

Significance
Mutations in SMARCAL1, a DNA remodeling protein fundamental to genome integrity during replication, lead to the developmental disorder Schimke immuno-osseous dysplasia (SIOD). SMARCAL1-deficient cells exhibit collapsed replication forks, S-phase cell cycle arrest, increased chromosomal breaks, hypersensitivity to genotoxic agents, and chromosomal instability. SMARCAL1 facilitates replication restart by catalyzing ATP-dependent fork regression and branch migration, the mechanism of which is poorly understood. Here we provide structural and biochemical evidence for a conserved substrate recognition domain in DNA repair enzymes that couples ATP hydrolysis to remodeling of a variety of DNA structures, and provide insight into the role of this domain in replication fork stability and genome integrity.
Keywords: replication restart, fork reversal
Abstract
SMARCAL1, a DNA remodeling protein fundamental to genome integrity during replication, is the only gene associated with the developmental disorder Schimke immuno-osseous dysplasia (SIOD). SMARCAL1-deficient cells show collapsed replication forks, S-phase cell cycle arrest, increased chromosomal breaks, hypersensitivity to genotoxic agents, and chromosomal instability. The SMARCAL1 catalytic domain (SMARCAL1CD) is composed of an SNF2-type double-stranded DNA motor ATPase fused to a HARP domain of unknown function. The mechanisms by which SMARCAL1 and other DNA translocases repair replication forks are poorly understood, in part because of a lack of structural information on the domains outside of the common ATPase motor. In the present work, we determined the crystal structure of the SMARCAL1 HARP domain and examined its conformation and assembly in solution by small angle X-ray scattering. We report that this domain is conserved with the DNA mismatch and damage recognition domains of MutS/MSH and NER helicase XPB, respectively, as well as with the putative DNA specificity motif of the T4 phage fork regression protein UvsW. Loss of UvsW fork regression activity by deletion of this domain was rescued by its replacement with HARP, establishing the importance of this domain in UvsW and demonstrating a functional complementarity between these structurally homologous domains. Mutation of predicted DNA-binding residues in HARP dramatically reduced fork binding and regression activities of SMARCAL1CD. Thus, this work has uncovered a conserved substrate recognition domain in DNA repair enzymes that couples ATP-hydrolysis to remodeling of a variety of DNA structures, and provides insight into this domain’s role in replication fork stability and genome integrity.
Accurate DNA replication is essential for genomic stability (1). Aberrant DNA structures, protein barriers, and chemically modified DNA thwart progression of the replication fork and lead to mutations, cytotoxicity, and increased chromosomal rearrangements (2–5). Uncoupling of polymerase and helicase activities at a stalled fork (6) results in an accumulation of single-stranded (ss) DNA (Fig. 1A), rendering the genome susceptible to nuclease cleavage and thereby increasing the probability of genetic rearrangements (7–9). Failure to stabilize stalled forks can lead to replisome dissociation and fork degradation or collapse. DNA damage response (DDR) pathways maintain genomic integrity by rectifying or protecting stalled or collapsed forks and regulating DNA repair (9–11). In humans, accumulation of the ssDNA-binding protein Replication Protein A (RPA) at stalled forks signals recruitment of the S-phase checkpoint kinase ATR (12–15), consequently triggering recruitment of proteins that promote sister chromatid cohesion and stabilize the histone/chromatin structure (16).
Fig. 1.

Structure of the SMARCAL1 HARP1 domain. (A) SMARCAL1 catalyzes regression of a stalled replication fork into a four-stranded “chicken foot” DNA structure. (B) SMARCAL1 domain organization. Patient mutations are marked by x (sense) and o (nonsense). HARP domains are shaded green, the RPA-binding domain (RBD) is purple, RecA domains are dark blue and red, and SNF2-specific inserted helical domains (HD) are light blue and pink. The HARP2-ATPase catalytic domain is denoted by a gold bar. (C) Two protomers (green and silver) of mouse HARP1 as observed in the crystal structure. The position of the twofold noncrystallographic symmetry axis is shown in orange. (D) SAXS model of monomeric HARP1 (green) superimposed on the GASBOR ab initio molecular envelope (gray). (E) Superposition of HARP1 (green) and the Archaeoglobus fulgidus XPB DRD (PDB ID code 2FWR; silver). (F) AfXPB (ATPase, blue/red; DRD, green). (G) T4 phage UvsW (PDB ID code 2OCA; ATPase, blue/red; HARP-like, green).
Stabilization, repair, and restart of stalled forks can involve regression of the fork into a four-stranded “chicken foot” structure, in which the nascent DNA strands are unwound from their respective leading and lagging strand templates and reannealed to one another (Fig. 1A) (17). SMARCAL1 (SWI/SNF-related, matrix associated, actin-dependent regulator of chromatin, subfamily a-like 1), also known as HARP (HepA-related protein), is one of several ATP-dependent motor proteins capable of fork regression and important for genetic stability, including Rad54, RecQ paralogs, BLM, WRN, FANCM, ZRANB3, HLTF/Rad5, T4 bacteriophage UvsW, archaeal HelQ/Hel308/Hjm, and Escherichia coli RecG (18–26). SMARCAL1 is a distant SNF2 family member of dsDNA translocating chromatin remodeling proteins (27) with a binding preference for branched DNA structures, and has been shown to catalyze ATP-dependent regression of model replication forks (Fig. 1A), branch migration of Holliday junctions, and reannealing of RPA-coated plasmids (28–30). RPA binding recruits SMARCAL1 to stalled replication forks in cells and regulates its specificity to promote regression of stalled forks caused by stalling of the leading strand polymerase as well as the reverse reaction to restore normal replication forks with lagging-strand ssDNA (31).
Depletion of SMARCAL1 in cells leads to MUS81-dependent double-strand (ds) breaks and cellular sensitivity to replication stress agents (29). Inherited, biallelic loss of function mutations in SMARCAL1 result in the disease Schimke immunoosseous dysplasia (SIOD), characterized by pleiotropic developmental problems and cancer predisposition (32–35). Mutations associated with SIOD are localized to the N-terminal RPA-binding domain, two HARP domains (HARP1 and HARP2), and the SNF2 (DEXD/HELICc) ATPase region (Fig. 1B).
Despite the conservation of the SMARCAL1 ATPase domain to the SNF2 family of chromatin remodelers (27, 36), the HARP sequences are unique, and thus how these accessory domains contribute to DNA remodeling activities of SMARCAL1 is unclear. Here we show that the HARP domain is structurally homologous to the substrate recognition domains of MutS and XPB and the putative DNA specificity motif of UvsW. We demonstrate the importance of this domain to catalytic activity by showing that loss of UvsW activity by deleting this domain is restored by a HARP-UvsW chimera, and that mutations in SMARCAL1 HARP2 abrogate fork regression activity by preferentially reducing SMARCAL1 binding to DNA junctions that resemble replication forks. Together with SAXS analysis of the protein–DNA complex, these data illustrate how the HARP-ATPase catalytic core binds DNA junctions to promote stabilization of stalled replication forks.
Results
HARP Is a Conserved DNA-Remodeling Domain.
The SMARCAL1 catalytic domain (SMARCAL1CD; residues 325–870) consists of the HARP2 and ATPase motifs (Fig. 1B), which together are sufficient for DNA binding and fork regression activities (29). HARP2 is required for activity and folding of the catalytic domain (29), and thus we set out to determine the crystal structure of the HARP motif to better understand its contribution to SMARCAL1 activity. In the absence of the ATPase domain, HARP2 was insoluble, consistent with a unified HARP2-ATPase domain. Therefore, we determined the crystal structure of HARP1 from Mus musculus SMARCAL1, which shares 88% and 76% sequence similarity to human HARP1 and HARP2, respectively. The X-ray crystal structure was phased by single-wavelength anomalous dispersion (SAD) using selenomethionine (SeMet)-substituted HARP1 and refined to 1.9 Å using native diffraction data to a crystallographic residual of 16.9% (Rfree = 19.7%) (SI Appendix, Table S1 and Fig. S1).
HARP1 crystallized as a dimer with an N-terminal two-stranded β-sheet (β1-β2) exchanged between two protomers related by a noncrystallographic symmetry axis (Fig. 1C). Dimerization of HARP1 in crystals and the presence of tandem HARP1 and HARP2 sequences in SMARCAL1 raised the hypothesis that the HARP domains may form intramolecular or intermolecular dimers. However, SMARCAL1 and SMARCAL1CD are both monomeric, as measured by size-exclusion chromatography (SEC) (SI Appendix, Fig. S2) and small-angle X-ray scattering (SAXS) (29), and no interaction between HARP1 and SMARCAL1CD was observed using affinity chromatography. In addition, SAXS analysis of HARP1 indicated a clear monomeric protein in solution (SI Appendix, Fig. S1C), with a solution state mass of 7.76 kDa (actual, 7.54 kDa). Thus, it is likely that HARP1 dimerization is a crystallographic artifact. Therefore, we constructed a model of the HARP1 monomer by exchanging the coordinates of the two symmetry-related β1-β2 sheets. The resulting compact globular fold consists of a five-stranded β-sheet packed against two α-helices. The model of the monomeric HARP1 motif showed excellent agreement with the SAXS profile and molecular envelope (Fig. 1D and SI Appendix, Fig. S1C), and Kratky analysis of the HARP1 scattering data was indicative of a compact, globular fold (Fig. 1D).
Our monomeric HARP model was further validated by searching for structural homologs to known protein folds using the Dali server (37). The resulting list of HARP homologs was dominated by DNA- and RNA-binding domains (SI Appendix, Table S2). The top-scoring hits were DNA-remodeling proteins XPB and UvsW, each with an rmsd of 2.1 Å and a z-score of 6.9 (Fig. 1E and SI Appendix, Fig. S3) (38–40). XPB helicase acts in opening DNA for transcription and nucleotide excision repair (41), and UvsW is a fork regression enzyme important for replication and repair in T4 phage (42–44). The HARP-like domains of both XPB and UvsW are located immediately N-terminal to an ATPase motor in a manner reminiscent of SMARCAL1CD (SI Appendix, Fig. S4) (29). This domain in XPB provides specificity for abasic sites and cyclopyrimidine dimers in DNA, and has been termed the damage recognition domain (DRD) based its similarity to the mismatch recognition domain (MRD) of MutS (38). Similarly, this N-terminal domain in UvsW is believed to bind DNA and provide specificity for branched structures based on its structural homology to the T4 transcription factor MotA (39, 45), although to our knowledge, the significance of this domain in UvsW has not yet been tested.
Given the conservation between the HARP folds and positions at the N-terminal ends of ATPase domains in SMARCAL1CD and UvsW, we hypothesized that the HARP motif is necessary for UvsW fork regression activity. To test this, we deleted the N-terminal domain from UvsW and replaced it with HARP1 from human SMARCAL1 to generate a HARP-UvsW chimera (Fig. 2 A and B), and measured fork regression using an established assay (29). The isolated UvsW ATPase domain, although soluble, exhibited no fork regression activity, whereas addition of HARP to the UvsW ATPase restored fork regression activity back to ∼75% of that of wild-type UvsW (Fig. 2C and SI Appendix, Fig. S5). Introduction of a K141R Walker A mutation, which abrogates UvsW ATPase and branch migration in wild-type UvsW (42, 46), also rendered the HARP-UvsW chimera inactive, indicating that the restored fork regression activity of the chimera is ATP-dependent. Thus, the structure and position of the HARP motif against the ATPase motor is conserved among DNA-remodeling proteins SMARCAL1, UvsW, and XPB and is integral to overall fork regression activities of SMARCAL1 and UvsW.
Fig. 2.

The SMARCAL1 HARP domain can operate with the UvsW motor domain. (A) Design of a HARP-UvsW chimera, in which the N-terminal substrate recognition domain of UvsW (yellow) is replaced with HARP1 from SMARCAL1 (green). The RecA lobes of the ATPase domain are in red and blue. (B) Schematic of the four constructs tested, with domains colored as in A. The position of the K141R Walker A mutation, which impairs UvsW ATPase and fork regression activities, is denoted by a white X. (C) Fork regression activity of wild-type UvsW (yellow circles), HARP-UvsW chimera (green squares), HARP-UvsWK141R mutant (black triangles), and UvsWATPase domain (gray diamonds). Raw data are provided SI Appendix, Fig. S5.
The HARP DNA-Binding Interface Is Necessary for Fork Regression.
After XPB and UvsW, the next-closest HARP homologs identified were eukaryotic translation initiation factor 1 (eIF1), the MRD of the human MutSα MSH6 and MSH3 subunits, with all MutS orthologs populating the list, and the T4 phage transcription factor MotA (SI Appendix, Table S2 and Fig. S3). Structures of MutS and eIF1 bound to their respective DNA and RNA substrates provide a structural hypothesis for how the HARP fold may interact with nucleic acids (SI Appendix, Figs. S6 and S7). In structures of MutSα (MSH6-MSH2), MutSβ (MSH3-MSH2), and bacterial and archaeal MutS bound to mismatch-containing DNA, the MRD from the MSH6 or MSH3 subunit engages the mismatch region through residues in the β1-β2 loop, the β2/β5 sheet, the coil preceding β5, and along β5 and α6, which widen the minor groove and bend the DNA (SI Appendix, Fig. S6B) (47–52). In contrast, the same domain from MSH2 makes nonspecific DNA contacts. Interestingly, the eIF1 protein is composed entirely of the HARP fold (53) and binds to 18S RNA in the 40S subunit of the ribosome in a nearly identical manner to the MRD–DNA interaction (SI Appendix, Figs. S6C and S7) (54). Thus, this structural analysis of the SMARCAL1 HARP domain identifies a generic binding architecture that is suited to a variety of nucleic acid targets.
Our previous analyses suggested that the HARP2 domain is required for SMARCAL1 DNA binding (29). Entire domain deletions or domain destabilizing mutants were used in those studies, however, making it difficult to discern whether the DNA-binding defects of the mutants were related to disruption of DNA interactions rather than to disruption of the SMARCAL1 fold. Furthermore, another group has reported that the HARP domains are not involved in DNA binding (55). Thus, to test whether and how the SMARCAL1 HARP domain contacts DNA, we designed a series of HARP2 mutations based on the MRD structural alignment (Fig. 3A and SI Appendix, Fig. S7). Three cluster mutants were designed to disrupt interactions in the β1-β2 and β4-β5 loops (R340G/K368D), the termini of α3 and α6 (D361P/E377N), and the β2/β5 sheet (E344Q/D346L/K371T), with substitutions to the corresponding HARP1 residues chosen to maintain the HARP fold. Mutations were placed in the context of SMARCAL1CD, and the structural integrity of each protein was verified by circular dichroism spectroscopy (SI Appendix, Fig. S7). We first examined binding of the HARP mutants to various branched DNA structures—overhang, splayed arm, flap, fork, and four-way junction—using fluorescence polarization. Wild-type SMARCAL1CD bound the five different DNA structures with near-equivalent efficiency (Fig. 3C and SI Appendix, Fig. S7E). The R340G/K368D mutant bound overhang, splayed arm, and flap structures similarly to wild type, showed a threefold decrease in affinity for the fork, and did not bind to a four-way junction. The D361P/E377N and E344Q/D346L/K371T mutants showed a 10-fold decrease in binding affinity to overhang and splayed arm DNA, and no measurable affinity for flap, fork, or four-way junction structures. Thus, the mutations had the greatest effect on binding to junctions containing an increasing number of dsDNA arms. Together with the structural homology to the MutS MRD, these results indicate that the HARP domain binds DNA in a structure-specific manner.
Fig. 3.
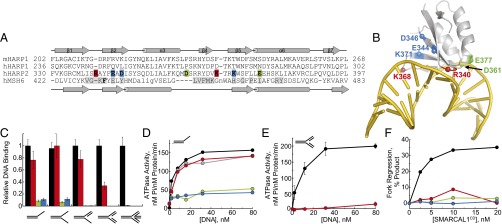
HARP2 is a DNA-binding domain. (A) Structure-based sequence alignment of the HARP1 domain from M. musculus (m) SMARCAL1 and the MRD of human (h) MSH6, together with sequences of human HARP1 and HARP2 aligned to mHARP1. Residues mutated in hHARP2 are highlighted in red, blue, and green, and hMSH6 DNA-binding residues are highlighted in gray. (B) Model of HARP bound to DNA based on superposition with the crystal structure of hMSH6-MRD (PDB ID code 2O8E) bound to a DNA mismatch. Positions of HARP2 mutations are indicated by colored spheres and labeled. (C) Relative DNA-binding affinities of wild-type (black) and mutant (red, green, and blue) SMARCAL1CD for overhang, splayed arm, flap, fork, and four-way junction DNA structures. Red, R340G/K368D; green, D361P/E377N; blue, E344Q/D346L/K371T. (D and E) DNA-dependent ATPase activity of SMARCAL1 (gray), SMARCAL1CD (black), and SMARCAL1CD mutants (red, green, and blue) against overhang (D) and fork (E) substrates. (F) Fork regression activities of wild-type and mutant SMARCAL1CD. Binding isotherms and fork regression data are shown in SI Appendix, Fig. S7.
We next measured the effect of the HARP2 mutations on DNA-dependent ATP hydrolysis and fork regression activities of SMARCAL1CD. ATPase activity of SMARCAL1CD was identical to that of the full-length enzyme (Fig. 3D). Consistent with their inability to bind DNA, the D361P/E377N and E344Q/D346L/K371T mutants were unable to hydrolyze ATP in the presence of any DNA substrate or to regress a model replication fork (Figs. 3 D and F). Likewise, the R340G/K368D mutant retained full ATPase activity in the presence of overhang DNA (Fig. 3D), but did not hydrolyze ATP in the presence of fork DNA (Fig. 3E), consistent with its reduced affinity for fork DNA. Consequently, fork regression of the R340G/K368D mutant was strongly impaired (Fig. 3F and SI Appendix, Fig. S7F). Taken together, these results demonstrate that DNA binding by the HARP2 domain is important for SMARCAL1 branch migration activity and couples DNA binding to ATP hydrolysis.
Solution States of the SMARCAL1CD/DNA Complex.
To gain structural insight into how the HARP–DNA interaction provides specificity during catalysis, we performed multiphase SAXS studies with SMARCAL1CD in the presence of DNA and ATP analogs (SI Appendix, Fig. S8). Complexes were assembled with a 25-mer DNA duplex containing a 10-nt ssDNA overhang (a simple DNA substrate that mimics a partial junction anticipated in DNA migration pathways) and either ATP substrate (ADPNP) or potential transition state (ADP⋅BeFx) mimetics. The scattering profiles of the ADPNP and ADP⋅BeFx states indicate significant differences in conformation of the two complexes (Fig. 4A).
Fig. 4.

SAXS analysis of DNA junction binding by the SMARCAL1 catalytic domain. (A) P(r) distribution for SMARCAL1CD bound to DNA containing a 25-bp duplex region and 10-nt ssDNA overhang in the presence of ADPNP (black) and ADP⋅BeFx (red). (B and C) Averaged MONSA density models of SMARCAL1CD (purple) complexed with overhang DNA (orange) in the presence of ADPNP (B) and ADP⋅BeFx (C). The thickness and length of the DNA density are consistent with a 25-bp duplex. Note the asymmetric bend in the protein-bound region of the DNA phase. (D and E) Schematic models for branch migration by SMARCAL1CD (D) and RecG (E). SF2 ATPase domains are in blue and red, and HARP (SMARCAL1) and wedge (RecG) substrate recognition domains (SRD) are in green. Arrows denote the direction of DNA movement.
We determined the molecular envelopes for each complex, with the density for the DNA and protein uniquely identified, by simultaneous analysis of SAXS data collected on the protein alone, DNA alone, and the complexes using MONSA. The SAXS envelopes for the DNA in both ADPNP and ADP⋅BeFx complexes show a linear conformation with an irregular structure at one end, presumably representative of the ssDNA overhang (Fig. 4 B and C). In the ADPNP complex, the protein envelope shows a two-domain globular structure bound to one end of the DNA (Fig. 4B). A kink in the DNA phase at the point of contact with the protein suggests that the protein is specifically bound to the ssDNA–dsDNA junction. Although we could not discern the precise orientation of the protein with respect to the DNA, atomistic models for both the catalytic core and dsDNA placed in the molecular envelopes are consistent with the DNA-dependent proteolysis protection pattern of SMARCAL1CD and resemble the cross-link repair helicase Hel308/HelQ bound to a splayed arm substrate (56–58) (SI Appendix, Fig. S9).
In contrast to the ADPNP complex, both domains of the protein in the ADP⋅BeFx complex engage the DNA (Fig. 4C). A larger protein domain, presumably representing the ATPase motor domain, contacts the middle of the DNA, whereas a smaller domain wraps around to contact one end of the DNA, which likely corresponds to the ssDNA given its irregular structure (Fig. 4C). The smaller protein lobe appears to correspond to the HARP domain in contact with the ssDNA or the ssDNA–dsDNA junction, which would place the larger ATPase domain in contact with the dsDNA, consistent with dsDNA translocase activity of the SNF2 superfamily (59, 60). Thus, the SAXS data are consistent with a DNA-binding function of the HARP domain and a conformational change that effects how the catalytic domain engages the DNA in the presence of nucleotide.
Discussion
This work establishes the HARP fold as a structurally conserved nucleic acid-binding domain that contributes to DNA remodeling by SMARCAL1 and UvsW through direct interaction with DNA. We had previously demonstrated that alanine substitution of invariant Trp and Phe residues in the interior of both HARP1 (W277A/F279A) and HARP2 (W372A/F374A) reduced DNA binding and ATPase activity (29), likely by disrupting the protein fold. Here, the structural homology between HARP and known nucleic acid-binding domains that we identified allowed us to assess DNA binding directly by creating surface mutations that retain protein folding.
Because HARP2 is integral to the fold of the catalytic domain (29), we cannot rule out the possibility that these mutations have an indirect effect on DNA binding, ATP hydrolysis, and fork regression by disrupting the HARP2–ATPase interface. Nonetheless, the subtle and specific effects of the R340G/K368D mutant on fork-dependent DNA binding and ATPase activity strongly support a direct HARP2–DNA interaction consistent with the extended DNA interaction surface visible in the ADP⋅BeFx SAXS density. Likewise, both SIOD point mutations in the HARP2 domain (E377Q and H379P) reside near the putative DNA-binding surface, suggesting that these mutations disrupt DNA binding by SMARCAL1 (SI Appendix, Fig. S10).
The preference of SMARCAL1 for substrates containing two regions of double-stranded character with an intervening gap suggests that the HARP domain senses a specific junction conformation or a disruption in the duplex structure. The analogous substrate recognition domains from MutS and XPB recognize DNA mismatches and modified nucleotides that disrupt the DNA duplex; thus, it is reasonable to hypothesize that the HARP domain recognizes the distortion in DNA found at the branch point of a stalled or regressed replication fork. Our mutational analysis of binding to different types of junctions implies that this specificity may be a result of the extra rigidity imposed by duplex DNA at a full DNA junction compared with a flap or splayed arm; that is, the HARP domain may be less important for binding to the flap structure because of the flexibility generated by the presence of ssDNA, whereas an additional dsDNA arm in the fork substrate reduces the degrees of freedom of the DNA and, consequently, renders the HARP domain more important for binding.
Full-length SMARCAL1 has two HARP domains, HARP1 and HARP2. We have shown that deletion of HARP1 from SMARCAL1 or the introduction of destabilizing HARP1 mutations has a modest effect on binding to splayed arm substrates, ATPase activity, and fork regression activities compared with deletion of HARP2 (29), but deletion of HARP1 greatly diminishes binding to a gapped DNA molecule. Thus, although HARP1 is not essential for translocation activity of the catalytic domain, it plays a role in substrate recognition and likely provides an additional level of specificity for a fork or a regression intermediate through weaker or more transient contacts to undistorted regions of the junction. In this way, HARP1 may be analogous to the MRD/domain I of MSH2, which aids mismatch recognition by MSH3 or MSH6 by stabilizing a sharp bend in the DNA through nonspecific contacts to dsDNA immediately adjacent to the damage (47, 48). Consistent with a DNA-binding role for HARP1, one of the two SIOD mutations in HARP1 (R247P) resides on the putative DNA-binding surface (SI Appendix, Fig. S10).
The relatively enigmatic structure-based mechanism for how SF2 translocases process various DNA substrates is clarified by these SMARCAL1 structural and mutational results coupled to existing structural information. E. coli RecG binds to a three-way junction with the fork arms disposed around a wedge domain (61). To regress a fork, RecG may pull on the template arm to direct the junction across the wedge domain (Fig. 4E). Displacement of the nascent DNA strands from the template and their adjacency would allow base pairing to form a four-way junction. These observations suggest a mechanism whereby the HARP domains provide specificity for a fork and stabilize the branch point during translocation, thereby guiding the displaced nascent strands to form a new dsDNA arm (Fig. 4D). SMARCAL1 recruitment to stalled forks with an orientation provided by binding RPA would endow SMARCAL1 with specific polarity and fork movement (31).
Collectively, the SMARCAL1 HARP domain and the damage recognition domains of MutS and XPB reveal related substrate recognition domains that couple specific DNA remodeling activities onto ATPase motor domains. These unified views of specific functional recognition of distorted DNA provide a framework and focus for many ongoing studies on replication fork stability and genome integrity.
Experimental Procedures
Detailed experimental procedures are available in the SI Appendix.
Protein Production.
Mus musculus (m) HARP1 (amino acids 197–268) and T4 phage UvsW proteins were expressed as His6-GST-fusion proteins from E. coli and purified by Ni-NTA affinity chromatography (NAC). After removal of the His6-GST-tag by protease cleavage, mHARP1 was further purified by ion exchange (IEX) and SEC, and UvsW was purified by heparin Sepharose chromatography. SeMet-mHARP1 was produced by methionine metabolic inhibition and purified as for native mHARP1. The HARP-UvsW chimera (human SMARCAL1 amino acids 237–302 + UvsW amino acids 83–503) was constructed by restriction-free cloning, and HARP-UvsWK141R was generated using QuikChange (Stratagene). UvsWATPase (amino acids 83–503) was subcloned from the wild-type vector. All mutant and chimera UvsW proteins were purified as for wild-type UvsW, but with an additional SEC finishing step. Human SMARCAL1 and SMARCAL1CD (amino acids 325–870) were purified from baculovirus-infected insect cells by NAC, IEX, and SEC. SMARCAL1 cluster mutants were constructed by replacing the DNA-encoding HARP2 (amino acids 325–397) with synthetic HARP2 cassettes that contained mutations, and the proteins were purified as for SMARCAL1CD.
Crystallization and X-Ray Data Collection.
mHARP1 crystals were grown by vapor diffusion at 21 °C from solutions containing ammonium sulfate and PEG 5K monomethyl ether at pH 6.5 (native) or ammonium sulfate and sodium acetate at pH 4.5 (SeMet). Native and Se-SAD X-ray diffraction data were collected from cryocooled crystals at beamline 21-ID at the Advanced Photon Source in Argonne, IL. X-ray phases were obtained from the positions of eight Se atoms in the asymmetric unit. The resulting 3.3-Å SAD electron density map was used to build polypeptide chains for all four HARP1 molecules in the asymmetric unit, which then served as a molecular replacement search model against 1.9-Å native diffraction data. The model was refined against a maximum likelihood target, with changes to the model made after each round of refinement using 2Fo-Fc composite omit and Fo-Fc maps. The coordinates and diffraction data have been deposited in the Protein Data Bank (PDB ID code 4O66).
Biochemical Assays.
Fork regression assays were performed as described previously (29) with minor modifications, as described in SI Appendix. Reactions were performed for 20 min at 37 °C in 40 mM Hepes (pH 7.6), 20 mM KCl, 5 mM MgCl2, 1 mM TCEP, 0.1 mg/mL BSA, 2 mM ATP, and 1 nM 32P-labeled DNA fork substrate. DNA-dependent ATPase activity was determined with a high-throughput colorimetric ATPase assay (Innova Biosciences) using 8 nM protein and 2–80 nM DNA in 100 mM Tris pH 7.4, 5 mM MgCl2, and 1 mM ATP. DNA binding was assayed by fluorescence polarization using 6-carboxyfluorescein–labeled DNA substrates containing 20-nt arms (SI Appendix, Table S3). Reactions were incubated for 20 min at 25 °C and contained 10–3,000 nM protein, 25 nM DNA, 20 mM Hepes (pH 7.6), 100 mM KCl, 5 mM MgCl2, 0.25 mg/mL BSA, 0.05 mM EDTA, 0.5 mM DTT, and 0.01% Tween 20.
SAXS.
SAXS data were collected and processed at the SIBYLS beamline at the Advanced Light Source (62–64). The DNA overhang substrate is shown in SI Appendix, Table S3. Samples were purified immediately before SAXS measurements by SEC using a Shodex KW-402 column in 20 mM Hepes (pH 7.5), 1% glycerol, 0.01% sodium azide, 2 mM TCEP, and either 150 mM NaCl (mHARP1) or 200 mM NaCl and 2 mM MgCl2 (SMARCAL1CD). For the nucleotide experiments, purification was performed with SEC buffer supplemented with either 1 mM ADPNP or 500 µM ADP, 500 µM BeCl2, and 5 mM NaF (ADP⋅BeFx). Sample homogeneity and complex formation were confirmed by inline SEC-coupled multiangle light scattering (65). Individual components were concentrated to 5 mg/mL for SEC purification, and for each sample, the peak fraction (40 μL) was obtained for SAXS analysis, with the corresponding SEC flow-through taken at 1.2 column volumes as the SAXS buffer blank (64).
For each fraction, four different SAXS exposures were measured as a 2/3 dilution series. Preliminary data analysis and reduction were performed with the ScÅtter program (SIBYLS beamline). Concentration-independent SAXS profiles were used directly for multiphase bead modeling by MONSA (66). For each complex, 10 independent MONSA runs were performed and averaged using DAMAVERM. The resulting averaged models were converted to volumetric density maps with the SITUS program suite (67) and then visualized with VMD (68).
Supplementary Material
Acknowledgments
We thank Ken Kreuzer for the UvsW expression plasmid and David Smith for help with remote X-ray data collection. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences (Contract DE-AC02-06CH11357). Use of the LS-CAT/Sector 21 beamline was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The SIBYLS beamline and SAXS efforts on protein–DNA complexes are supported in part by National Institutes of Health (NIH) Grant MINOS GM10540, by the US Department of Energy program on Integrated Diffraction Analysis Technologies, and by Structural Cell Biology of DNA Repair Machines (SBDR) NIH Grant P01 CA092584. This work was supported by a pilot grant from the Vanderbilt Center of Molecular Toxicology (to B.F.E.) and NIH Grant R01 CA136933 to (D.C.). A.C.M. was funded by an American Cancer Society postdoctoral fellowship (PF-12-220-01) and the Vanderbilt Training Program in Environmental Toxicology (T32 ES07028). Core facilities were supported by SBDR NIH Grant P01 CA092584, Vanderbilt Center in Molecular Toxicology Grant P30 ES000267, and Vanderbilt-Ingram Cancer Center Grant P30 CA068485.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4O66).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1324143111/-/DCSupplemental.
References
- 1.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 2.Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121(5):689–702. doi: 10.1016/j.cell.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 3.Rothstein R, Michel B, Gangloff S. Replication fork pausing and recombination or “gimme a break.”. Genes Dev. 2000;14(1):1–10. [PubMed] [Google Scholar]
- 4.Branzei D, Foiani M. The DNA damage response during DNA replication. Curr Opin Cell Biol. 2005;17(6):568–575. doi: 10.1016/j.ceb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Hyrien O. Mechanisms and consequences of replication fork arrest. Biochimie. 2000;82(1):5–17. doi: 10.1016/s0300-9084(00)00344-8. [DOI] [PubMed] [Google Scholar]
- 6.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19(9):1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297(5581):599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 8.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297(5581):552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 9.Schlacher K, et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145(4):529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seviour EG, Lin SY. The DNA damage response: Balancing the scale between cancer and ageing. Aging (Albany, NY Online) 2010;2(12):900–907. doi: 10.18632/aging.100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22(1):106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cimprich KA, Cortez D. ATR: An essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci USA. 2004;101(27):10078–10083. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J Biol Chem. 2006;281(51):39517–39533. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- 15.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300(5625):1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 16.Ciccia A, Elledge SJ. The DNA damage response: Making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol. 2010;11(10):683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 18.Bugreev DV, Mazina OM, Mazin AV. Rad54 protein promotes branch migration of Holliday junctions. Nature. 2006;442(7102):590–593. doi: 10.1038/nature04889. [DOI] [PubMed] [Google Scholar]
- 19.Hickson ID. RecQ helicases: Caretakers of the genome. Nat Rev Cancer. 2003;3(3):169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 20.Blastyák A, Hajdú I, Unk I, Haracska L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol. 2010;30(3):684–693. doi: 10.1128/MCB.00863-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blastyák A, et al. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell. 2007;28(1):167–175. doi: 10.1016/j.molcel.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gari K, Décaillet C, Stasiak AZ, Stasiak A, Constantinou A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell. 2008;29(1):141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 23.McGlynn P, Lloyd RG. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell. 2000;101(1):35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 24.Yusufzai T, Kadonaga JT. Annealing helicase 2 (AH2), a DNA-rewinding motor with an HNH motif. Proc Natl Acad Sci USA. 2010;107(49):20970–20973. doi: 10.1073/pnas.1011196107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Long DT, Kreuzer KN. Fork regression is an active helicase-driven pathway in bacteriophage T4. EMBO Rep. 2009;10(4):394–399. doi: 10.1038/embor.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, et al. Hjm/Hel308A DNA helicase from Sulfolobus tokodaii promotes replication fork regression and interacts with Hjc endonuclease in vitro. J Bacteriol. 2008;190(8):3006–3017. doi: 10.1128/JB.01662-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flaus A, Martin DM, Barton GJ, Owen-Hughes T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006;34(10):2887–2905. doi: 10.1093/nar/gkl295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hockensmith JW, Wahl AF, Kowalski S, Bambara RA. Purification of a calf thymus DNA-dependent adenosinetriphosphatase that prefers a primer-template junction effector. Biochemistry. 1986;25(24):7812–7821. doi: 10.1021/bi00372a005. [DOI] [PubMed] [Google Scholar]
- 29.Bétous R, et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26(2):151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yusufzai T, Kadonaga JT. HARP is an ATP-driven annealing helicase. Science. 2008;322(5902):748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bétous R, et al. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3(6):1958–1969. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boerkoel CF, et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet. 2002;30(2):215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 33.Baradaran-Heravi A, et al. SMARCAL1 deficiency predisposes to non-Hodgkin lymphoma and hypersensitivity to genotoxic agents in vivo. Am J Med Genet A. 2012;158A(9):2204–2213. doi: 10.1002/ajmg.a.35532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll C, et al. Schimke Immunoosseous Dysplasia associated with undifferentiated carcinoma and a novel SMARCAL1 mutation in a child. Pediatr Blood Cancer. 2013;60(9):E88–E90. doi: 10.1002/pbc.24542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simon AJ, et al. Novel SMARCAL1 bi-allelic mutations associated with a chromosomal breakage phenotype in a severe SIOD patient. J Clin Immunol. 2013 doi: 10.1007/s10875-013-9957-3. [DOI] [PubMed] [Google Scholar]
- 36.Thomä NH, et al. Structure of the SWI2/SNF2 chromatin-remodeling domain of eukaryotic Rad54. Nat Struct Mol Biol. 2005;12(4):350–356. doi: 10.1038/nsmb919. [DOI] [PubMed] [Google Scholar]
- 37.Holm L, Rosenstrom P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan L, et al. Conserved XPB core structure and motifs for DNA unwinding: Implications for pathway selection of transcription or excision repair. Mol Cell. 2006;22(1):27–37. doi: 10.1016/j.molcel.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 39.Kerr ID, et al. Crystallographic and NMR analyses of UvsW and UvsW.1 from bacteriophage T4. J Biol Chem. 2007;282(47):34392–34400. doi: 10.1074/jbc.M705900200. [DOI] [PubMed] [Google Scholar]
- 40.Sickmier EA, Kreuzer KN, White SW. The crystal structure of the UvsW helicase from bacteriophage T4. Structure. 2004;12(4):583–592. doi: 10.1016/j.str.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 41.Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst) 2011;10(7):697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carles-Kinch K, George JW, Kreuzer KN. Bacteriophage T4 UvsW protein is a helicase involved in recombination, repair and the regulation of DNA replication origins. EMBO J. 1997;16(13):4142–4151. doi: 10.1093/emboj/16.13.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manosas M, et al. RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat Commun. 2013;4:2368. doi: 10.1038/ncomms3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manosas M, Perumal SK, Croquette V, Benkovic SJ. Direct observation of stalled fork restart via fork regression in the T4 replication system. Science. 2012;338(6111):1217–1220. doi: 10.1126/science.1225437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li N, Sickmier EA, Zhang R, Joachimiak A, White SW. The MotA transcription factor from bacteriophage T4 contains a novel DNA-binding domain: The “double wing” motif. Mol Microbiol. 2002;43(5):1079–1088. doi: 10.1046/j.1365-2958.2002.02809.x. [DOI] [PubMed] [Google Scholar]
- 46.Webb MR, Plank JL, Long DT, Hsieh TS, Kreuzer KN. The phage T4 protein UvsW drives Holliday junction branch migration. J Biol Chem. 2007;282(47):34401–34411. doi: 10.1074/jbc.M705913200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warren JJ, et al. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26(4):579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 48.Gupta S, Gellert M, Yang W. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat Struct Mol Biol. 2012;19(1):72–78. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lebbink JH, et al. Magnesium coordination controls the molecular switch function of DNA mismatch repair protein MutS. J Biol Chem. 2010;285(17):13131–13141. doi: 10.1074/jbc.M109.066001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamers MH, et al. ATP increases the affinity between MutS ATPase domains: Implications for ATP hydrolysis and conformational changes. J Biol Chem. 2004;279(42):43879–43885. doi: 10.1074/jbc.M406380200. [DOI] [PubMed] [Google Scholar]
- 51.He Z, Henricksen LA, Wold MS, Ingles CJ. RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature. 1995;374(6522):566–569. doi: 10.1038/374566a0. [DOI] [PubMed] [Google Scholar]
- 52.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407(6805):703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 53.Reibarkh M, et al. Eukaryotic initiation factor (eIF) 1 carries two distinct eIF5-binding faces important for multifactor assembly and AUG selection. J Biol Chem. 2008;283(2):1094–1103. doi: 10.1074/jbc.M708155200. [DOI] [PubMed] [Google Scholar]
- 54.Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 2011;331(6018):730–736. doi: 10.1126/science.1198308. [DOI] [PubMed] [Google Scholar]
- 55.Ghosal G, Yuan J, Chen J. The HARP domain dictates the annealing helicase activity of HARP/SMARCAL1. EMBO Rep. 2011;12(6):574–580. doi: 10.1038/embor.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adelman CA, et al. HELQ promotes RAD51 paralogue-dependent repair to avert germ cell loss and tumorigenesis. Nature. 2013;502(7471):381–384. doi: 10.1038/nature12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takata K, Reh S, Tomida J, Person MD, Wood RD. Human DNA helicase HELQ participates in DNA interstrand crosslink tolerance with ATR and RAD51 paralogs. Nat Commun. 2013;4:2338. doi: 10.1038/ncomms3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Büttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol. 2007;14(7):647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 59.Dürr H, Flaus A, Owen-Hughes T, Hopfner KP. Snf2 family ATPases and DExx box helicases: differences and unifying concepts from high-resolution crystal structures. Nucleic Acids Res. 2006;34(15):4160–4167. doi: 10.1093/nar/gkl540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dürr H, Körner C, Müller M, Hickmann V, Hopfner KP. X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell. 2005;121(3):363–373. doi: 10.1016/j.cell.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 61.Singleton MR, Scaife S, Wigley DB. Structural analysis of DNA replication fork reversal by RecG. Cell. 2001;107(1):79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]
- 62.Classen S, et al. Implementation and performance of SIBYLS: A dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J Appl Cryst. 2013;46(Pt 1):1–13. doi: 10.1107/S0021889812048698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hura GL, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6(8):606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rambo RP, Tainer JA. Bridging the solution divide: Comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering. Curr Opin Struct Biol. 2010;20(1):128–137. doi: 10.1016/j.sbi.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rambo RP, Tainer JA. Improving small-angle X-ray scattering data for structural analyses of the RNA world. RNA. 2010;16(3):638–646. doi: 10.1261/rna.1946310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Svergun DI. Restoring low-resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76(6):2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wriggers W. Using Situs for the integration of multi-resolution structures. Biophys Rev. 2010;2(1):21–27. doi: 10.1007/s12551-009-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.