

Abstract
The membrane spanning domain (MSD) of human immunodeficiency virus 1 (HIV-1) envelope glycoprotein gp41 is important for fusion and infection. We used molecular dynamics (MD) simulations (3.4 μs total) to relate membrane and peptide properties that lead to water solvation of the α-helical gp41 MSD’s midspan arginine in pure dipalmitoylphosphatidylcholine (DPPC) and in 50/50 DPPC/cholesterol membranes. We find that the midspan arginine is solvated by water that penetrates the inner leaflet, leading to a so-called water defect. The water defect is surprisingly robust across initial conditions and membrane compositions, but the presence of cholesterol modulates its behavior in several key ways. In the cholesterol-containing membranes, fluctuations in membrane thickness and water penetration depth are localized near the midspan arginine, and the MSD helices display a tightly regulated tilt angle. In the cholesterol-free membranes, thickness fluctuations are not as strongly correlated to the peptide position and tilt angles vary significantly depending on protein position relative to boundaries between domains of differing thickness. Cholesterol in an HIV-1 viral membrane is required for infection. Therefore, this work suggests that the colocalized water defect and membrane thickness fluctuations in cholesterol-containing viral membranes play an important role in fusion by bringing the membrane closer to a stability limit that must be crossed for fusion to occur.
Keywords: Midspan arginine, Molecular dynamics, Metadynamics, Transmembrane, Membrane simulation, Snorkeling
1. Introduction
One of the proteins responsible for mediating fusion of human immunodeficiency virus 1 (HIV-1) particles to target cells is known as gp41. It resides in the viral membrane in so-called envelope spikes, which are trimers of noncovalently-attached dimers of envelope glycoproteins gp120 and gp41 [1,2]. In the native spike, gp41 spans the viral membrane as the spike’s anchor and is thought to be hidden in a meta-stable conformation by gp120. The membrane spanning domain (MSD) of gp41 is of great interest as it is responsible for both anchoring gp41 in the bilayer and for playing a role in fusion, according to truncation studies [3,4]. Mutagenesis has also shown that the specific sequence of the twenty-seven residue MSD is important for membrane fusion [5–11]. The MSD houses the GXXXG motif, postulated to mediate transmembrane helical interactions, and the charged residues K681, R694, R705, and R707 (Table 1) [3–14].
Table 1.
HIV-1 gp41 MSD peptide sequence (NL4-3 consensus).
| Sequence | K | L | F | I | M | I | V | G | G | L | V | G | L | R | I | V | F | A | V | L | S | I | V | N | R | V | R |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Residue index | 681 | . | . | 684 | . | . | 687 | . | . | 690 | . | . | . | 694 | . | . | . | 698 | . | . | 701 | . | . | 704 | . | . | 707 |
| Localindex | 1 | . | . | 4 | . | . | 7 | . | . | 10 | . | . | . | 14 | . | . | . | 18 | . | . | 21 | . | . | 24 | . | . | 27 |
The gp41 MSD is predicted to be membrane-spanning based on mutagenesis and to be α-helical based on both bioinformatic and CD analyses [10,15–17]. Recently, our group supported this conclusion using all-atom metadynamics simulations and further showed that the membrane-spanning, α-helical MSD’s midspan arginine (R694) likely “snorkels” in the inner leaflet by allowing the guanidino-group access to the layer of lipid headgroups [18]. The snorkeling configuration is not surprising partially because a charged residue in an ideally structured hydrophobic lipid bilayer core is highly energetically unfavorable [19]. Therefore, transmembrane arginines probably have mechanisms to compensate for the charge in the middle of a lipid bilayer, and those in the middle of membrane-spanning α-helices can nevertheless be solvated through a so-called “water-defect” [19–24]. Indeed, previous calculations of the free energy of insertion from solvent to lipid bilayer of a midspan-arginine-containing transmembrane peptide (not gp41 MSD) by molecular dynamics and experiments ultimately determined the energetic cost of insertion to be only a few kcal/mol with defect solvation [19,25,26].
Bilayer water defects are characterized by water molecules and often lipid headgroups that have encroached deep into the bilayer hydrophobic core in order to interact directly with polar/charged residues. Relatively large water defects have been studied by solid-state NMR and have also been observed in various simulation systems involving guanidinium ions, arginine analogs, and arginine containing transmembrane proteins (both β-barrels and α-helices) in various lipids [20,21,23,26–31]. It has not been previously investigated if HIV-1 gp41 MSD displays a water defect in the native spike complex, but an arginine at position 694 (HXBc2 sequence) is required for full fusion [11,4]. Therefore, it stands to reason that if a water defect is present due to R694, the defect may play a role in fusion as well. Additionally, the HIV-1 membrane is rich in cholesterol (up to 50 mol-%), which is well known to increase the thickness and to more order model bilayers in the liquid phase [32,33]. Cholesterol seems to have multiple roles during the HIV lifecycle. It is important for Gag assembly at the plasma membrane, co-receptor binding and of course, fusion [34–39]. Since cholesterol-depletion compromises fusion [36,40], it is of interest to investigate the influence of cholesterol on solvation by defect near HIV-1 gp41 MSD.
In this paper, as part of a segment-based approach to investigating structure/function relationships in gp41, molecular simulations are used to study particular aspects of R694-solvation of the α-helical gp41 MSD monomeric peptide in a model lipid bilayer. Our major finding is that solvation of R694 invariably requires penetration of water into the inner leaflet, establishing a classical water defect. This feature of R694 solvation is robust with respect to the peptide’s local lipid composition, including the presence of cholesterol, and it is initial-condition-independent. However, we find that cholesterol appears to modulate the behavior of the defect. For instance, the presence of cholesterol appears to focus bilayer thickness and water penetration fluctuations at the midspan-arginine-containing protein, whereas these fluctuations are not strongly correlated to protein position in the cholesterol-free bilayers. Based on these findings, we offer some speculation that a robust water defect near a gp41 trimer perhaps contributes to the metastability of the spike and viral membrane, bringing it closer to a stability limit that must be crossed for virus–cell fusion.
2. Methods
2.1. General
All-atom molecular dynamics (MD) simulations were performed with NAMD 2.8 [41] along with VMD 1.9 [42], the CHARMM force field [43,44] with recent lipid-based corrections [45,46], and explicit TIP3P water.
Preliminary systems were run for 100 ns with configuration parameters based on those recommended by the CHARMM-GUI Membrane Builder (grid spacing of 1 Å, nonbonded shifted from 10 to 12 Å, 2 fs timestep) [47]. These simulations were compared to the same systems run with more aggressive configuration parameters (grid spacing of 2 Å, nonbonded shifted from 8 to 9 Å, 1 fs timestep) to determine if we could decrease computational time without sacrificing accuracy. We were able to use these more aggressive parameters for the heterogeneous, cholesterol-containing simulations (WT1, etc., see below) but not for the homogeneous dipalmitoylphosphatidylcholine (DPPC) simulations (WT1ΔChol, etc., see below). The WT1 simulation run with less aggressive parameters (known as WT1c) is also analyzed for various observables in Supplemental Figures 1, 2E/F, 3A, 7, and 8, for comparison to the WT1 system. In all cases, the Langevin thermostat was set at a temperature of 310 K and the Langevin piston Nosé-Hoover barostat was set at 1 atm. The z-cell dimension was allowed to fluctuate independently of x and y, which were kept at a constant ratio. Periodic boundary conditions were employed. All simulations were run on TACC XSEDE resources.
2.2. Choice of lipids
Cholesterol-containing bilayers are used to represent a model of the HIV-1 viral membrane. A major lipidic component in the HIV lipidome is sphingomyelin, a fully saturated lipid, and HIV envelopes contain up to 50% cholesterol [34]. Because no CHARMM parameters for sphingomyelin exist in the open literature, we chose to work with DPPC, also a fully saturated lipid. DPPC and SM undergo their main phase transition at nearly the same temperature [48,49]. At the physiologically relevant temperature of 310 K, 50/50 DPPC/cholesterol bilayers will exist in the liquid-ordered phase [50,51]. To the best of our knowledge, no one has measured the main phase transition temperature (Tm) of a pure DPPC simulation using the CHARMM36 lipid force field parameters. Using other force fields, Tm has been suggested to be between 300 and 320 K [52,53], which compares fairly well to the experimental temperatures of 314–315 K [54,55]. Our pure DPPC membranes are gel-like and order parameters show the DPPC bilayers to be slightly less ordered than the cholesterol-containing bilayers (see Supplemental Figure 3).
2.3. System setup
We generated initial states for the WT1, WT2, and WT3 systems using a steered-MD (SMD) approach to insert an α-helical gp41 MSD model in three distinct locations of an equilibrated lipid bilayer composed of 50% DPPC and of 50% cholesterol, and each system was subjected to several rounds of equilibration following the procedure detailed previously [18] before launching individual 300 ns NPT production runs. Because lipid diffusion is so slow on a 300 ns timescale, the lipids cannot rearrange enough for ergodic sampling of all possible lipid arrangements; hence local compositions around each protein are largely determined by the choice of the initial insertion point. Therefore, the three WT simulation systems differed mainly in the local bilayer composition near the protein (Supplemental Figure 4). The same SMD approach was used to insert an α-helical gp41 MSD model into an equilibrated lipid bilayer composed purely of DPPC. We refer to this system as WT1ΔChol, and it was subjected to 300 ns NPT production MD after equilibration. Two other replicas, WT2ΔChol and WT3ΔChol, were branched off from the WT1ΔChol trajectory (at 0 ns and preproduction). The protein backbone was restrained and the lipids were allowed to further equilibrate around the peptide for 25 ns before starting production runs. Both protein-free bilayers were initially generated using the CHARMM-GUI membrane-builder and equilibrated [47]. For comparison of some observables, the membranes (with no peptide) were also run for 300 ns NPT MD; these systems are referred to as DPPC and DPPC/Chol systems. The R694L mutant system was generated using the VMD mutator plug-in operating on the equilibrated WT1 system, and this mutant system was subjected to both metadynamics (detailed below) and 300 ns production MD. Post-production, R694L was mutated back to WT, generating the R694LR system, which was subjected to 300 ns NPT production MD. All systems were neutralized and brought to 0.1 M NaCl before production runs. The cholesterol-containing systems have ~58,000 atoms, including 183 cholesterol and 151 DPPC molecules. These systems are roughly 80 × 80 × 80 Å3. The pure DPPC systems have ~89,000 atoms, including 334 lipid molecules. These systems are roughly 90 × 90 × 100 Å3.
2.4. Metadynamics of R694L MSD
Following our previous work on WT MSD [18], we used metadynamics [56–59] to test whether or not the R694L MSD’s minimum free-energy state is α-helical in a 50% DPPC and 50% cholesterol bilayer membrane. Briefly, in metadynamics, the MD trajectory is biased by a history-dependent potential that is the sum of Gaussians deposited along a chosen collective variable (CV); the forces arising from these Gaussians allow high-probability regions along CV space to be explored first and discourage the system from revisiting the same CV values. As before, we used as a CV the RMSD of the backbone (C, Cα, N, O) atoms of the peptide with respect to a perfect α-helix, an average over a total of 109 atoms. The lower boundary, an RMSD of 0 Å, indicates a perfect helix, and the upper boundary, an RMSD of 10 Å, indicates a partially unfolded peptide. The metadynamics parameters included a Gaussian width of 0.01 Å, a weight of 0.15 kcal/mol, and a deposition frequency of 1 ps.
2.5. Observables computed in the production molecular dynamics simulations
The mass density as a function of position along the global membrane normal direction, z, of various components [protein, cholesterol, lipid headgroups, lipid tails, water (local and global)] averaged over the last 100 ns of the trajectory was determined after shifting the x,y-origin to the peptide center of mass, shifting the z-origin to the bilayer center of mass, and rewrapping all coordinates into the central image. Data for the WT MSD in the mixed bilayer was averaged over replicas WT1, WT2, and WT3, while the data for the WT MSD in the pure DPPC membrane was averaged over replicas WT1ΔChol, WT2ΔChol, and WT3ΔChol. The number of water molecules uniquely associated to each residue was determined by partitioning every water molecule within 4 Å of any protein atom into cells of a Voronoi tessellation defined by the locations of the α-carbons (i.e., a water belongs to the residue to whose Cα it is closest). Helix tilt was defined by the angle between the vector aligned along the helix and the global z-axis. The tilt was averaged over 3 replicas for the WT systems in a mixed and pure DPPC bilayer and only included statistics for the last 100 ns. Maps of membrane thickness L(x,y) were determined by dividing the x,y-plane into squares of size 4 × 4 Å2, calculating the z-distance between the center of mass of the lipid headgroups in the outer and inner leaflets within each square and averaging over the trajectory. Also mapped in the plane of the membrane was the average-minimum distance sampled by any water molecule on the inner side to the global membrane midplane, at the same resolution as L.
In general for this paper, if an observable is averaged over the trajectory for a single system, we report the standard deviation. If an observable is averaged between replicas (ex. WT1, WT2, and WT3), we report the standard error. We label these as SD and SE, respectively.
3. Results
3.1. HIV-1 gp41 MSD and its R694L mutant span the cholesterol-containing DPPC bilayer as α-helices
The result for the R694L MSD metadynamics is very similar to that seen for the WT1 MSD, [18] namely, the most stable state is an α-helix. The primary output of the metadynamics calculation of the R694L MSD is the potential of mean force (PMF) as a function of RMS distance from an ideal α-helix (chosen to be the collective variable, CV). The cumulative PMFs averaged from the last 20 ns for the WT1 (computed previously) [18] and R694L systems are shown in Fig. 1. Metadynamics required approximately 320 ns to achieve diffusive sampling of the CV and convergence of the PMF, as seen in Supplemental Figures 5 and 6. It is interesting to note that multiple unfolding–refolding events were observed.
Fig. 1.
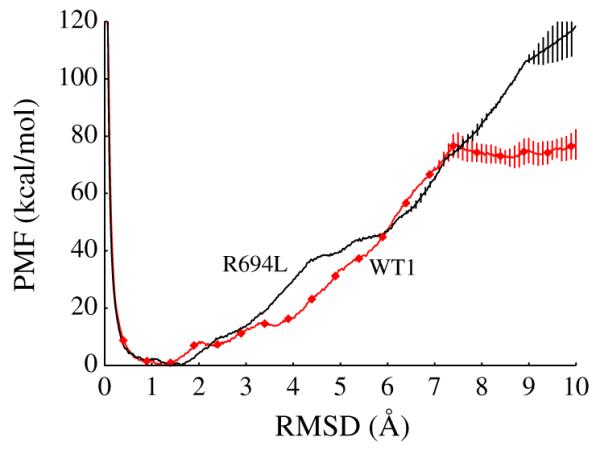
Final cumulative, conformational potential of mean force (PMF) in kcal/mol vs. collective variable (RMSD of backbone compared to perfect helix) in Å from the R694L peptide metadynamics, in black. The PMF vs. CV from the WT1 peptide metadynamics from our previous study is also shown here, in red diamonds [18]. Error bars represent standard deviations of the last 20 ns.
The 300 ns production NPT MD simulations also support the conclusion that the α-helical state is stable for both the WT1 and R694L systems. In Fig. 2A and B, we show traces of the metadynamics CV observed during equilibrium MD launched from helical initial states for both systems, along with CV histograms. Both systems remain close to perfectly α-helical. The WT1 peptide seems to have greater flexibility than the R694L peptide, as indicated by its wider RMSD distribution. In Fig. 2C, we show similar data for the WT1ΔChol system, which indicates that the WT peptide is helical in a pure DPPC membrane.
Fig. 2.

Collective variable (RMSD of backbone compared to perfect helix) in Å vs. simulation time in ns and histogram of RMSD vs. frequency for equilibrium MD of (A) WT1, (B) R694L, and (C) WT1ΔChol systems.
Production MD further shows that these stable α-helices remain in a classical membrane-spanning orientation. In Fig. 3, we show mass–density profiles, averaged over the last 100 ns, for various components in the WT (averaged over 3 replicas), R694L, and WTΔChol (averaged over 3 replicas) systems. The right-hand panels in this figure show global density profiles that establish the z-span (membrane normal) of the bilayer, and the accompanying left-hand panels show the scaled local density of protein and water within 4 Å of the protein. Clearly all proteins span the bilayer. Also highlighted in the left-hand panels of Fig. 3 are the mass density distribution of atoms in the 694 residue; we see that R694 density overlaps with water density within 4 Å, indicating the R694 is solvated in both the WT and WTΔChol systems. R694L is clearly not solvated, as expected.
Fig. 3.

(A, C, and E) Mass density in 102 amu/Å3 along membrane normal in Å during equilibrium MD for various components of the system: protein, local water (defined as within 4 Å of protein), and midspan residue (arginine or leucine). (B, D, and F) Mass density in amu/Å3 along membrane normal in Å during equilibrium MD for various components of the system: lipid tails, lipid headgroups, water, and cholesterol. A and B are averages for WT1,WT2, and WT3 systems. E and F are averages for WT1ΔChol, WT2ΔChol, and WT3ΔChol systems. All statistics are from the last 100 ns of each trajectory and error bars in A, B, E and F represent standard error.
3.2. The midspan arginine is solvated by a robust water defect
Fig. 4 shows end-point snapshots from the WT1, R694L, WT1ΔChol, and WT3ΔChol systems, which illustrate some of the major differences among these systems. The major difference between the WT1 and R694L configurations shown here is that the WT1 has a classical water defect (highlighted by cyan van der Waals spheres) that connects the R694 side-chain with the bulk solvent through the inner leaflet, while no such defect exists in the R694L system. Noticeable in the snapshots of the WT1ΔChol and WT3ΔChol configurations are the overall gel-like alignment of the DPPC tails except near the proteins, which also display classical water defects.
Fig. 4.

Representative system configurations from equilibrium MD at 300 ns rendered in VMD [42] for the WT1, R694L, WT1ΔChol, and WT3ΔChol systems. Lipid headgroups are in red vdW, lipid tails in white vdW, cholesterol in yellow vdW, water in cyan isosurface, peptide in orange new cartoon, and R694 or R694L in orange vdW. Water with oxygens within 4 Å of the protein and located on the inner leaflet of the membrane are shown in cyan vdW. For clarity, lipids, cholesterol, and water in the foreground of all four configurations are not shown.
Solvation of R694 is a robust feature of the entire set of production MD runs on the WT MSD. In Fig. 5A, we show the number of water molecules uniquely associated with each residue of the MSD for the three wild-type systems in the mixed bilayer (WT1, WT2, and WT3, averaged over the final 100 ns of each simulation). The data show that the midspan arginine (local residue index 14) directly interacts with 4.8 ± 0.03 (SE) water molecules throughout the trajectory. Peaks every fourth residue thereafter signify the amphipathic nature of the sequence and the fact that the waters predominantly solvate one side of the helix and, importantly, connect to the bulk interior-side water. It is interesting to note that the uniformity in the solvation profile in the three systems exists in spite of large differences in the local membrane composition near the protein in the three systems, illustrated in Supplemental Figure 4.
Fig. 5.
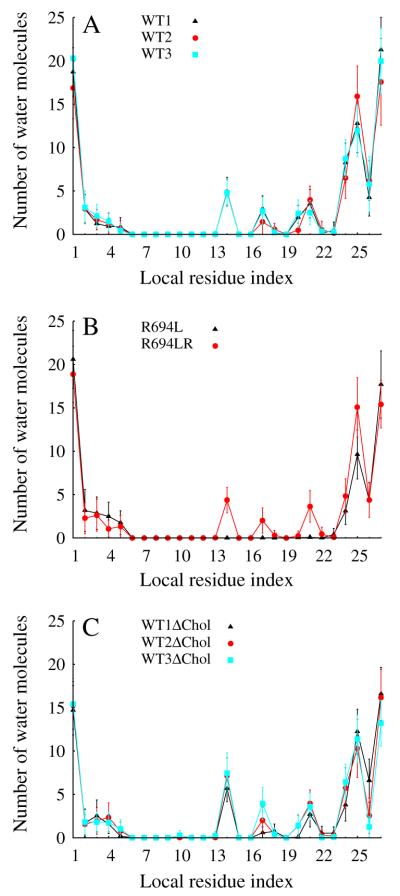
Number of water molecules within 4 Å of protein vs. amino acid that each water molecule is uniquely attributed to during equilibrium MD. All statistics are from the last 100 ns of each trajectory and error bars represent standard deviation.
Fig. 5B shows that the midspan leucine mutant has no water defect. Since the water defect in the WT1 system may have resulted from waters solvating R694 prior to the production run (see System Setup), a second WT system was generated by spontaneously “undoing” the mutation of R694L to generate the R694LR system which was allowed to equilibrate and accumulate 300 ns of production MD. Water molecules enter the membrane within 9 ns and the water defect re-establishes itself on a roughly 100 ns timescale, as shown in mass density plots in Supplemental Figure 2C, and D. In Fig. 5B, we show the number of water molecules uniquely associated with each residue of the MSD for the R694LR system, which is nearly identical to that of the original WT1 system. Because the R694L system has no water defect, the waters that form the defect in R694LR spontaneously entered the membrane upon mutation and production MD. As shown in Fig. 5C, the midspan arginine interacts with 6.7 ± 0.44 (SE) waters in the pure DPPC bilayer with an amphipathic solvation signature similar to those in the mixed bilayer.
Fig. 6A shows the tilt angle of the MSD helical axis with respect to the global z axis as a function of simulation time in the three mixed-bilayer WT simulations. Though strongly fluctuating, all MSDs in the mixed bilayer sample around a tilt angle of 19.5° ± 1.4 (SE) over the last 100 ns. The data in Fig. 6B show that the midspan leucine mutant displays a lower tilt angle than the WT with an average of 14.3° ± 3.1 (SD) and the R694LR system has an average of 24.7° ± 3.8 (SD) for the last 100 ns. Taken together, this suggests that the water defect is correlated with larger tilt angles. In contrast to the uniformity of tilt angle observed for the cholesterol-containing systems, Fig. 6C illustrates that the tilt angles for the MSDs in the pure DPPC bilayers can vary significantly (averages of 7.2° ± 3.0, 22.6° ± 3.0, 39.5° ± 3.7 (SD) for WT1ΔChol, WT2 ΔChol, WT3ΔChol, respectively). For example, WT1ΔChol shows a nearly vertical MSD helix, a fact also illustrated by the snapshot of this system shown in Fig. 4. At the other extreme, WT3ΔChol samples tilt angles between 30 and 45°. Yet all three MSDs in pure DPPC show classical water defects, as indicated by the water counts in Fig. 5C. The apparent relationship between the water defect and helix tilt is therefore dependent on the membrane cholesterol content: only in the mixed bilayer is larger tilt unambiguously associated with the defect. To explain this, we next turn to local maps of membrane thickness.
Fig. 6.
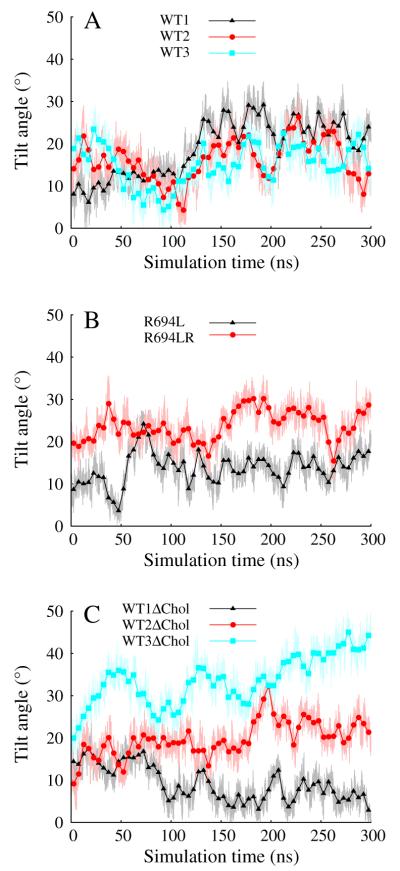
Tilt angle in degrees vs. simulation time in ns.
3.3. Defect-mediated local thinning is cholesterol-dependent
The two left-hand columns of Fig. 7 show maps of membrane thickness L (see Methods) and its standard deviation σL averaged over each of the three contiguous 100 ns intervals of the 300 ns production runs for the mixed-bilayer system WT1 and the leucine mutant R694L. L is nearly identical between the two mixed-bilayer systems, except for the area within about 10 Å from the protein in which L is significantly lower for the WT1 MSD. Fluctuations in L are essentially absent in the leucine-mutant system, while fluctuations of more than 7 Å appear correlated with the position of the water defect in the WT1 system. The time-course of these maps for both the mixed-bilayer systems is essentially constant, indicating that the uniformity of membrane thickness and the correlation of strong fluctuations with the water defect site are stable phenomena.
Fig. 7.

Maps of membrane thickness, L, (top) in Å and standard deviation, σL, (bottom) in Å for the WT1, R694L, WT1ΔChol, and WT3ΔChol systems during 100 ns intervals of equilibrium MD. Overlaid on the maps are the x and y positions of the non-hydrogen atoms of the peptide (black circles) and the 694 residue (white circles) from the last frame of the trajectories. The N- and C-termini are labeled.
In the two right-hand columns of Fig. 7 we show L and σL in the same manner as in the left-hand columns, but for two of the pure-DPPC systems, WT1ΔChol and WT3ΔChol. L is much less uniform over the entire system for the pure DPPC bilayers, which display large regions of distinctly lower thickness associated with lipid tails that are not ordered. Bilayer thinning local to the peptide is also much less severe in WT1ΔChol than in the mixed-bilayer system WT1, which is consistent with the observation of near-zero tilt of the MSD helix in the pure DPPC system. However, we still observe that the strongest fluctuations in L are correlated with the water defect, but that these fluctuations in L require at least 100 ns to equilibrate. In contrast to WT1ΔChol, the MSD helix in WT3ΔChol is strongly tilted and lies in a locally thin region of the membrane. Nevertheless, fluctuations in membrane thickness are localized around the defect and these fluctuations also require at least 100 ns to equilibrate. Peristaltic motions, similar to our definition of thickness fluctuations here, have been measured experimentally and computationally in pure DPPC bilayers and the thickness fluctuation amplitude was found to be 3.5–5 Å (at various temperatures) [60–62]. These agree fairly well with our observations.
Because membrane thickness depends on locations of lipid headgroups in both leaflets, it does not necessarily reflect the true effect of water penetration into one bilayer. In Fig. 8 we show maps of the average-minimum distance of any water molecule on the inner side to the global membrane midplane (z = 0), and its standard deviation, at 4 × 4 Å2 resolution, for three representative systems (WT1, WT1ΔChol and WT3ΔChol). In the mixed-bilayer, water penetration is strongly localized at the protein and also strongly fluctuating. For the two pure DPPC cases, the areas of greatest water penetration are evidently unions of areas of lowest membrane thickness and the local water defect. That is, the midspan arginine is not solely responsible for bringing water close to the midplane; thickness fluctuations do so as well. It appears that the main effect of cholesterol is to localize thinning of the membrane and contain the water defect.
Fig. 8.
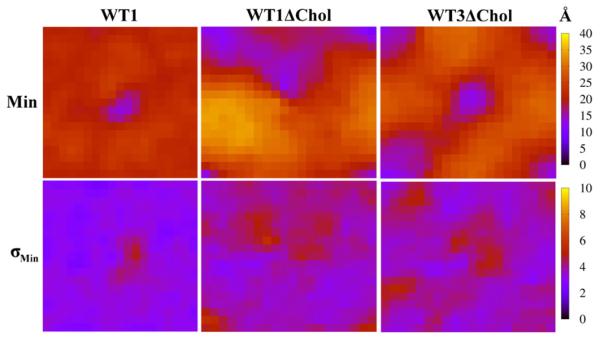
Maps of average minimum distance along membrane normal in the inner leaflet of water molecules from global center of mass of lipid bilayer, Min (top), in Å and standard deviation, σMin, (bottom) in Å for WT1, WT1ΔChol, and WT3ΔChol systems.
Maps of membrane thickness and average minimum water penetration for systems not shown in Figs. 7 and 8 are in Supplemental Figures 7 and 8. For example, in Supplemental Figure 7, the protein-free, cholesterol-containing bilayer, DPPC/Chol, has a similar uniform thickness of 48 Å compared to the protein-free areas of the water-defect-free R694L system. This thickness is consistent with previous experimental and computational studies on DPPC/Chol systems and is larger than cholesterol-free DPPC membranes in the liquid phase due to the ordering effect of cholesterol [32,33]. Also, the protein-free bilayer, DPPC, is consistent with previous studies on pure DPPC bilayers in the solid phase; this system has an area per lipid of 48.8 Å2 comparable to the experimental value of 47.2 Å2 [63].
4. Conclusion
We have used an extensive set of MD simulations to probe the nature of the water defect that solvates the midspan arginine (R694) in the HIV-1 gp41 MSD and its relationship to cholesterol. Our results support the previous finding that the thermodynamically preferred state of the excised gp41 MSD in a cholesterol-containing-bilayer is as a membrane-spanning α-helix, and further metadynamics results indicate that the R694L mutant is also stable as a membrane-spanning α-helix. The water defect solvating R694 is a robust feature of all simulations of wild-type gp41 MSD, independent of local membrane composition and initial conditions. Cholesterol’s major effect seems to be localizing membrane thinning to the water defect, which appears to more tightly regulate the tilt angles displayed by membrane-spanning MSD helices. The defect-local membrane thinning conferred by cholesterol is strongly fluctuating.
The insights gained from these simulations allow us to embark on some informed speculation that we hope will stimulate further experimental work aimed at understanding structure–function relationships in spike-membrane systems. First, we speculate that tightly regulated tilt angles for the gp41 MSDs are important for stabilizing trimeric complexes that involve direct interaction of MSDs, because an axisymmetric trimer with a three-fold axis normal to the bilayer plane would require its monomeric constituents (individual MSD helices) to have the same tilt angle. This may mean that cholesterol is in fact important in conferring stability to the HIV-1 envelope spike.
Second, the effect of the water defect on the stability of the membrane against poration or fusion could also be important. An interesting question to pursue now is, which represents a “weaker” membrane in the context of the HIV-1 gp41 MSD: the cholesterol-containing or cholesterol-free bilayer? Although we have not done the calculations to answer this question, it may be worth considering that cholesterol-depleted HIV-1 is unable to infect, and that this effect is reversible. It is possible that strong water fluctuations along membrane normal in the inner leaflet hydrophobic core near membrane spikes on HIV-1 disappear as cholesterol is depleted, and can easily resume as cholesterol is resupplied. It is reasonable to think that with the gp41 trimer in native HIV-1 spikes, the monomer water defects have merged and represent a local metastability of the membrane that can be harnessed during fusion. We speculate that these localized water fluctuations are important for metastability and therefore are important for entry.
Supplementary Material
Acknowledgements
Financial support from the National Institutes of Health (grant No. R01 AI-084117-01) is acknowledged. This research was supported in part by the National Science Foundation through XSEDE resources under grant No. TG-MCB070073N.
Footnotes
Appendix A. Supplementary data Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbamem.2014.01.009.
References
- [1].Frankel AD, Young JA. HIV-1: Fifteen proteins and an RNA, Annu. Rev. Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- [2].Gallo S. The HIV env-mediated fusion reaction, Biochim. Biophys. Acta. 2003;1614(1):36–50. doi: 10.1016/s0005-2736(03)00161-5. [DOI] [PubMed] [Google Scholar]
- [3].Yue L, Shang L, Hunter E. Truncation of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein defines elements required for fusion, incorporation, and infectivity. J. Virol. 2009;83(22):11588–11598. doi: 10.1128/JVI.00914-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Owens RJ, Burke C, Rose JK. Mutations in the membrane-spanning domain of the human immunodeficiency virus envelope glycoprotein that affect fusion activity. J. Virol. 1994;68(1):570–574. doi: 10.1128/jvi.68.1.570-574.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Helseth E, Olshevsky U, Gabuzda D, Ardman B, Haseltine W, Sodroski J. Changes in the transmembrane region of the human immunodeficiency virus type 1 gp41 envelope glycoprotein affect membrane fusion. J. Virol. 1990;64(12):6314–6318. doi: 10.1128/jvi.64.12.6314-6318.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Miyauchi K, Komano J, Yokomaku Y, Sugiura W, Yamamoto N, Matsuda Z. Role of the specific amino acid sequence of the membrane-spanning domain of human immunodeficiency virus type 1 in membrane fusion. J. Virol. 2005;79(8):4720–4729. doi: 10.1128/JVI.79.8.4720-4729.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miyauchi K, Curran R, Matthews E, Komano J, Hoshino T, Engelman DM, Matsuda Z. Mutations of conserved glycine residues within the membrane-spanning domain of human immunodeficiency virus type 1 gp41 can inhibit membrane fusion and incorporation of Env onto virions. Jpn. J. Infect. Dis. 2006;59(2):77–84. [PubMed] [Google Scholar]
- [8].Shang L, Yue L, Hunter E. Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell–cell fusion and virus infection. J. Virol. 2008;82(11):5417–5428. doi: 10.1128/JVI.02666-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shang L, Hunter E. Residues in the membrane-spanning domain core modulate conformation and fusogenicity of the HIV-1 envelope glycoprotein. J. Virol. 2010;404(2):158–167. doi: 10.1016/j.virol.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miyauchi K, Curran AR, Long Y, Kondo N, Iwamoto A, Engelman DM, Matsuda Z. The membrane-spanning domain of gp41 plays a critical role in intracellular trafficking of the HIV envelope protein. Retrovirology. 2010;7(1):95. doi: 10.1186/1742-4690-7-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Long Y, Meng F, Kondo N, Iwamoto A, Matsuda Z. Conserved arginine residue in the membrane-spanning domain of HIV-1 gp41 is required for efficient membrane fusion. Protein Cell. 2011;2(5):369–376. doi: 10.1007/s13238-011-1051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix–helix association. J. Mol. Biol. 2000;296(3):911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- [13].Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 2005;102(40):14278–14283. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Welman M, Lemay G, Cohen EA. Role of envelope processing and gp41 membrane spanning domain in the formation of human immunodeficiency virus type 1 (HIV-1) fusion-competent envelope glycoprotein complex. Virus Res. 2007;124(1–2):103–112. doi: 10.1016/j.virusres.2006.10.009. [DOI] [PubMed] [Google Scholar]
- [15].Andreassen H, Bohr H, Bohr J, Brunak S, Buggae T, Cotterill RMJ, Jacobsen C, Kusk P, Lautrup B, Petersen SB, et al. Analysis of the secondary structure of the human immunodeficiency virus (HIV) proteins p17, gp120, and gp41 by computer modeling based on neural network methods. J. Acquir. Immune Defic. Syndr. 1990;3(6):615–622. [PubMed] [Google Scholar]
- [16].Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987;237(4820):1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- [17].Berman PW, Nunes WM, Haffar OK. Expression of membrane-associated and secreted variants of gp160 of human immunodeficiency virus type 1 in vitro and in continuous cell lines. J. Virol. 1988;62(9):3135–3142. doi: 10.1128/jvi.62.9.3135-3142.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gangupomu VK, Abrams CF. All-atom models of the membrane-spanning domain of HIV-1 gp41 from metadynamics. Biophys. J. 2010;99(10):3438–3444. doi: 10.1016/j.bpj.2010.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schow EV, Freites JA, Cheng P, Bernsel A, von Heijne G, White SH, Tobias DJ. Arginine in membranes: the connection between molecular dynamics simulations and translocon-mediated insertion experiments. J. Membr. Biol. 2011;239(1–2):35–48. doi: 10.1007/s00232-010-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fleming PJ, Freites JA, Moon CP, Tobias DJ, Fleming KG. Outer membrane phospholipase A in phospholipid bilayers: a model system for concerted computational and experimental investigations of amino acid side chain partitioning into lipid bilayers. Biochim. Biophys. Acta. 2012;1818(2):126–134. doi: 10.1016/j.bbamem.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].MacCallum JL, Bennett WFD, Tieleman DP. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J. 2008;94(9):3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Johansson ACV, Lindahl E. Amino-acid solvation structure in transmembrane helices from molecular dynamics simulations. Biophys. J. 2006;91(12):4450–4463. doi: 10.1529/biophysj.106.092767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vostrikov VV, Hall BA, Greathouse DV, Koeppe RE, Sansom MSP. Changes in transmembrane helix alignment by arginine residues revealed by solid-state NMR experiments and coarse-grained MD simulations. J. Am. Chem. Soc. 2010;132(16):5803–5811. doi: 10.1021/ja100598e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vostrikov VV, Hall BA, Sansom MSP, Koeppe RE. Accommodation of a central arginine in a transmembrane peptide by changing the placement of anchor residues. J. Phys. Chem. B. 2012;116(43):12980–12990. doi: 10.1021/jp308182b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Moon CP, Fleming KG. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl. Acad. Sci. U. S. A. 2011;108(25):10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gumbart J, Roux B. Determination of membrane-insertion free energies by molecular dynamics simulations. Biophys. J. 2012;102(4):795–801. doi: 10.1016/j.bpj.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Freites JA, Tobias DJ, von Heijne G, White SH. Interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. U. S. A. 2005;102(42):15059–15064. doi: 10.1073/pnas.0507618102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Herrera FE, Bouchet A, Lairion F, Disalvo EA, Pantano S. Molecular dynamics study of the interaction of arginine with phosphatidylcholine and phosphatidylethanolamine bilayers. J. Phys. Chem. B. 2012;116(15):4476–4483. doi: 10.1021/jp2096357. [DOI] [PubMed] [Google Scholar]
- [29].Krepkiy D, Mihailescu M, Freites JA, Schow EV, Worcester DL, Gawrisch K, Tobias DJ, White SH, Swartz KJ. Structure and hydration of membranes embedded with voltage-sensing domains. Nature. 2009;462(7272):473–479. doi: 10.1038/nature08542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li LB, Vorobyov I, Allen TW. The role of membrane thickness in charged protein–lipid interactions. Biochim. Biophys. Acta. 2012;1818(2):135–145. doi: 10.1016/j.bbamem.2011.10.026. [DOI] [PubMed] [Google Scholar]
- [31].Li S, Su Y, Luo W, Hong M. Water–protein interactions of an arginine-rich membrane peptide in lipid bilayers investigated by solid-state nuclear magnetic resonance spectroscopy. J. Phys. Chem. B. 2010;114(11):4063–4069. doi: 10.1021/jp912283r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sankaram MB, Thompson TE. Modulation of phospholipid acyl chain order by cholesterol. a solid-state 2H nuclear magnetic resonance study. Biochemistry. 1990;29(47):10676–10684. doi: 10.1021/bi00499a015. [DOI] [PubMed] [Google Scholar]
- [33].Cournia Z, Ullmann GM, Smith JC. Differential effects of cholesterol, ergosterol and lanosterol on a dipalmitoyl phosphatidylcholine membrane: a molecular dynamics simulation study. J. Phys. Chem. B. 2007;111(7):1786–1801. doi: 10.1021/jp065172i. [DOI] [PubMed] [Google Scholar]
- [34].Brügger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Kräusslich HG. The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. U. S. A. 2006;103(8):2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Campbell S, Gaus K, Bittman R, Jessup W, Crowe S, Mak J. The raft-promoting property of virion-associated cholesterol, but not the presence of virion-associated Brij 98 rafts, is a determinant of human immunodeficiency virus type 1 infectivity. J. Virol. 2004;78(19):10556–10565. doi: 10.1128/JVI.78.19.10556-10565.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guyader M, Kiyokawa E, Abrami L, Turelli P, Trono D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J. Virol. 2002;76(20):10356–10364. doi: 10.1128/JVI.76.20.10356-10364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. U. S. A. 2001;98(24):13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Waheed AA, Freed EO. Lipids and membrane microdomains in HIV-1 replication. Virus Res. 2009;143(2):162–176. doi: 10.1016/j.virusres.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhukovsky MA, Lee P-H, Ott A, Helms V. Putative cholesterol-binding sites in human immunodeficiency virus (HIV) coreceptors CXCR4 and CCR5. Proteins Struct. Funct. Bioinf. 2013;81(4):555–567. doi: 10.1002/prot.24211. [DOI] [PubMed] [Google Scholar]
- [40].Campbell SM, Crowe SM, Mak J. Virion-associated cholesterol is critical for the maintenance of HIV-1 structure and infectivity. AIDS. 2002;16(17):2253–2261. doi: 10.1097/00002030-200211220-00004. [DOI] [PubMed] [Google Scholar]
- [41].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics. 1996;14(1):33–38. 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- [43].MacKerell AD, Jr., Bashford D, Bellott M, Dunbrack RL, Jr., Evanseck JD, Field MJ, Fischer S, Gao d J., Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenk rich, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;5647(97):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- [44].Feller SE, MacKerell AD. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B. 2000;104(31):7510–7515. [Google Scholar]
- [45].Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pastor RW, MacKerell AD. Development of the CHARMM force field for lipids. J. Phys. Chem. Lett. 2011;2(13):1526–1532. doi: 10.1021/jz200167q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009;97(1):50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Guo W, Kurze V, Huber T, Afdhal NH, Beyer K, Hamilton JA. A solid-state NMR study of phospholipid–cholesterol interactions: sphingomyelin–cholesterol binary systems. Biophys. J. 2002;83(3):1465–1478. doi: 10.1016/S0006-3495(02)73917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].McIntosh TJ, Simon SA, Needham D, Huang CH. Structure and cohesive properties of sphingomyelin/cholesterol bilayers. Biochemistry. 1992;31(7):2012–2020. doi: 10.1021/bi00122a017. [DOI] [PubMed] [Google Scholar]
- [50].Chiang Y-W, Costa-Filho AJ, Freed JH. Dynamic molecular structure and phase diagram of DPPC-cholesterol binary mixtures: a 2D-ELDOR study. J. Phys. Chem. B. 2007;111(38):11260–11270. doi: 10.1021/jp0732110. [DOI] [PubMed] [Google Scholar]
- [51].Redondo-Morata L, Giannotti MI, Sanz F. Influence of cholesterol on the phase transition of lipid bilayers: a temperature-controlled force spectroscopy study. Langmuir. 2012;28(35):12851–12860. doi: 10.1021/la302620t. [DOI] [PubMed] [Google Scholar]
- [52].Leekumjorn S, Sum AK. Molecular studies of the gel to liquid-crystalline phase transition for fully hydrated DPPC and DPPE bilayers. Biochim. Biophys. Acta. 2007;1768(2):354–365. doi: 10.1016/j.bbamem.2006.11.003. [DOI] [PubMed] [Google Scholar]
- [53].Schubert T, Schneck E, Tanaka M. First order melting transitions of highly ordered dipalmitoyl phosphatidylcholine gel phase membranes in molecular dynamics simulations with atomistic detail. J. Chem. Phys. 2011;135(5):055105. doi: 10.1063/1.3615937. [DOI] [PubMed] [Google Scholar]
- [54].Janiak M, Small D, Shipley G. Nature of the thermal pretransition of synthetic phospholipids: dimyristoyl-and dipalmitoyllecithin. Biochemistry. 1976;15(21):4575–4580. doi: 10.1021/bi00666a005. [DOI] [PubMed] [Google Scholar]
- [55].Wu F-G, Jia Q, Wu R-G, Yu Z-W. Regional cooperativity in the phase transitions of dipalmitoylphosphatidylcholine bilayers: the lipid tail triggers the isothermal crystallization process. J. Phys. Chem. B. 2011;115(26):8559–8568. doi: 10.1021/jp200733y. [DOI] [PubMed] [Google Scholar]
- [56].Laio A, Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A. 2002;99(20):12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Laio A, Rodriguez-Fortea A, Gervasio FL, Ceccarelli M, Parrinello M. Assessing the accuracy of metadynamics. J. Phys. Chem. B. 2005;109(14):6714–6721. doi: 10.1021/jp045424k. [DOI] [PubMed] [Google Scholar]
- [58].Bussi G, Laio A, Parrinello M. Equilibrium free energies from nonequilibrium metadynamics. Phys. Rev. Lett. 2006;96:090601. doi: 10.1103/PhysRevLett.96.090601. [DOI] [PubMed] [Google Scholar]
- [59].Laio A, Gervasio FL. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008;71(12):126601. [Google Scholar]
- [60].Lindahl E, Edholm O. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecular dynamics simulations. Biophys. J. 2000;79(1):426–433. doi: 10.1016/S0006-3495(00)76304-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Brannigan G, Brown FLH. A consistent model for thermal fluctuations and protein-induced deformations in lipid bilayers. Biophys. J. 2006;90(5):1501–1520. doi: 10.1529/biophysj.105.075838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Woodka AC, Butler PD, Porcar L, Farago B, Nagao M. Lipid bilayers and membrane dynamics: insight into thickness fluctuations. Phys. Rev. Lett. 2012;109(5):058102. doi: 10.1103/PhysRevLett.109.058102. [DOI] [PubMed] [Google Scholar]
- [63].Tristram-Nagle S, Zhang R, Suter RM, Worthington CR, Sun WJ, Nagle JF. Measurement of chain tilt angle in fully hydrated bilayers of gel phase lecithins. Biophys. J. 1993;64(4):1097–1109. doi: 10.1016/S0006-3495(93)81475-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.