chimeras significantly increases GVHD while markedly reducing GVT effects against A20, compared to the chimeras receiving wildtype DLI (31). Together, these studies demonstrate that IFN-γ promotes GVT effects of both CD4+ and CD8+ T cells, while inhibiting GVHD following allo-HCT.
IFN-γ has also been shown to enhance tumor antigen-specific GVT effects. Post-transplant vaccination with tumor antigen-pulsed DCs achieved significant prostate tumor regression in mice receiving DLI following hematopoietic stem cell transplantation, and this GVT activity requires IFN-γ production by donor cells (101). In addition, recipient lymphocyte infusion (RLI) in established mixed allogeneic chimeras demonstrated anti-tumor responses without the risk of GVHD (24). In this model, the anti-tumor response is likely to be mediated by tumor antigen-specific immune responses, although RLI-induced host-versus-graft response is essential for the induction of anti-tumor effects following RLI. Interestingly, IFN-γ is also critical for RLI-mediated anti-tumor effects (106). Together, these studies demonstrate that IFN-γ is required for the optimal induction of GVT effects while inhibiting GVHD in various models involving different conditioning, transplants, and tumor cells.
Mechanisms by which IFN-γ selectively promotes GVT effects in allo-HCT
The following are possible mechanisms for IFN-γ to promote GVT effects in allo-HCT recipients (Fig. 3).
Fig. 3. Mechanisms of interferon-γ (IFN-γ)-mediated graft-versus-tumor effects.
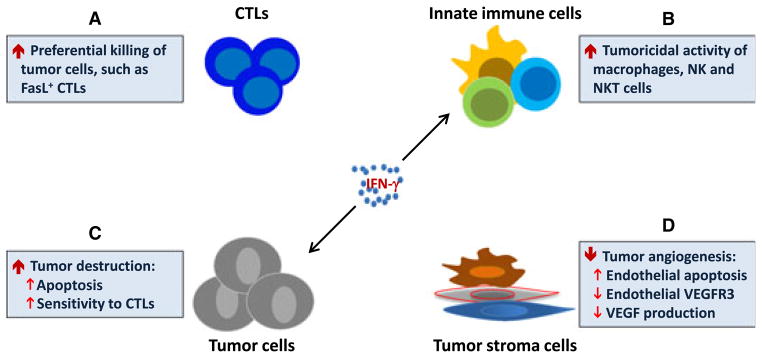
IFN-γ produced by activated T lymphocytes and innate immune cells in the lymphoid system and the tumor promotes anti-tumor/leukemia responses by: (A) promoting cytotoxic T lymphocytes (CTLs) with FasL expression that preferentially destroy tumor and leukemia cells and (B) activating the tumoricidal activity of macrophages, natural killer and natural killer T cells; (C) direct signaling in tumor cells that induces apoptosis and upregulates major histocompatibility complex antigen and Fas expression leading to increased sensitivity to CTLs and (D) inhibiting tumor angiogenesis.
IFN-γ promotes LGVHR
LGVHR is defined as the GVH response that selectively eliminates recipient hematopoietic cells. Following HCT, selective elimination of recipient hematopoietic cells should be safe if donor HSC engraftment is successful (Fig. 1). Studies emerging from animal models and clinical scenarios have shown that conversion from mixed to full allogeneic donor chimerism may occur with limited GVHD. This indicates that LGVHR can be selectively preserved, while the capability to mediate tissue GVHD is suppressed (24, 58, 107, 108). Both animal studies and clinical data have shown that such alloresponses directed against host lymphohematopoietic cells can eliminate host leukemic cells, leading to long-term remissions (24, 107, 108). By promoting LGVHR while inhibiting epithelial tissue injury, IFN-γ shows paradoxical effects on alloreactive T cells (31). Elimination of IFN-γ leads to augmentation of tissue GVHD and mortality, but paradoxically, to reduction in LGVHR and GVT effects. Thus, IFN-γ-mediated preferential promotion of LGVHR is an important mechanism for its ability to enhance the GVT effect while inhibiting GVHD. Thus far, the mechanism for selective promotion or preservation of LGVHR by IFN-γ is not completely understood. IFN-γ signaling-induced PD-L1 upregulation and the associated elimination of alloreactive donor T cells in GVHD target tissues are likely to contribute to the confinement of alloresponses in the lymphohematopoietic compartment.
IFN-γ may also promote LGVHR through direct signaling in hematopoietic cells. IFN-γ production has been associated with bone marrow failure in patients with chronic inflammation (109–113). It has been demonstrated that hematopoietic progenitor cells from patients with aplastic anemia were able to regain the capacity of proliferation in vitro following blockade of IFN-γ (114). Direct cytotoxicity of IFN-γ on bone marrow stem cells was indicated by the observation that rejection of wildtype HSCs occurs in IFN-γR-deficient, but not IFN-γ/IFN-γR double-deficient, syngeneic recipients (115). Studies using IFN-γR-deficient mice indicate that GVHD-related bone marrow failure and lymphoid hypoplasia is also largely dependent on IFN-γ signaling in recipient hematopoietic cells (116, 117). A recent study demonstrated that bone marrow failure following transplantation of allogeneic naive CD4+ T cells was associated with increased CD4+ Th1 cell development within bone marrow and lymphoid tissues, and adoptive transfer of Th1 cells generated during GVHD induced bone marrow failure in the secondary recipients (116). These Th1 cells express CXCR4, which was associated with their accumulation in the lymphohematopoietic tissues. Importantly, bone marrow failure induced by alloreactive Th1 cells was a result of IFN-γ-mediated toxicity (116).
IFN-γ enhances anti-tumor cytotoxic T lymphocyte (CTL) activity
Fas-FasL pathway has been shown to be a predominant mechanism for alloreactive T cells to eliminate recipient hematopoietic cells (56), including leukemia cells (118). FasL also mediates GVT effects against solid tumor cells (101). Elimination of IFN-γ causes impairment of FasL expression on alloreactive CTLs in allo-HCT recipients (57). These studies support the possibility that IFN-γ promotes LGVHR and GVT effects by facilitating FasL-mediated cytotoxicity of alloreactive donor T cells.
IFN-γ signaling in tumor and non-hematopoietic tumor stromal cells
IFN-γ mediates anti-tumor effects by directly inhibiting tumor cell growth as well as by inducing T-cell-mediated anti-tumor responses (119–121). The potent anti-tumor effects of IFN-γ in IFN-γ R-deficient mice inoculated with syngeneic melanoma cells demonstrated a direct in vivo cytotoxicity of IFN-γ on tumors (122, 123). In addition, IFN-γ signaling in tumor cells sensitizes tumor cells to cytotoxic T cells by upregulating the expression of Fas and MHC molecules (124–127). However, IFN-γ signaling in tumor cells may not always favor an anti-tumor response. Although a large body of literature revealed an inhibitory effect of IFN-γ on hematopoiesis in the setting of allo-HCT (109–117), this cytokine has been shown to promote the proliferation of HSCs in the setting of infection (128, 129). Likewise, a recent study showed that leukemia-reactive CTL-released IFN-γ may promote leukemia stem cell (LSC) proliferation (130). It has also been suggested that IFN-γ signaling in tumor cells may lead to a large increase in non-cognate peptide/MHC-I complexes, which in turn reduces the potential of tumor cells to be recognized by tumor antigen-specific CD8+ CTLs (131). Recent studies in mouse and human melanoma revealed that in addition to increasing tumor cell proliferation, IFN-γ also contributes to the tumor immunosuppressive microenvironment by upregulating indole-amine-2,3-dioxygenase and PD-L1 expression (132–134). It is reasonable to assume that the effects of IFN-γ on tumor cells can be influenced by several other factors, such as tumor types, the timing and level of IFN-γ and the presence of other inflammatory cytokines or growth factors. Therefore, further studies are needed to determine the role of IFN-γ in tumor cell proliferation and/or tumor immunoresistance in recipients of allo-HCT.
Many tumor cells, however, are not sensitive to the direct cytotoxic effect of IFN-γ. As discussed earlier, IFN-γ can significantly improve GVT effects against EL4 cells, which are insensitive to IFN-γ-mediated anti-proliferative or cytotoxic effects (42). Although IFN-γ upregulates the expression of Fas and MHC class I on EL4 cells, upon treatment with IFN-γ, EL4 cells show only a moderate increase in susceptibility to the cytotoxicity of allogeneic CD8+ T cells (42). Similarly, the requirement for IFN-γ in RLI-induced anti-tumor effects against A20 leukemia is unlikely to be mediated by direct cytotoxicity of IFN-γ on the tumor cells (106). These results indicate that IFN-γ may promote GVT effects via mechanisms independent of its direct interaction with the tumor cells. To address this question, using virally transduced IFN-γR-deficient HSCs overexpressing Notch 1, we have recently established a primary IFN-γ-unresponsive T-cell leukemia model. Using this model, we have demonstrated that IFN-γ is capable of promoting GVT effects without directly interacting with leukemia cells (135).
It is generally accepted that tumor establishment and growth need support from stromal components, including both hematopoietic cells (e.g. macrophages, granulocytes, B cells, and T cells) and non-hematopoietic cells (e.g. fibroblasts and endothelial cells). IFN-γ has been shown to enhance anti-tumor responses by its signaling in non-hematopoietic tumor stromal cells. Studies using a B16-OVA melanoma model showed that tumor rejection requires expression of IFN-γR in the host cells (136, 137). Growing evidence demonstrates that IFN-γ mediates anti-tumor effects by inhibiting angiogenesis (124, 138, 139). Both endothelial cells and fibroblasts express high levels of IFN-γR (140, 141). IFN-γ-mediated apoptosis of angiogenic endothelial cells and disruption of the tumor vasculature can be induced by its direct signaling in endothelial cells (142). Recently, it was reported that IFN-γ produced within the tumor may suppress the expression of vascular endothelial growth factor receptor 3 in the tumor vessel endothelial cells, thereby inhibiting tumor angiogenesis (143). IFN-γ signaling may also inhibit angiogenesis by suppressing VEGF production by tumor-associated stromal fibroblasts (144). More recently, it was reported that the direct interaction between IFN-γ and stromal cells as well as IFN-γ-induced Fas expression on tumor stroma play an important role in tumor rejection by adoptively transferred tumor-specific T cells (145).
Role of IFN-γ in GVT effects of innate immune cells
NK cells, an important innate immune cell population (146), have the potential to mediate GVL effects while inhibiting GVHD in recipients of allo-HCT (147, 148). IFN-γ mobilizes NK cells and promotes their accumulation in the tumors, thereby reducing tumor metastasis (149, 150). This effect is mediated by direct interaction of IFN-γ with NK cells and requires CXCR3 expression by NK cells (149, 150). Experiments using murine models have shown that CD4/CD8 double negative (DN) spleen cells, which do not mediate detectable anti-tumor activity when injected alone, are required for the optimal induction of GVL effects, which is dependent on donor CD8+ T cells (42). These data indicate that donor DN splenocytes act synergistically with donor CD8+ T cells to augment anti-leukemic alloreactivity. This phenomenon possibly reflects the anti-tumor effects of NK and/or NKT cells. NKT cells straddle the interface of innate and adaptive immunity (146) and play a pivotal role in tumor surveillance (151). IFN-γ may possibly enhance anti-tumor responses by activating NKT cells (152). In addition, both activated NK and NKT cells may aid in separation of GVT effects from GVHD via IFN-γ production. It has been shown that NK cell-produced IFN-γ is essential to anti-tumor immunity of tumor antigen-specific T cells (153). Our studies showed that early IFN-γ production by NK cells contributes to GVHD protection by IL-12 (Wang H, Dey B, Sykes M, Yang YG, unpublished observation).
Macrophages, a major phagocytic cell population mediating innate immunity, exert direct and indirect tumoricidal functions following stimulation with IFN-γ and LPS (154, 155). CD47 serves as a ‘marker of self’ for macrophages, and its interaction with the inhibitory receptor SIRPα (signal regulatory protein α) on macrophages prevents engulfment of autologous hematopoietic cells (156–158). Macrophage activation is controlled by the balance between activating and inhibitory receptor signaling. Interestingly, leukemia cells avoid phagocytosis by upregulating CD47 (159–161). A recent study suggested that CD47-SIRPα interaction also protects solid tumors from phagocytosis (162). These studies underscore the importance of phagocytic cells such as macrophages, in tumor cell rejection. Thus, the action of IFN-γ toward augmentation of phagocytic activity of macrophages (163, 164) may contribute to IFN-γ-mediated GVT effects following allo-HCT.
Macrophages undergo classical activation (M1) upon stimulation by Toll-like receptor ligands and IFN-γ or alternative activation (M2) while subjected to an anti-inflammatory condition in the absence of IFN-γ (165). Tumor-associated macrophages (TAMs) exhibit an alternatively activated macrophage (Type 2 or M2)-like phenotype and are thought to favor tumor growth by downregulating immunity and facilitating angiogenesis (166). It has been shown that products of Th1 (e.g. IFN-γ) and Th2 (e.g. IL-4) responses downregulate M2 and M1 activity, respectively (167). Thus, downregulation of M2 activity by IFN-γ might be an additional mechanism contributing to IFN-γ-mediated anti-tumor responses.
Concluding remarks
IFN-γ and its widely expressed receptor signaling regulate the function of a variety of cells, including hematopoietic and non-hematopoietic cells, innate and adaptive immune cells, as well as normal and malignant cells, and play a complex role in both GVHD and GVT effects. Elimination of this cytokine in murine models markedly diminishes GVT effects but exacerbates GVHD-associated parenchymal tissue injuries and mortality. Although the underlying mechanisms remain partly undefined, inhibition of GVHD by IFN-γ is largely mediated by promoting apoptosis and suppressing proliferation of alloreactive T cells, as well as by signaling in parenchymal tissue cells that upregulate PD-L1 expression, leading to elimination of alloreactive T cells within the GVHD target tissues. The role of IFN-γ in Tregs is controversial. However, it is generally accepted that lack of IFN-γ leads to impaired Treg function, thereby contributing to the exacerbation of GVHD. On the other hand, promotion of GVT effects by IFN-γ may be due to the enhancement of LGVHR by IFN-γ, which mediates GVT or GVL effects without severe GVHD. In addition to its direct cytotoxic effect on hematopoietic and tumor cells, IFN-γ promotes the development of alloreactive CTLs expressing FasL that preferentially kill hematopoietic and tumor cells. IFN-γ may also promote GVT effects by upregulation of MHC antigen expression on cancer cells, leading to increased sensitivity to CTLs, stimulation of anti-tumor innate immune responses, and inhibition of tumor angiogenesis.
Acknowledgments
We thank Dr. Rémi J. Creusot for critical reading of the manuscript. This study was supported in part by grants from the National Institutes of Health (P01CA111519, P01AI045897, R01 AI064569, and RC1 HL100117) and American Cancer Society (RSG-03-227-01-LIB).
Footnotes
The authors declare no conflicts of interest.
References
- 1.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 2.Huang S, et al. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 3.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 4.Refaeli Y, Van Parijs L, Alexander SI, Abbas AK. Interferon gamma is required for activation-induced death of T lymphocytes. J Exp Med. 2002;196:999–1005. doi: 10.1084/jem.20020666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu CQ, Wittmer S, Dalton DK. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:123–128. doi: 10.1084/jem.192.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tau GZ, et al. Interferon gamma signaling alters the function of T helper type 1 cells. J Exp Med. 2000;192:977–986. doi: 10.1084/jem.192.7.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernabei P, et al. Interferon-gamma receptor 2 expression as the deciding factor in human T, B, and myeloid cell proliferation or death. J Leukoc Biol. 2001;70:950–960. [PubMed] [Google Scholar]
- 8.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 9.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 10.Afkarian M, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 11.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-γ and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 14.Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science. 2005;307:430–433. doi: 10.1126/science.1103336. [DOI] [PubMed] [Google Scholar]
- 15.Lazarevic V, et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat Immunol. 2011;12:96–104. doi: 10.1038/ni.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou M, Zhang Y, Ardans JA, Wahl LM. Interferon-gamma differentially regulates monocyte matrix metalloproteinase-1 and -9 through tumor necrosis factor-alpha and caspase 8. J Biol Chem. 2003;278:45406–45413. doi: 10.1074/jbc.M309075200. [DOI] [PubMed] [Google Scholar]
- 17.Haring JS, Corbin GA, Harty JT. Dynamic regulation of IFN-γ signaling in antigen-specific CD8+ T cells responding to infection. J Immunol. 2005;174:6791–6802. doi: 10.4049/jimmunol.174.11.6791. [DOI] [PubMed] [Google Scholar]
- 18.Novelli F, et al. Switching on of the proliferation or apoptosis of activated human T lymphocytes by IFN-gamma is correlated with the differential expression of the alpha- and beta-chains of its receptor. J Immunol. 1996;157:1935–1943. [PubMed] [Google Scholar]
- 19.Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 20.Novelli F, et al. Expression and role in apoptosis of the alpha- and beta-chains of the IFN-gamma receptor on human Th1 and Th2 clones. J Immunol. 1997;159:206–213. [PubMed] [Google Scholar]
- 21.Rigamonti L, et al. Surface expression of the IFN-gamma R2 chain is regulated by intracellular trafficking in human T lymphocytes. J Immunol. 2000;164:201–207. doi: 10.4049/jimmunol.164.1.201. [DOI] [PubMed] [Google Scholar]
- 22.Regis G, Conti L, Boselli D, Novelli F. IFNgammaR2 trafficking tunes IFNgamma-STAT1 signaling in T lymphocytes. Trends Immunol. 2006;27:96–101. doi: 10.1016/j.it.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Burman AC, et al. IFNgamma differentially controls the development of idiopathic pneumonia syndrome and GVHD of the gastrointestinal tract. Blood. 2007;110:1064–1072. doi: 10.1182/blood-2006-12-063982. [DOI] [PubMed] [Google Scholar]
- 24.Li HW, Sykes M. Emerging concepts in haematopoietic cell transplantation. Nat Rev Immunol. 2012;12:403–416. doi: 10.1038/nri3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jenq RR, van den Brink MRM. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer. 2010;10:213–221. doi: 10.1038/nrc2804. [DOI] [PubMed] [Google Scholar]
- 26.Krenger W, Ferrara JL. Graft-versus-host disease and the Th1/Th2 paradigm. Immunol Res. 1996;15:50–73. doi: 10.1007/BF02918284. [DOI] [PubMed] [Google Scholar]
- 27.Hill GR, Ferrara JL. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood. 2000;95:2754–2759. [PubMed] [Google Scholar]
- 28.Ellison CA, Natuik SA, McIntosh AR, Scully SA, Danilenko DM, Gartner JG. The role of interferon-gamma, nitric oxide and lipopolysaccharide in intestinal graft-versus-host disease developing in F1-hybrid mice. Immunology. 2003;109:440–449. doi: 10.1046/j.1365-2567.2003.01663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi T, et al. Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood. 2009;114:3101–3112. doi: 10.1182/blood-2009-05-219402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asavaroengchai W, et al. An essential role for IFN-gamma in regulation of alloreactive CD8 T cells following allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2007;13:46–55. doi: 10.1016/j.bbmt.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, et al. Paradoxical effects of IFN-gamma in graft-versus-host disease reflect promotion of lymphohematopoietic graft-versus-host reactions and inhibition of epithelial tissue injury. Blood. 2009;113:3612–3619. doi: 10.1182/blood-2008-07-168419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sykes M, Szot G, Nguyen P, Pearson D. Interleukin-12 inhibits murine graft-versus-host disease. Blood. 1995;86:2429–2438. [PubMed] [Google Scholar]
- 33.Yang YG, Sykes M. The role of interleukin-12 in preserving the graft-versus-leukemia effect of allogeneic CD8 T cells independently of GVHD. Leukemia Lymphoma. 1999;33:409–420. doi: 10.3109/10428199909058446. [DOI] [PubMed] [Google Scholar]
- 34.Dey BR, Yang Y-G, Szot GL, Pearson DA, Sykes M. Interleukin-12 inhibits graft-versus-host disease through an Fas-mediated mechanism associated with alterations in donor T-cell activation and expansion. Blood. 1998;91:3315–3322. [PubMed] [Google Scholar]
- 35.Yang YG, Dey B, Sergio JJ, Sykes M. Interleukin-12 prevents severe acute graft-versus-host disease (GVHD) and GVHD-associated immune dysfunction in a fully major histocompatibility complex haplotype-mismatched murine bone marrow transplantation model. Transplantation. 1997;64:1343–1352. doi: 10.1097/00007890-199711150-00018. [DOI] [PubMed] [Google Scholar]
- 36.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 37.Via CS, Rus V, Gately MK, Finkelman FD. IL-12 stimulates the development of acute graft-versus-host disease in mice that normally would develop chronic, autoimmune graft-versus-host disease. J Immunol. 1994;153:4040–4047. [PubMed] [Google Scholar]
- 38.Williamson E, Garside P, Bradley JA, More IA, Mowat AM. Neutralizing IL-12 during induction of murine acute graft-versus-host disease polarizes the cytokine profile toward a Th2-type alloimmune response and confers long term protection from disease. J Immunol. 1997;159:1208–1215. [PubMed] [Google Scholar]
- 39.Yang YG, Dey BR, Sergio JJ, Pearson DA, Sykes M. Donor-derived interferon gamma is required for inhibition of acute graft-versus-host disease by interleukin 12. J Clin Invest. 1998;102:2126–2135. doi: 10.1172/JCI4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy WJ, et al. Differential effects of the absence of interferon-gamma and IL-4 in acute graft-versus-host disease after allogeneic bone marrow transplantation in mice. J Clin Invest. 1998;102:1742–1748. doi: 10.1172/JCI3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welniak LA, Blazar BR, Anver MR, Wiltrout RH, Murphy WJ. Opposing roles of interferon-gamma on CD4+ T cell-mediated graft-versus-host disease: effects of conditioning. Biol Blood Marrow Transplant. 2000;6:604–612. doi: 10.1016/s1083-8791(00)70025-5. [DOI] [PubMed] [Google Scholar]
- 42.Yang YG, Qi J, Wang MG, Sykes M. Donor-derived interferon gamma separates graft-versus-leukemia effects and graft-versus-host disease induced by donor CD8 T cells. Blood. 2002;99:4207–4215. doi: 10.1182/blood.v99.11.4207. [DOI] [PubMed] [Google Scholar]
- 43.Reddy P, et al. Interleukin-18 regulates acute graft-versus-host disease by enhancing Fas-mediated donor T cell apoptosis. J Exp Med. 2001;194:1433–1440. doi: 10.1084/jem.194.10.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reddy P, et al. Interleukin 18 preserves a perforin-dependent graft-versus-leukemia effect after allogeneic bone marrow transplantation. Blood. 2002;100:3429–3431. doi: 10.1182/blood-2002-04-1252. [DOI] [PubMed] [Google Scholar]
- 45.Mauermann N, et al. Interferon-gamma regulates idiopathic pneumonia syndrome, a Th17+CD4+ T-cell-mediated graft-versus-host disease. Am J Respir Crit Care Med. 2008;178:379–388. doi: 10.1164/rccm.200711-1648OC. [DOI] [PubMed] [Google Scholar]
- 46.Gowdy KM, et al. Protective role of T-bet and Th1 cytokines in pulmonary graft-versus-host disease and peribronchiolar fibrosis. Am J Respir Cell Mol Biol. 2012;46:249–256. doi: 10.1165/rcmb.2011-0131OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 47.Villarino AV, Gallo E, Abbas AK. STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J Immunol. 2010;185:6461–6471. doi: 10.4049/jimmunol.1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun K, et al. IFN-gamma receptor-deficient donor T cells mediate protection from graft-versus-host disease and preserve graft-versus-tumor responses after allogeneic bone marrow transplantation. J Immunol. 2012;189:2033–2042. doi: 10.4049/jimmunol.1102853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi J, et al. IFNgammaR signaling mediates alloreactive T-cell trafficking and GVHD. Blood. 2012;120:4093–4103. doi: 10.1182/blood-2012-01-403196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, et al. Interferon γ stabilizes the T helper cell type 1 phenotype. J Exp Med. 2001;194:165–172. doi: 10.1084/jem.194.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Via CS, Shustov A, Rus V, Lang T, Nguyen P, Finkelman FD. In vivo neutralization of TNF-alpha promotes humoral autoimmunity by preventing the induction of CTL. J Immunol. 2001;167:6821–6826. doi: 10.4049/jimmunol.167.12.6821. [DOI] [PubMed] [Google Scholar]
- 52.Rus V, Svetic A, Nguyen P, Gause WC, Via CS. Kinetics of Th1 and Th2 cytokine production during the early course of acute and chronic murine graft-versus-host disease. Regulatory role of donor CD8+ T cells. J Immunol. 1995;155:2396–2406. [PubMed] [Google Scholar]
- 53.Ellison CA, Fischer JM, HayGlass KT, Gartner JG. Murine graft-versus-host disease in an F1-hybrid model using IFN-gamma gene knockout donors. J Immunol. 1998;161:631–640. [PubMed] [Google Scholar]
- 54.Ellison CA, Bradley DS, Fischer JM, Hayglass KT, Gartner JG. Murine graft-versus-host disease induced using interferon-gamma-deficient grafts features antibodies to double-stranded DNA, T helper 2-type cytokines and hypereosinophilia. Immunology. 2002;105:63–72. doi: 10.1046/j.0019-2805.2001.01353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shustov A, Nguyen P, Finkelman F, Elkon KB, Via CS. Differential expression of Fas and Fas ligand in acute and chronic graft-versus-host disease: up-regulation of Fas and Fas ligand requires CD8+ T cell activation and IFN-gamma production. J Immunol. 1998;161:2848–2855. [PubMed] [Google Scholar]
- 56.Baker MB, Riley RL, Podack ER, Levy RB. Graft-versus-host-disease-associated lymphoid hypoplasia and B cell dysfunction is dependent upon donor T cell-mediated Fas-ligand function, but not perforin function. Proc Natl Acad Sci USA. 1997;94:1366–1371. doi: 10.1073/pnas.94.4.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puliaev R, Nguyen P, Finkelman FD, Via CS. Differential requirement for IFN-gamma in CTL maturation in acute murine graft-versus-host disease. J Immunol. 2004;173:910–919. doi: 10.4049/jimmunol.173.2.910. [DOI] [PubMed] [Google Scholar]
- 58.Pelot MR, et al. Lymphohematopoietic graft-vs.-host reactions can be induced without graft-vs-host disease in murine mixed chimeras established with a cyclophosphamide-based nonmyeloablative conditioning regimen. Biol Blood Marrow Transplant. 1999;5:133–143. doi: 10.1053/bbmt.1999.v5.pm10392959. [DOI] [PubMed] [Google Scholar]
- 59.Chakraverty R, et al. An inflammatory checkpoint regulates recruitment of graft-versus-host reactive T cells to peripheral tissues. J Exp Med. 2006;203:2021–2031. doi: 10.1084/jem.20060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whitmire JK, Eam B, Benning N, Whitton JL. Direct interferon-γ signaling dramatically enhances CD4+ and CD8+ T cell memory. J Immunol. 2007;179:1190–1197. doi: 10.4049/jimmunol.179.2.1190. [DOI] [PubMed] [Google Scholar]
- 61.Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 62.Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. 2003;4:835–842. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 63.C-y W, et al. Distinct lineages of TH1 cells have differential capacities for memory cell generation in vivo. Nat Immunol. 2002;3:852–858. doi: 10.1038/ni832. [DOI] [PubMed] [Google Scholar]
- 64.Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med. 2005;11:748–756. doi: 10.1038/nm1257. [DOI] [PubMed] [Google Scholar]
- 65.Koyanagi M, Fukada K, Uchiyama T, Yagi J, Arimura Y. Long-term exposure to superantigen induces p27Kip1 and Bcl-2 expression in effector memory CD4+ T cells. Cell Immunol. 2007;248:77–85. doi: 10.1016/j.cellimm.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 66.Jatzek A, Tejera MM, Singh A, Sullivan JA, Plisch EH, Suresh M. p27Kip1 Negatively regulates the magnitude and persistence of CD4 T cell memory. J Immunol. 2012;189:5119–5128. doi: 10.4049/jimmunol.1201482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Veiga-Fernandes H, Rocha B. High expression of active CDK6 in the cytoplasm of CD8 memory cells favors rapid division. Nat Immunol. 2004;5:31–37. doi: 10.1038/ni1015. [DOI] [PubMed] [Google Scholar]
- 68.Singh A, Jatzek A, Plisch EH, Srinivasan R, Svaren J, Suresh M. Regulation of memory CD8 T-cell differentiation by cyclin-dependent kinase inhibitor p27Kip1. Mol Cell Biol. 2010;30:5145–5159. doi: 10.1128/MCB.01045-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takami K, et al. Interferon- γ inhibits hepatocyte growth factor-stimulated cell proliferation of human bronchial epithelial cells. Am J Resp Cell Mol Biol. 2002;26:231–238. doi: 10.1165/ajrcmb.26.2.4643. [DOI] [PubMed] [Google Scholar]
- 70.Yamazaki T, Akiba H, Koyanagi A, Azuma M, Yagita H, Okumura K. Blockade of B7-H1 on macrophages suppresses CD4+ T cell proliferation by augmenting IFN-γ-induced nitric oxide production. J Immunol. 2005;175:1586–1592. doi: 10.4049/jimmunol.175.3.1586. [DOI] [PubMed] [Google Scholar]
- 71.Tewari K, Nakayama Y, Suresh M. Role of direct effects of IFN-gamma on T cells in the regulation of CD8 T cell homeostasis. J Immunol. 2007;179:2115–2125. doi: 10.4049/jimmunol.179.4.2115. [DOI] [PubMed] [Google Scholar]
- 72.Sercan O, Hammerling GJ, Arnold B, Schuler T. Innate immune cells contribute to the IFN-gamma-dependent regulation of antigen-specific CD8+ T cell homeostasis. J Immunol. 2006;176:735–739. doi: 10.4049/jimmunol.176.2.735. [DOI] [PubMed] [Google Scholar]
- 73.Sercan O, Stoycheva D, Hammerling GJ, Arnold B, Schuler T. IFN-gamma receptor signaling regulates memory CD8+ T cell differentiation. J Immunol. 2010;184:2855–2862. doi: 10.4049/jimmunol.0902708. [DOI] [PubMed] [Google Scholar]
- 74.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blazar BR, et al. Blockade of programmed death-1 engagement accelerates graft-versus-host disease lethality by an IFN-gamma-dependent mechanism. J Immunol. 2003;171:1272–1277. doi: 10.4049/jimmunol.171.3.1272. [DOI] [PubMed] [Google Scholar]
- 76.Yang HR, et al. Mechanistic insights into immunomodulation by hepatic stellate cells in mice: a critical role of interferon-gamma signaling. Hepatology. 2009;50:1981–1991. doi: 10.1002/hep.23202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bolinger B, et al. IFN-gamma-receptor signaling ameliorates transplant vasculopathy through attenuation of CD8+ T-cell-mediated injury of vascular endothelial cells. Eur J Immunol. 2010;40:733–743. doi: 10.1002/eji.200939706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wysocki CA, Panoskaltsis-Mortari A, Blazar BR, Serody JS. Leukocyte migration and graft-versus-host disease. Blood. 2005;105:4191–4199. doi: 10.1182/blood-2004-12-4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 80.Liu L, Callahan MK, Huang D, Ransohoff RM. Chemokine receptor CXCR3: an unexpected enigma. Curr Top Dev Biol. 2005;68:149–181. doi: 10.1016/S0070-2153(05)68006-4. [DOI] [PubMed] [Google Scholar]
- 81.Nakajima C, et al. Induction of the chemokine receptor CXCR3 on TCR-stimulated T cells: dependence on the release from persistent TCR-triggering and requirement for IFN-gamma stimulation. Eur J Immunol. 2002;32:1792–1801. doi: 10.1002/1521-4141(200206)32:6<1792::AID-IMMU1792>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 82.Ichiba T, et al. Early changes in gene expression profiles of hepatic GVHD uncovered by oligonucleotide microarrays. Blood. 2003;102:763–771. doi: 10.1182/blood-2002-09-2748. [DOI] [PubMed] [Google Scholar]
- 83.Croudace JE, et al. Chemokine-mediated tissue recruitment of CXCR3+ CD4+ T cells plays a major role in the pathogenesis of chronic GVHD. Blood. 2012;120:4246–4255. doi: 10.1182/blood-2012-02-413260. [DOI] [PubMed] [Google Scholar]
- 84.Feng G, et al. Exogenous IFN-gamma ex vivo shapes the alloreactive T-cell repertoire by inhibition of Th17 responses and generation of functional Foxp3+ regulatory T cells. Eur J Immunol. 2008;38:2512–2527. doi: 10.1002/eji.200838411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kelchtermans H, et al. Defective CD4+CD25+ regulatory T cell functioning in collagen-induced arthritis: an important factor in pathogenesis, counter-regulated by endogenous IFN-gamma. Arthritis Res Ther. 2005;7:R402–R415. doi: 10.1186/ar1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nishibori T, Tanabe Y, Su L, David M. Impaired development of CD4+ CD25+ regulatory T cells in the absence of STAT1: increased susceptibility to autoimmune disease. J Exp Med. 2004;199:25–34. doi: 10.1084/jem.20020509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ma H, et al. Absence of Stat1 in donor CD4(+) T cells promotes the expansion of Tregs and reduces graft-versus-host disease in mice. J Clin Invest. 2011;121:2554–2569. doi: 10.1172/JCI43706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei B, Baker S, Wieckiewicz J, Wood KJ. IFN-gamma triggered STAT1-PKB/AKT signalling pathway influences the function of alloantigen reactive regulatory T cells. Am J Transplant. 2010;10:69–80. doi: 10.1111/j.1600-6143.2009.02858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol. 2013;34:74–80. doi: 10.1016/j.it.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 90.Hall AO, et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity. 2012;37:511–523. doi: 10.1016/j.immuni.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity. 2012;37:501–510. doi: 10.1016/j.immuni.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sawitzki B, Kingsley CI, Oliveira V, Karim M, Herber M, Wood KJ. IFN-gamma production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J Exp Med. 2005;201:1925–1935. doi: 10.1084/jem.20050419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Z, et al. Role of IFN-gamma in induction of Foxp3 and conversion of CD4+ CD25− T cells to CD4+ Tregs. J Clin Invest. 2006;116:2434–2441. doi: 10.1172/JCI25826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koenecke C, et al. IFN-γ production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol. 2012;189:2890–2896. doi: 10.4049/jimmunol.1200413. [DOI] [PubMed] [Google Scholar]
- 96.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 97.Yang YG, Sergio JJ, Pearson DA, Szot GL, Shimizu A, Sykes M. Interleukin-12 preserves the graft-versus-leukemia effect of allogeneic CD8 T cells while inhibiting CD4-dependent graft-versus-host disease in mice. Blood. 1997;90:4651–4660. [PubMed] [Google Scholar]
- 98.Sykes M, Abraham VS, Harty MW, Pearson DA. IL-2 reduces graft-versus-host disease and preserves a graft-versus-leukemia effect by selectively inhibiting CD4+ T cell activity. J Immunol. 1993;150:197–205. [PubMed] [Google Scholar]
- 99.Korngold R, Sprent J. Surface markers of T cells causing lethal graft-vs-host disease to class I vs class II H-2 differences. J Immunol. 1985;135:3004–3010. [PubMed] [Google Scholar]
- 100.Palathumpat V, Dejbakhsh-Jones S, Strober S. The role of purified CD8+ T cells in graft-versus-leukemia activity and engraftment after allogeneic bone marrow transplantation. Transplantation. 1995;60:355–361. doi: 10.1097/00007890-199508270-00010. [DOI] [PubMed] [Google Scholar]
- 101.Ramirez-Montagut T, et al. IFN-γ and Fas ligand are required for graft-versus-tumor activity against renal cell carcinoma in the absence of lethal graft-versus-host disease. J Immunol. 2007;179:1669–1680. doi: 10.4049/jimmunol.179.3.1669. [DOI] [PubMed] [Google Scholar]
- 102.Zhan Y, Corbett AJ, Brady JL, Sutherland RM, Lew AM. CD4 help-independent induction of cytotoxic CD8 cells to allogeneic P815 tumor cells is absolutely dependent on costimulation. J Immunol. 2000;165:3612–3619. doi: 10.4049/jimmunol.165.7.3612. [DOI] [PubMed] [Google Scholar]
- 103.Meguro A, et al. Lack of IL-21 signal attenuates graft-versus-leukemia effect in the absence of CD8 T-cells. Bone Marrow Transplant. 2011;46:1557–1565. doi: 10.1038/bmt.2010.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Glimcher LH, Kim KJ, Green I, Paul WE. Ia antigen-bearing B cell tumor lines can present protein antigen and alloantigen in a major histocompatibility complex-restricted fashion to antigen-reactive T cells. J Exp Med. 1982;155:445–459. doi: 10.1084/jem.155.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sun K, Li M, Sayers TJ, Welniak LA, Murphy WJ. Differential effects of donor T-cell cytokines on outcome with continuous bortezomib administration after allogeneic bone marrow transplantation. Blood. 2008;112:1522–1529. doi: 10.1182/blood-2008-03-143461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rubio MT, Kim YM, Sachs T, Mapara M, Zhao G, Sykes M. Antitumor effect of donor marrow graft rejection induced by recipient leukocyte infusions in mixed chimeras prepared with nonmyeloablative conditioning: critical role for recipient-derived IFN-gamma. Blood. 2003;102:2300–2307. doi: 10.1182/blood-2002-12-3949. [DOI] [PubMed] [Google Scholar]
- 107.Mapara MY, Kim Y-M, Marx J, Sykes M. Donor lymphocyte infusion-mediated graft-versus-leukemia effects in mixed chimeras established with a nonmyeloablative conditioning regimen: extinction of graft-versus-leukemia effects after conversion to full donor chimerism. Transplantation. 2003;76:297–305. doi: 10.1097/01.TP.0000072014.83469.2D. [DOI] [PubMed] [Google Scholar]
- 108.Spitzer TR, et al. Intentional induction of mixed chimerism and achievement of antitumor responses after nonmyeloablative conditioning therapy and HLA-matched donor bone marrow transplantation for refractory hematologic malignancies. Biol Blood Marrow Transplant. 2000;6:309–320. doi: 10.1016/s1083-8791(00)70056-5. [DOI] [PubMed] [Google Scholar]
- 109.Dufour C, et al. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood. 2003;102:2053–2059. doi: 10.1182/blood-2003-01-0114. [DOI] [PubMed] [Google Scholar]
- 110.Zoumbos NC, Gascon P, Djeu JY, Young NS. Interferon is a mediator of hematopoietic suppression in aplastic anemia in vitro and possibly in vivo. Proc Natl Acad Sci USA. 1985;82:188–192. doi: 10.1073/pnas.82.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Iwasaki T, et al. Graft-versus-host-disease-associated donor cell engraftment in an F1 hybrid model is dependent upon the Fas pathway. Immunology. 2000;99:94–100. doi: 10.1046/j.1365-2567.2000.00919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bloom ML, Wolk AG, Simon-Stoos KL, Bard JS, Chen J, Young NS. A mouse model of lymphocyte infusion-induced bone marrow failure. Exp Hematol. 2004;32:1163–1172. doi: 10.1016/j.exphem.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 113.Chen J, Lipovsky K, Ellison FM, Calado RT, Young NS. Bystander destruction of hematopoietic progenitor and stem cells in a mouse model of infusion-induced bone marrow failure. Blood. 2004;104:1671–1678. doi: 10.1182/blood-2004-03-1115. [DOI] [PubMed] [Google Scholar]
- 114.Laver J, et al. In vitro interferon-gamma production by cultured T-cells in severe aplastic anaemia: correlation with granulomonopoietic inhibition in patients who respond to anti-thymocyte globulin. Br J Haematol. 1988;69:545–550. doi: 10.1111/j.1365-2141.1988.tb02413.x. [DOI] [PubMed] [Google Scholar]
- 115.Rottman M, et al. IFN-gamma mediates the rejection of haematopoietic stem cells in IFN-gammaR1-deficient hosts. PLoS Med. 2008;5:e26. doi: 10.1371/journal.pmed.0050026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chewning JH, Zhang W, Randolph DA, Swindle CS, Schoeb TR, Weaver CT. Allogeneic Th1 cells home to host bone marrow and spleen and mediate IFNgamma-dependent aplasia. Biol Blood Marrow Transplant. 2013;19:876–887. doi: 10.1016/j.bbmt.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Delisle JS, Gaboury L, Belanger MP, Tasse E, Yagita H, Perreault C. Graft-versus-host disease causes failure of donor hematopoiesis and lymphopoiesis in interferon-gamma receptor-deficient hosts. Blood. 2008;112:2111–2119. doi: 10.1182/blood-2007-12-130534. [DOI] [PubMed] [Google Scholar]
- 118.van den Brink MR, Burakoff SJ. Cytolytic pathways in haematopoietic stem-cell transplantation. Nat Rev Immunol. 2002;2:273–281. doi: 10.1038/nri775. [DOI] [PubMed] [Google Scholar]
- 119.Nastala CL, et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol. 1994;153:1697–1706. [PubMed] [Google Scholar]
- 120.Sayers TJ, Wiltrout TA, McCormick K, Husted C, Wiltrout RH. Antitumor effects of α-interferon and γ-interferon on a murine renal cancer (Renca) in vitro and in vivo. Cancer Res. 1990;50:5414–5420. [PubMed] [Google Scholar]
- 121.Tan J, et al. Interferon-gamma-inducing factor elicits antitumor immunity in association with interferon-gamma production. J Immunother. 1998;21:48–55. doi: 10.1097/00002371-199801000-00006. [DOI] [PubMed] [Google Scholar]
- 122.Rodrigues EG, Garofalo AS, Travassos LR. Endogenous accumulation of IFN-gamma in IFN-gamma-R(−/−) mice increases resistance to B16F10-Nex2 murine melanoma: a model for direct IFN-gamma anti-tumor cytotoxicity in vitro and in vivo. Cytokines Cell Mol Ther. 2002;7:107–116. doi: 10.1080/13684730310000121. [DOI] [PubMed] [Google Scholar]
- 123.Chawla-Sarkar M, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 124.Meunier M-C, Delisle J-S, Bergeron J, Rineau V, Baron C, Perreault C. T cells targeted against a single minor histocompatibility antigen can cure solid tumors. Nat Med. 2005;11:1222–1229. doi: 10.1038/nm1311. [DOI] [PubMed] [Google Scholar]
- 125.Ikeda H, Old LJ, Schreiber RD. The roles of IFNγ in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13:95–109. doi: 10.1016/s1359-6101(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 126.Weber JS, Rosenberg SA. Modulation of murine tumor major histocompatibility antigens by cytokines in vivo and in vitro. Cancer Res. 1988;48:5818–5824. [PubMed] [Google Scholar]
- 127.Seliger B, Ruiz-Cabello F, Garrido F. IFN inducibility of major histocompatibility antigens in tumors. Adv Cancer Res. 2008;101:249–276. doi: 10.1016/S0065-230X(08)00407-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao X, et al. Brief report: interferon-gamma induces expansion of Lin(−)Sca-1(+)C-Kit(+) Cells. Stem Cells. 2010;28:122–126. doi: 10.1002/stem.252. [DOI] [PubMed] [Google Scholar]
- 130.Schurch C, Riether C, Amrein MA, Ochsenbein AF. Cytotoxic T cells induce proliferation of chronic myeloid leukemia stem cells by secreting interferon-gamma. J Exp Med. 2013;210:605–621. doi: 10.1084/jem.20121229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cho HI, Lee YR, Celis E. Interferon gamma limits the effectiveness of melanoma peptide vaccines. Blood. 2011;117:135–144. doi: 10.1182/blood-2010-08-298117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zaidi MR, et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature. 2011;469:548–553. doi: 10.1038/nature09666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Spranger S, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Landsberg J, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490:412–416. doi: 10.1038/nature11538. [DOI] [PubMed] [Google Scholar]
- 135.Yang Y, et al. IFN-gamma promotes graft-versus-leukemia effects without directly interacting with leukemia cells in mice after allogeneic hematopoietic cell transplantation. Blood. 2011;118:3721–3724. doi: 10.1182/blood-2010-05-283887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ibe S, Qin Z, Schuler T, Preiss S, Blankenstein T. Tumor rejection by disturbing tumor stroma cell interactions. J Exp Med. 2001;194:1549–1559. doi: 10.1084/jem.194.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schuler T, Blankenstein T. Cutting edge: CD8+ effector T cells reject tumors by direct antigen recognition but indirect action on host cells. J Immunol. 2003;170:4427–4431. doi: 10.4049/jimmunol.170.9.4427. [DOI] [PubMed] [Google Scholar]
- 138.Qin Z, et al. A critical requirement of interferon gamma-mediated angiostasis for tumor rejection by CD8+ T cells. Cancer Res. 2003;63:4095–4100. [PubMed] [Google Scholar]
- 139.Qin Z, Blankenstein T. CD4+ T cell–mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 2000;12:677–686. doi: 10.1016/s1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 140.Takaoka A. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- 141.Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ, Yokota SJ, Bankert RB. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol. 2007;178:5552–5562. doi: 10.4049/jimmunol.178.9.5552. [DOI] [PubMed] [Google Scholar]
- 142.Ruegg C, Yilmaz A, Bieler G, Bamat J, Chaubert P, Lejeune FJ. Evidence for the involvement of endothelial cell integrin αVβ3 in the disruption of the tumor vasculature induced by TNF and IFN-γ. Nat Med. 1998;4:408–414. doi: 10.1038/nm0498-408. [DOI] [PubMed] [Google Scholar]
- 143.Sorensen EW, Gerber SA, Frelinger JG, Lord EM. IL-12 suppresses vascular endothelial growth factor receptor 3 expression on tumor vessels by two distinct IFN-gamma-dependent mechanisms. J Immunol. 2010;184:1858–1866. doi: 10.4049/jimmunol.0903210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lu Y, et al. Responsiveness of stromal fibroblasts to IFN-gamma blocks tumor growth via angiostasis. J Immunol. 2009;183:6413–6421. doi: 10.4049/jimmunol.0901073. [DOI] [PubMed] [Google Scholar]
- 145.Listopad JJ, et al. Fas expression by tumor stroma is required for cancer eradication. Proc Natl Acad Sci USA. 2013;110:2276–2281. doi: 10.1073/pnas.1218295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 147.Palmer JM, Rajasekaran K, Thakar MS, Malarkannan S. Clinical relevance of natural killer cells following hematopoietic stem cell transplantation. J Cancer. 2013;4:25–35. doi: 10.7150/jca.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Asai O, et al. Suppression of graft-versus-host disease and amplification of graft-versus-tumor effects by activated natural killer cells after allogeneic bone marrow transplantation. J Clin Invest. 1998;101:1835–1842. doi: 10.1172/JCI1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wald O, et al. IFN-gamma acts on T cells to induce NK cell mobilization and accumulation in target organs. J Immunol. 2006;176:4716–4729. doi: 10.4049/jimmunol.176.8.4716. [DOI] [PubMed] [Google Scholar]
- 150.Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008;68:8437–8445. doi: 10.1158/0008-5472.CAN-08-1440. [DOI] [PubMed] [Google Scholar]
- 151.Smyth MJ, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661–668. doi: 10.1084/jem.191.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Janat-Amsbury MM, Yockman JW, Anderson ML, Kieback DG, Kim SW. Combination of local, non-viral IL12 gene therapy and systemic paclitaxel chemotherapy in a syngeneic ID8 mouse model for human ovarian cancer. Anticancer Res. 2006;26:3223–3228. [PubMed] [Google Scholar]
- 153.Li Z, Pradera F, Kammertoens T, Li B, Liu S, Qin Z. Cross-talk between T cells and innate immune cells is crucial for IFN-gamma-dependent tumor rejection. J Immunol. 2007;179:1568–1576. doi: 10.4049/jimmunol.179.3.1568. [DOI] [PubMed] [Google Scholar]
- 154.Brewington R, Chatterji M, Zoubine M, Miranda RN, Norimatsu M, Shnyra A. IFN-gamma-independent autocrine cytokine regulatory mechanism in reprogramming of macrophage responses to bacterial lipopolysaccharide. J Immunol. 2001;167:392–398. doi: 10.4049/jimmunol.167.1.392. [DOI] [PubMed] [Google Scholar]
- 155.Khare V, Sodhi A, Singh SM. Effect of aging on the tumoricidal functions of murine peritoneal macrophages. Nat Immun. 1996;15:285–294. [PubMed] [Google Scholar]
- 156.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 157.Blazar BR, et al. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med. 2001;194:541–549. doi: 10.1084/jem.194.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang H, Madariaga ML, Wang S, Van Rooijen N, Oldenborg PA, Yang YG. Lack of CD47 on nonhematopoietic cells induces split macrophage tolerance to CD47null cells. Proc Natl Acad Sci USA. 2007;104:13744–13749. doi: 10.1073/pnas.0702881104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jaiswal S, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Majeti R, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Theocharides AP, et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med. 2012;209:1883–1899. doi: 10.1084/jem.20120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Willingham SB, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nambu M, et al. Regulation of Fc gamma receptor expression and phagocytosis of a human monoblast cell line U937. Participation of cAMP and protein kinase C in the effects of IFN-gamma and phorbol ester. J Immunol. 1989;143:4158–4165. [PubMed] [Google Scholar]
- 164.Gruel N, Fridman WH, Teillaud JL. Bypassing tumor-specific and bispecific antibodies: triggering of antitumor immunity by expression of anti-FcgammaR scFv on cancer cell surface. Gene Ther. 2001;8:1721–1728. doi: 10.1038/sj.gt.3301575. [DOI] [PubMed] [Google Scholar]
- 165.Schmieder A, Michel J, Schonhaar K, Goerdt S, Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol. 2012;22:289–297. doi: 10.1016/j.semcancer.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 166.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 167.Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Imunol. 2012;32:463–488. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]