

Significance
Solid tumors contain large numbers of immune cells, including monocytes and monocyte-derived macrophages that promote tumor progression. During tumor development, monocytes accumulate in the spleen. However, the influence of spleen cells on tumor growth remains controversial. Here, we used novel methods for tracking intertissue migration and monitoring hematopoiesis to show that during tumor development the bone marrow dramatically accelerates production of monocytes, rapidly transferring many of these newly formed cells to a reservoir in the spleen. However, these spleen monocytes are less able than their bone marrow counterparts to enter the tumor and make only a minor contribution to the tumor-infiltrating monocyte population. These findings clarify the roles of the spleen and bone marrow in cancer development.
Keywords: cancer immunity, tumor-associated macrophage, monocytopoiesis, KikGR, Fucci
Abstract
Myeloid cells such as monocytes and monocyte-derived macrophages promote tumor progression. Recent reports suggest that extramedullary hematopoiesis sustains a sizable reservoir of tumor-infiltrating monocytes in the spleen. However, the influence of the spleen on tumor development and the extent to which spleen monocytes populate the tumor relative to bone marrow (BM) monocytes remain controversial. Here, we used mice expressing the photoconvertible protein Kikume Green-Red to track the redistribution of monocytes from the BM and spleen, and mice expressing fluorescent ubiquitination-based cell-cycle indicator proteins to monitor active hematopoiesis in these tissues. In mice bearing late-stage tumors, the BM, besides being the major site of monocyte production, supplied the expansion of the spleen reservoir, replacing 9% of spleen monocytes every hour. Deployment of monocytes was equally rapid from the BM and the spleen. However, BM monocytes were younger than those in the spleen and were 2.7 times more likely to migrate into the tumor from the circulation. Partly as a result of this intrinsic difference in migration potential, spleen monocytes made only a minor contribution to the tumor-infiltrating monocyte population. At least 27% of tumor monocytes had traveled from the BM in the last 24 h, compared with only 2% from the spleen. These observations highlight the importance of the BM as the primary hematopoietic tissue and monocyte reservoir in tumor-bearing mice, despite the changes that occur in the spleen monocyte reservoir during tumor development.
Myeloid cells such as monocytes infiltrate almost all solid tumors (1). The presence of tumor-associated macrophages, which are derived directly from monocytes, correlates with poor prognosis (2, 3). These cells influence tumor progression through direct interactions with tumor cells, by modulating adaptive immune responses to the tumor, and by creating conditions that support angiogenesis, invasion, and metastasis (4–6). Understanding the origins and characteristics of tumor-infiltrating myeloid cells may lead to novel approaches to cancer therapy.
In healthy adult humans and mice, all myeloid cell formation occurs in the bone marrow (BM) (7). However, in certain disease states, organs such as the liver and spleen become sites of extramedullary hematopoiesis (8, 9). In tumor-bearing mice myeloid cells accumulate in the spleen (10–12), making this organ a candidate source of tumor-infiltrating cells. Recent reports propose that a rapidly mobilized reservoir of monocytes in the spleen contributes significant numbers of infiltrating cells in acute and chronic inflammatory conditions including myocardial infarction (13–15), stroke (14), atherosclerosis (15), and cancer (16, 17). In cancer, extramedullary hematopoiesis in the spleen is suggested to sustain a continuous supply of monocytes to the tumor (16, 17). These reports challenge the classical view of the BM as the primary site of monocyte production and supply (18). Current approaches to measuring tissue origin and migration include BrdU labeling, adoptive transfer, tissue transplantation, and parabiosis experiments. However, it has remained difficult to assess accurately the relative contributions of different pools of myeloid cells to the tumor-infiltrating monocyte population (16).
In the present study, we developed methods using mice expressing the photoconvertible protein Kikume Green-Red (KikGR) (19, 20) or fluorescent ubiquitination-based cell cycle indicator (Fucci) proteins (21, 22), which enabled us to compare the BM and spleen monocyte pools directly in terms of supply and production. We demonstrate that spleen-pool monocytes make only a minor contribution to the tumor-infiltrating monocyte population, partly because BM-pool monocytes have an intrinsic migration advantage over their spleen-pool counterparts.
Results
Spleen Monocyte Cellularity Increases Only in Late-Stage Tumor Development.
To characterize the relationship between tumor growth and the expansion of the spleen monocyte pool in tumor-bearing mice, we inoculated Lewis lung carcinoma (3LL) tumor cells s.c. into the flanks of wild-type mice. This syngeneic transplant model yields reproducible nodular tumor growth in immunocompetent hosts with minimal metastasis, facilitating quantitative analysis of tumor tissue and infiltrating immune cells. Palpable solid tumors developed after 7 d with a dramatic increase in tumor growth after 28 d (Fig. S1 A and B) that coincided with increases in spleen weight (Fig. S1C), spleen monocyte cellularity (Fig. S1D), and blood monocyte numbers (Fig. S1E). We focused on the Ly6Chi “inflammatory” monocyte subset because these cells make up the majority of the tumor-infiltrating monocyte population (23). The number of monocytes in the tumor peaked at day 21 (Fig. S1F), at the same time that a transient decrease in BM monocytes was observed (Fig. S1D). These data show that expansion of the spleen monocyte pool occurs only in late-stage tumor development, prompting us to focus on day-35 tumors in subsequent experiments.
Tumor-Infiltrating Monocytes Are Supplied Primarily from the BM Pool.
To determine the contribution of the spleen to the tumor-infiltrating monocyte population, we used day-35 tumor-bearing BM chimeras in which myeloid cells expressed the green-to-red irreversibly photoconvertible protein KikGR (hereafter, “KikGR mice”) (19, 20). By adapting previously developed methods that involve surgically irradiating selected tissues with violet light (24–27), we were able to timestamp monocytes present in the left femur (for monocytes of BM origin) or spleen and then track their redistribution throughout the body over a 24-h period using flow cytometry (Fig. 1 A and B). The procedure resulted in photoconversion of 25–40% of monocytes in either pool (Fig. 1B and Fig. S2C) without affecting cell viability or migration potential (Fig. S3 A and C). KikGR-Red (KikRed) protein levels remained stable for the duration of the experiments (Fig. S3B), and none of the results obtained by the KikGR approach were attributable to acute fluctuations in monocyte numbers following surgery. Femur photoconversion data were adjusted to represent the contribution of the whole-body BM pool, and all data were adjusted to simulate 100% photoconversion of source tissues, thus correcting for variations in photoconversion efficiency (see Materials and Methods for details).
Fig. 1.
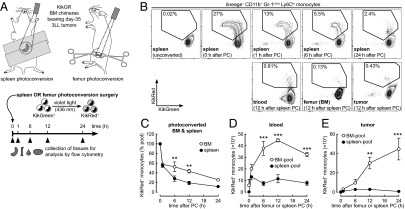
Tumor-infiltrating monocytes are supplied primarily from the BM pool. (A) Surgical procedure and timeline for photoconversion experiments. The spleen or left femur of KikGR BM chimeras bearing day-35 3LL tumors was irradiated with violet light, resulting in the photoconversion of myeloid cell KikGreen protein to KikRed. (B) Representative flow cytometry plots showing the egress of photoconverted (KikRed+) Ly6Chi monocytes from the spleens of tumor-bearing mice and their redistribution to the blood, unconverted BM, and tumor (see Fig. S2C for BM egress plots). (C) Egress of KikRed+ Ly6Chi monocytes from the BM and spleen after photoconversion. Data are expressed as a proportion of the initial pool of photoconverted cells in each tissue. (D and E) Accumulation of KikRed+ BM-pool and spleen-pool Ly6Chi monocytes in the blood and tumor. PC, photoconversion. **P ≤ 0.01; ***P ≤ 0.001 (BM-pool vs. spleen-pool). Data represent means ± SEM (n = 3) and are representative of two independent experiments.
After photoconversion, KikRed+ monocytes egressed rapidly from the BM and spleen (Fig. 1C) and appeared in the blood (Fig. 1D). Time-course graphs (Fig. 1 C–E) represent the redistribution of cells labeled KikRed+ at time 0 h as they move from source tissues (BM or spleen) to destination tissues (blood and tumor). A lesser proportion of photoconverted monocytes was retained in the spleen than in the BM (Fig. 1C; 6 h time point onwards), suggesting that monocyte replacement occurs more rapidly in the spleen pool. However, BM-pool KikRed+ monocytes outnumbered spleen-pool KikRed+ monocytes in the blood, in line with whole-body BM and spleen monocyte numbers (Table S1). Similar results were observed in non–tumor-bearing mice (Fig. S4 A and B).
Importantly, the KikGR approach enabled us to determine the extent of monocyte migration from pools in the BM and spleen to the tumor. BM-pool KikRed+ cells considerably outnumbered spleen-pool KikRed+ cells in the tumor, even at the peak of spleen-pool KikRed+ monocyte accumulation at 12 h (30 × 103 vs. 3 × 103) (Fig. 1E). From the peak numbers of KikRed+ cells that were detected in the tumor, we estimate that at least 27% (45 × 103) of tumor monocytes had traveled from the BM in the last 24 h compared with ∼2% (3 × 103) from the spleen, not taking cell death or exit from the tumor into account (Fig. 1E and Table S1). Taken together, these results indicate that spleen-pool monocytes make a relatively minor contribution to the tumor-infiltrating monocyte population.
BM-Pool Monocytes Have a Migration Advantage over Spleen-Pool Monocytes.
We next investigated possible causes for the dominance of BM-pool monocytes within the tumor. The accumulation of monocytes is influenced by the availability of monocytes from the BM or spleen, their ability to extravasate into the tumor, and the length of time that they are retained within the tumor. In tumor-bearing mice, the BM contains six times more monocytes than the spleen (Table S1). Surprisingly, however, our data still favored the accumulation in the tumor of BM-pool monocytes over spleen-pool monocytes, even when corrected for the greater availability of BM-pool monocytes in the body (Fig. 2A). This result suggested that additional factors might limit the potential of spleen-pool monocytes to accumulate in the tumor.
Fig. 2.

BM-pool monocytes have a migration advantage over spleen-pool monocytes. (A) Accumulation of KikRed+ monocytes in the tumor expressed relative to their abundance in respective source pools (see Fig. S4C for full time-course data). PC, photoconversion. (B and C) Weighted least squares regression analysis of KikRed+ monocyte data was used to calculate blood-to-tumor migration probability (B) and tumor residence time (C). (D) BM or spleen cells from day-14 tumor-bearing donors were enriched for monocytes by lineage depletion before adoptive transfer into tumor-bearing recipients. (E) Representative flow cytometry plots showing donor spleen-derived [carboxyfluorescein succinimidyl ester (CFSE)+ CD45.1−] and donor BM-derived (CFSE+ CD45.1+) monocytes before and after transfer. (F) Ratio of donor spleen-derived to BM-derived monocytes in recipient tissues 4 h after transfer. n.s., not significant; **P ≤ 0.01; ***P ≤ 0.001. Data represent means ± SEM (n = 3) in A and F or SE (30–39 degrees of freedom) in B and C and are representative of (A, E, and F) or are pooled from (B and C) two independent experiments.
We therefore adopted a mathematical approach to quantify and compare the migration properties of tumor-infiltrating monocytes. Using weighted least squares regression analysis, equations modeling the entry and exit of cells to and from the tumor were fitted to blood and tumor KikGR time-course data (see Materials and Methods for details). Migration probability, the probability per unit time that a cell would migrate from the blood to the tumor, was 2.7-fold higher for BM-pool monocytes than for spleen-pool monocytes (Fig. 2B), whereas no significant difference was detected between the tumor residence times of cells from either pool (Fig. 2C). These results show that an intrinsic difference in the ability of monocytes from different pools to migrate from the blood to the tumor is at least partly responsible for the dominance of BM-pool monocytes within the tumor.
As an independent approach to compare the migration potential of BM-pool and spleen-pool monocytes, we examined the redistribution of cells from both sources after adoptive transfer of an equal mixture of these cells directly into the blood of tumor-bearing mice (Fig. 2D). Four hours after transfer, there still were equal numbers of donor BM and spleen monocytes in the blood (Fig. 2 E and F), but there was a higher proportion of BM-pool cells than spleen-pool cells in the recipient tumor (Fig. 2 E and F). These results support our conclusion that BM-pool monocytes extravasate into the tumor more readily than their spleen-pool counterparts because of an intrinsic difference in their migration potential.
The BM Is the Major Site of Monocyte Production in Tumor-Bearing Mice.
Even if spleen-pool monocytes make only a minor contribution to the tumor-infiltrating monocyte population, it has been reported that the spleen plays a role in the production and development of monocytes in the tumor-bearing state (16, 17). To assess the relative contribution of the spleen to monocyte production in tumor-bearing mice, we used mice expressing Fucci proteins (hereafter, “Fucci mice”) (21, 22) and flow cytometry to compare the extent of hematopoiesis occurring in the BM and spleen (Fig. 3A). In Fucci mice, Fucci-orange (mKO2) and Fucci-green (mAG) are expressed reciprocally in the G1 and S/G2/M phases of the cell cycle, respectively. Because Fucci-green is degraded by ubiquitin-mediated proteolysis in late M phase, there is no carryover of green fluorescence to daughter cells. Numbers of actively proliferating (Fucci-green+) hematopoietic stem cells (HSCs), granulocyte-macrophage progenitor (GMP) cells (committed precursors to the monocyte and neutrophil lineages), and monocytes were 10- to 100-fold higher in the spleen and were fivefold higher in the BM of day-33 tumor-bearing mice than in non–tumor-bearing mice (Fig. 3B). However, actively proliferating HSCs, GMP cells, and monocytes in the BM far outnumbered those in the spleen in both non–tumor-bearing and tumor-bearing mice (Fig. 3B). Furthermore, in tumor-bearing mice, 65% of BM monocytes (up from 21% in non–tumor-bearing mice) were actively proliferating, compared with only 16% of spleen monocytes (Fig. 3C).
Fig. 3.

The BM is the major site of monocyte production in tumor-bearing mice. (A) Representative flow cytometry plots showing actively proliferating Ly6Chi monocytes (or promonocytes) in the BM and spleen of Fucci transgenic mice. (B) Actively proliferating (Fucci-green+) lineage-negative c-kit+ Sca-1+ HSCs, lineage-negative c-kit+ Sca-1– CD16/32+ CD34+ GMP cells, and Ly6Chi monocytes in the BM, spleen, and whole-body blood of non–tumor-bearing (NT) and tumor-bearing mice (see Fig. S2D for flow cytometry gating). (C) Proportions of actively proliferating Ly6Chi monocytes in the BM, spleen, and blood. (D) Expression levels (mean fluorescence intensity) of Fucci-orange in Fucci-green− monocytes from the BM, spleen, and blood of day-33 tumor-bearing mice. Fucci-orange accumulates during the G1 phase of the cell cycle, thus providing an indication of the time elapsed since last cell division. **P ≤ 0.01; ***P ≤ 0.001 (comparisons as indicated). ††P ≤ 0.01; †††P ≤ 0.001 (compared with non–tumor-bearing control). Data represent means ± SEM (n = 6–8) and are representative of two independent experiments.
Although mature monocytes are postmitotic (18), proliferating monocytes have been described in the BM of healthy mice and elsewhere in disease states (28–30). These immature cells have promonocyte morphology, express the cell surface proteins CD11b and Ly6C, and share functional similarities with mature monocytes. They are distinct from monoblast-like cells that do not express CD11b, including macrophage and dendritic cell precursors (“MDPs”) and common monocyte progenitors (“cMoPs”) (30, 31). Thus, the Fucci-green+ Ly6Chi cells observed in our experiments are likely to represent immature monocytes or promonocytes. Because the Fucci-orange fusion protein undergoes cell-cycle–dependent ubiquitination followed by proteolysis during the S/G2/M phases but accumulates during the G1 phase, it provides an indication of the time elapsed since last cell division in Fucci-green− cells (22). Expression levels of Fucci-orange were lower in BM monocytes than in spleen and blood monocytes, indicating that the BM contains relatively younger cells (Fig. 3 A and D). Taken together, these results demonstrate that the BM is the major site of monocyte production in tumor-bearing mice.
The Supply of BM-Pool Monocytes to the Spleen Increases in the Tumor-Bearing State.
The relatively low proportion of Fucci-green− (Fucci-orange+) G1/G0-phase monocyte progeny observed in the BM of tumor-bearing mice suggests that many monocytes or promonocytes exit the BM while in the S/G2/M phase of the cell cycle (Fig. 3C). The number and proportion of Fucci-green+ monocytes in the blood increased considerably in the tumor-bearing state (cell number increased by 100-fold) but remained much lower than in the spleen (Fig. 3 B and C). These data suggest either that monocytes/promonocytes are becoming Fucci-green+ in the spleen or that Fucci-green+ monocytes/promonocytes from the circulation are accumulating selectively in the spleen. Therefore we used femur photoconversion experiments to determine the extent to which the spleen pool is repopulated by BM monocytes. Monocytes moved continuously from the BM to the spleen, and in tumor-bearing mice, the accumulation of BM-pool KikRed+ monocytes in the spleen increased substantially (Fig. 4A). Given that the size of the BM pool does not increase in tumor-bearing mice, this result demonstrates that a greater quantity and proportion of BM-pool monocytes migrate to the spleen in the tumor-bearing state. Our data suggest that in tumor-bearing mice, at least 9% (0.4 × 106) of spleen monocytes are replaced by BM-pool monocytes every hour (Fig. S4D and Table S1), not taking recirculation from the spleen into account.
Fig. 4.

The supply of BM-pool monocytes to the spleen increases in the tumor-bearing state. (A) Accumulation of KikRed+ BM-pool Ly6Chi monocytes in the unconverted spleen. (B) Accumulation of KikRed+ BM-pool and spleen-pool Ly6Chi monocytes in the unconverted BM. BM-pool data indicate the reentry of cells egressing from the opposite (photoconverted) femur. PC, photoconversion. See Fig. S4 D and E for full time-course data. ***P ≤ 0.001. Data represent means ± SEM (n = 3) and are representative of two independent experiments.
Femur photoconversion experiments also allowed us to measure the reentry of cells into the BM, because KikRed+ monocytes that had egressed from the photoconverted femur could be detected in the opposite, unconverted femur. Fewer BM-pool KikRed+ monocytes reentered the BM in tumor-bearing mice than in non–tumor-bearing mice (Fig. 4B), suggesting that in the tumor-bearing state most BM-pool monocytes are consumed in peripheral tissues such as the tumor and spleen, rather than returning to the BM. Taken together, our results show that increased monocyte production in the BM of tumor-bearing mice supplies the expansion of the spleen pool, partly through the rapid transfer of immature monocytes or promonocytes that continue to divide in the spleen.
Discussion
Tumor-infiltrating myeloid cells are short lived and are sustained by constant supply from the circulation (23). In this study we used KikGR and Fucci mice to compare the contributions of the BM and spleen as sources of tumor-infiltrating monocytes. We provide definitive evidence that monocytes from both the BM and spleen pools are deployed rapidly and continuously to the tumor. However, the vast majority of tumor-infiltrating monocytes were supplied from the BM pool, partly because BM-pool monocytes had an intrinsic migration advantage over spleen-pool monocytes. Moreover, the BM was the major site of monocyte production in tumor-bearing mice, and the rapid transfer of BM monocytes supplied the expansion of the spleen monocyte pool.
Our results demonstrate that, despite its fixed physical capacity, the BM is able to accelerate production of monocytes dramatically to meet demand. This accelerated monocytopoiesis occurs without any detectable increase in BM total cell number, suggesting that the newly produced cells overflow into the circulation rapidly. Short-pulse (≤12 h) BrdU labeling studies in tumor-bearing mice have shown that most myeloid cells populate peripheral tissues, including the spleen, only after expansion and mobilization from the BM (23). Our KikGR data confirm the rapid transfer of monocytes from the BM to the spleen and suggest that accelerated monocytopoiesis in the BM drives expansion of the spleen pool. Thus, although the spleen provides overflow capacity for the BM, it does not supplant the BM as the primary site of monocytopoiesis in tumor-bearing mice. The trigger for increased monocytopoiesis in the BM may be the increased consumption of monocytes by the tumor, because peak tumor monocyte infiltration at day 21 coincided with a transient drop in BM monocyte numbers. In addition, tumors can secrete long-range signals that activate hematopoietic cells in the BM (32, 33).
The basis for the difference in the migration potential of BM-pool and spleen-pool monocytes remains unclear. It seems unlikely that the splenic microenvironment influences monocyte function, because KikGR data show that the majority of monocytes in the spleen pass through only transiently. However, monocytes in the BM were younger than those in the spleen. Differences in the maturity of monocytes might result in differences in the expression of chemokine receptors, adhesion molecules, and survival factors that influence migration into and survival within inflammatory sites (34). For example, the chemokine receptor CCR2 mediates monocyte recruitment to the tumor (23, 35) and is required for monocyte egress from the BM but not from the spleen (13, 36). Selection for expression of CCR2 upon monocyte egress from the BM could provide these cells with a migration advantage over older spleen-pool monocytes that do not face similar selection upon egress to the circulation. Future studies should clarify the mechanistic basis for differences in the migration potential of monocytes from different pools.
KikGR and Fucci mice offer powerful, quantitative means for tracking intertissue cell migration and monitoring cell proliferation under steady-state conditions. Unlike adoptive transfer experiments in which cells prepared ex vivo are reinjected into the circulation en masse, the KikGR approach enables visualization of changes in supply over time, by revealing the gradual egress of specific cell populations from their source tissues and their accumulation in the circulation. Moreover, unlike tissue transplantation and parabiosis experiments, KikGR experiments cause minimal disruption of physiological conditions. By analyzing KikGR time-course data from destination tissues and the blood, we were able to determine the migration potential of specific cell populations. This approach provides new insights into cell migration in vivo and builds upon previous attempts to model monocyte tissue kinetics (14). In addition, Fucci mice provide a sensitive, real-time indication of progress through the cell cycle with no carryover of signal to daughter cells. Future studies might use the KikGR and Fucci approaches to investigate the relative contributions of BM and spleen monocytes in other inflammatory diseases.
Our findings highlight the importance of the BM as the primary hematopoietic tissue and monocyte reservoir in tumor-bearing mice. Targeting excessive monocytopoiesis in the BM of tumor-bearing mice may limit not only the supply of tumor-infiltrating monocytes but also the expansion of the spleen monocyte pool. Therapies that limit the number of monocytes and macrophages that are available to the tumor may improve the prognosis of patients with cancer.
Materials and Methods
Methods for cell preparation, flow cytometry, adoptive transfer, and in vitro migration assays are provided in SI Materials and Methods. The antibodies used for flow cytometry are listed in Table S2.
Animals.
Male C57BL/6J wild-type mice (CD45.2+) were purchased from CLEA Japan or Japan SLC. Rosa26CAG::KikGR knockin mice (C57BL/6 background), which express KikGR in all cell types, were generated at RIKEN. KikGR BM chimeras were generated by lethally irradiating 6-wk-old male C57BL/6J host mice with a split dose of 9.5 Gy (OM-150HT X-ray system; OHMiC), followed by i.v. injection with 5 × 106 syngeneic BM cells pooled from the femurs, tibias, and spines of Rosa26CAG::KikGR donors. Chimeras were used for tumor inoculation or experiments at least 5 wk after BM transfer, at which stage >99% chimerism was observed in monocytes from the BM, blood, and spleen. B6.SJL-Ptprca Pepcb/BoyJ congenic mice (CD45.1+) were purchased from The Jackson Laboratory. These mice were crossbred with C57BL/6J mice to generate CD45.1+ CD45.2+ mice for adoptive transfer experiments. Fucci double transgenic mice were generated by crossbreeding FucciG1 (line no. 639) and FucciS/G2/M (line no. 474) animals (obtained from A. Miyawaki, RIKEN BioResource Center, Tsukuba, Japan) as described previously (21). These mice express the fusion proteins mKO2-hCdt1(30/120) and mAG-hGem(1/110) under the CAG promoter; the no. 639 and no. 474 mouse lines were used to overcome the poor expression in hematopoietic cells that is observed in other Fucci lines (22). KikGR and Fucci mice were backcrossed to C57BL/6 mice for at least 10 generations before use. Mice were 6–12 wk of age (9–15 wk for BM chimeras) when used for tumor inoculation or experiments. All mice were bred and maintained under specific pathogen-free conditions in animal facilities at the University of Tokyo, and all animal procedures were approved by and conducted in accordance with the guidelines of the Animal Care Committee of the Graduate School of Medicine, the University of Tokyo.
Tumor Inoculation.
The 3LL tumor cell line (a subclone of the C57BL mouse-derived Lewis lung carcinoma line) was obtained from F. Abe (Nihon Kayaku, Tokyo). Mice were anesthetized with sodium pentobarbital (50 mg/kg, i.p.), the fur around the inoculation site was shaved to improve tumor visualization, and a 50-µL suspension of 5 × 105 tumor cells was injected s.c. into the right flank. Tumor dimensions were measured using digital calipers, and tumor volume (in cubic millimeters) was estimated using the formula volume = length × (width)2 × 0.5, which is derived from the formula for a prolate spheroid.
Photoconversion Surgery.
Photoconversion procedures were adapted from methods developed previously (24–27) and are depicted in Fig. 1A. KikGR BM chimeras were anesthetized with isoflurane [1–3% (vol/vol) by inhalation], the fur around the spleen or left femur was shaved, and the skin disinfected with 70% (vol/vol) ethanol.
For spleen photoconversion, an ∼1.5-cm horizontal incision was made in the skin on the left side of the abdomen, the spleen visualized, and an ∼0.5-cm incision was made in the abdominal wall, through which the spleen was externalized gently. Two pieces of sterile aluminum foil (folded to remove sharp edges that might cause injury) were arranged on either side of the spleen, shielding the skin and abdominal cavity. The spleen was irradiated with violet light (three sides × 3 min, 150 mW/cm2) using spot UV curing equipment (SP9-250VB; USHIO) fitted with a 436-nm g-line band-pass filter, fiber optic cable (SF101-NQ), and square lens (X057). Light intensity was measured with a UIT-250 digital photometer (UVD-S405 detector; USHIO), and light source distance was set accordingly. During irradiation of the visceral surfaces, the spleen was held in place with a piece of sterile nonadhesive plastic film, moistened with saline, which did not affect light transmission. After photoconversion, the spleen was replaced, the abdominal cavity was closed with 6–0 nylon suture, and the skin was closed with 9-mm wound clips.
For femur photoconversion, an ∼1.5-cm vertical incision was made in the skin on the dorsal side of the left thigh, and jeweler’s forceps were used to separate the vastus lateralis and biceps femoris muscles gently, exposing the femur. This approach minimized interference with the sciatic nerve and blood vessels running through the thigh. Bone reduction forceps were used to hold the muscle clear of the femur while it was irradiated with violet light (5 min, 500 mW/cm2). After photoconversion, the muscle surrounding the femur was replaced, and the skin was closed with 9-mm wound clips. Throughout and after both photoconversion procedures the spleen and femur were irrigated with sterile saline solution. Surgery and photoconversion were conducted concurrently on different mice, allowing one mouse to be photoconverted every 10 min. Mice were allowed to recover on a heating mat (37 °C) and regained full mobility within 30 min.
Mice were subjected to either spleen or femur photoconversion, never both. Unconverted mice (no surgery, to calibrate flow cytometry gates) and 0-h group mice (killed before photoconversion by terminal cardiac puncture and cervical dislocation, to assess photoconversion efficiency) were included in each experiment. Although surgery itself induced acute leukocytosis (peaking within 3 h), sham photoconversion experiments (in which surgery was performed with or without irradiation) confirmed that exposure to violet light did not affect myeloid cell distribution, as is consistent with previous findings (25).
For in vitro photoconversion, single-cell suspensions of BM cells (2.5 × 106 cells in 100 µL in transparent microcentrifuge tubes) were irradiated with violet light (3 min, 150 mW/cm2). Cells either were analyzed immediately or were incubated for 24 h in RPMI culture medium/10% FBS at 37 °C (5% CO2 humidified atmosphere) before analysis. In vitro photoconversion efficiency was >99%.
Calculation of Migration Probability and Residence Time for Tumor-Infiltrating Monocytes.
Monocytes enter the tumor from the blood and leave the tumor (by dying or by exiting back to the blood) at uniform rates. Accordingly, migration into and exit from the tumor can be described by the differential equation, d(NT rT)/dt = NB rB p − NT rT /τ (Eq. 1). p (/h) represents the probability per unit time that a cell will migrate from the blood to the tumor (migration probability). The units of p are scalable such that, for example, p = 0.60/h = 0.01/min. τ (h) represents the average time that a cell remains in the tumor before dying or exiting (residence time). Changes in tumor monocyte number caused by in situ proliferation were assumed to be negligible based on the very low proportions of actively proliferating monocytes detected in the tumors of Fucci mice. rB and rT (expressed as a function of time) represent the proportion of KikRed+ cells in the blood or tumor, respectively, within the population of interest (e.g., BM-pool monocytes). NB and NT represent the total number of monocytes (both KikGreen+ and KikRed+) in the blood or tumor, respectively, and are assumed to remain constant over the duration of each 24-h experiment (Table S1). Monocyte accumulation in the blood was modeled by the equation, rB(t) = aB [exp(−kB1t) − exp(−kB2t)] (Eq. 2). After substituting Eq. 2 into Eq. 1 and taking m = (NB/N)p and k = 1/τ, the solution of Eq. 1 under the initial condition rT(0) = 0 gives rT(t) = aBm{[exp(−kB1t) −exp(–kt)]/(k − kB1) – [exp(−kB2t) − exp(−kt)]/(k − kB2)} (Eq. 3). Eqs. 2 and 3 were fitted to blood and tumor data obtained from KikGR mouse photoconversion experiments using weighted least squares regression to obtain estimates for aB, kB1, kB2, m, and k, with weights given by the reciprocal of the variance of each measurement. The SE associated with each of the estimated parameters was determined by the law of error propagation. Finally, m and k (± SE) were used to calculate p and τ (± SE). Calculations were performed using the statistical software R (version 3.0.1; www.r-project.org).
Data Analyses.
Blood and BM cell numbers were adjusted to represent whole-body values based on the assumptions that the total blood volume of a mouse is 7% of its body weight and that a single mouse femur comprises 1/15 of total body BM cells (37). Photoconversion of 100% of myeloid cells in the femur or spleen was not possible by the procedures used in this study, and, unlike the spleen, a single femur represents only a fraction of the whole-body BM pool. To correct for differences in the extent of photoconversion of each myeloid cell pool, the proportion of KikRed+ cells in each tissue was multiplied by 100/(% photoconversion efficiency) and, in mice undergoing femur photoconversion, by a factor of 15. Thus, KikGR data represent the contribution of the spleen or whole-body BM if all myeloid cells within these pools were photoconverted at time 0 h. The number of KikRed+ cells was calculated by multiplying the adjusted proportion of KikRed+ cells in each tissue by mean cell counts from nonphotoconverted or 0-h group mice (Table S1), thus ensuring that results did not reflect acute fluctuations in total cell numbers following surgery. Because variations in tumor size might have influenced infiltration by KikRed+ cells, tumor cell numbers were normalized by tumor weight, i.e., multiplication by (mean tumor weight for experiment)/(tumor weight for individual mouse). Finally, to correct for differences in the whole-body abundances of spleen-pool and BM-pool cells, which could account for differences in their redistribution after photoconversion, some data for KikRed+ cell numbers also were expressed as a proportion of their respective source pool (% pool).
Statistics.
Results are expressed as mean ± SEM for n independent observations, where n represents the number of individual mice in each treatment group per time point. All data are representative of results obtained in at least two independent experiments. Where appropriate, data were statistically analyzed using GraphPad Prism software (version 6.0b; GraphPad Software) as follows: two-tailed unpaired t test (Fucci percentage data, KikGR in vitro photoconversion data, and migration probability and residence time data); one-way ANOVA with Dunnett’s posttest (wild-type mouse tumor kinetics data, adoptive transfer data); one-way ANOVA with Tukey posttest (Fucci mean fluorescence intensity data); two-way ANOVA with Sidak posttest (KikGR time-course data); and two-way ANOVA with Tukey posttest (Fucci cell number data). P values less than 0.05 were considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank A. Miyawaki and the RIKEN BioResource Center for providing FucciG1 and FucciS/G2/M mice; C. Kasahara and A. Hata for animal care; S. Aoki and S. Fujita for expert technical assistance; and I. Bertoncello for helpful discussions. This work was supported by the Japan Science and Technology Agency CREST program; Grants-in-Aid for Scientific Research (C) 25460491 (to S.U.) and (B) 25293113 (to K.M.) from the Japanese Ministry of Education, Culture, Sports, Science and Technology; an Australian Government Prime Minister’s Australia Asia Endeavour Award (to F.H.W.S.); and an E & V Puzey Foundation Scholarship (to F.H.W.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. F.G. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1402914111/-/DCSupplemental.
References
- 1.Hanahan D, Coussens LM. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 2.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 3.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 5.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 6.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luis TC, Killmann NM, Staal FJ. Signal transduction pathways regulating hematopoietic stem cell biology: Introduction to a series of Spotlight Reviews. Leukemia. 2012;26(1):86–90. doi: 10.1038/leu.2011.260. [DOI] [PubMed] [Google Scholar]
- 8.Kim CH. Homeostatic and pathogenic extramedullary hematopoiesis. J Blood Med. 2010;1:13–19. doi: 10.2147/JBM.S7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Malley DP, et al. Morphologic and immunohistochemical evaluation of splenic hematopoietic proliferations in neoplastic and benign disorders. Mod Pathol. 2005;18(12):1550–1561. doi: 10.1038/modpathol.3800480. [DOI] [PubMed] [Google Scholar]
- 10.Woodruff MF, Symes MO. The significance of splenomegaly in tumour-bearing mice. Br J Cancer. 1962;16:120–130. doi: 10.1038/bjc.1962.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young MR, Aquino S, Young ME. Differential induction of hematopoiesis and immune suppressor cells in the bone marrow versus in the spleen by Lewis lung carcinoma variants. J Leukoc Biol. 1989;45(3):262–273. doi: 10.1002/jlb.45.3.262. [DOI] [PubMed] [Google Scholar]
- 12.Bronte V, et al. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96(12):3838–3846. [PMC free article] [PubMed] [Google Scholar]
- 13.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leuschner F, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209(1):123–137. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutta P, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cortez-Retamozo V, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci USA. 2012;109(7):2491–2496. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cortez-Retamozo V, et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity. 2013;38(2):296–308. doi: 10.1016/j.immuni.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med. 1968;128(3):415–435. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nowotschin S, Hadjantonakis AK. Use of KikGR a photoconvertible green-to-red fluorescent protein for cell labeling and lineage analysis in ES cells and mouse embryos. BMC Dev Biol. 2009;9:49. doi: 10.1186/1471-213X-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsutsui H, Karasawa S, Shimizu H, Nukina N, Miyawaki A. Semi-rational engineering of a coral fluorescent protein into an efficient highlighter. EMBO Rep. 2005;6(3):233–238. doi: 10.1038/sj.embor.7400361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakaue-Sawano A, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 22.Tomura M, et al. Contrasting quiescent G0 phase with mitotic cell cycling in the mouse immune system. PLoS ONE. 2013;8(9):e73801. doi: 10.1371/journal.pone.0073801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawanobori Y, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111(12):5457–5466. doi: 10.1182/blood-2008-01-136895. [DOI] [PubMed] [Google Scholar]
- 24.Tomura M, et al. Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci USA. 2008;105(31):10871–10876. doi: 10.1073/pnas.0802278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomura M, et al. Activated regulatory T cells are the major T cell type emigrating from the skin during a cutaneous immune response in mice. J Clin Invest. 2010;120(3):883–893. doi: 10.1172/JCI40926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kotani M, et al. Systemic circulation and bone recruitment of osteoclast precursors tracked by using fluorescent imaging techniques. J Immunol. 2013;190(2):605–612. doi: 10.4049/jimmunol.1201345. [DOI] [PubMed] [Google Scholar]
- 27.Tomura M, Kabashima K. Analysis of cell movement between skin and other anatomical sites in vivo using photoconvertible fluorescent protein “Kaede”-transgenic mice. Methods Mol Biol. 2013;961:279–286. doi: 10.1007/978-1-62703-227-8_18. [DOI] [PubMed] [Google Scholar]
- 28.Serbina NV, Hohl TM, Cherny M, Pamer EG. Selective expansion of the monocytic lineage directed by bacterial infection. J Immunol. 2009;183(3):1900–1910. doi: 10.4049/jimmunol.0900612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ugel S, et al. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep. 2012;2(3):628–639. doi: 10.1016/j.celrep.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Hettinger J, et al. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol. 2013;14(8):821–830. doi: 10.1038/ni.2638. [DOI] [PubMed] [Google Scholar]
- 31.Fogg DK, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science. 2006;311(5757):83–87. doi: 10.1126/science.1117729. [DOI] [PubMed] [Google Scholar]
- 32.Marigo I, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32(6):790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 33.McAllister SS, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133(6):994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sunderkötter C, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172(7):4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 35.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7(3):311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 37.Boggs DR. The total marrow mass of the mouse: A simplified method of measurement. Am J Hematol. 1984;16(3):277–286. doi: 10.1002/ajh.2830160309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.