

Abstract
It is controversial whether fast-folding proteins can form productive on-pathway intermediates that are more stable than the denatured state because noncovalent intermediates are usually evanescent. Here, we apply the classical criteria for the existence of intermediates: namely, the intermediates form and react rapidly enough to be on pathway and they can be isolated and characterized. The folding of the 71-residue, mainly α-helical FF domain from human HYPA/FBP11 fulfills these classical criteria, as was found for Im7. The FF domain folds in two phases, one on the μs and the other on the ms time scale. An engineered mutant folds only to a partly folded state, with some 20–40% of the native helical content. The kinetic properties of the mutant are identical to those found for the fast phase of the wild-type protein, and it is likely that the mutant folds just to the intermediate state. A full kinetic analysis of the folding of wild-type protein, using the amplitudes of its native and denatured states and the observed values for the mutant, rules out an off-pathway scheme but fits an on-pathway scheme, with a low energy intermediate that is modeled by the mutant. The experimental proof benchmarks a molecular dynamics method that identifies an obligatory intermediate observed in multiple simulations. The conformational space defining this intermediate is visited several times in the simulations, leading to high populations consistent with the presence of a low energy intermediate.
Keywords: FF domain, helix bundle, molecular dynamics, folding intermediate
It is of fundamental mechanistic importance to determine whether there are intermediates on protein folding pathways en route to the native state. We, for example, favor a mechanism for the folding of domains that at one extreme, nucleation-condensation (1–3), involves simultaneous formation of secondary and tertiary structure without any discreet intermediates, which can then slide to the classical framework or diffusion–collision mechanism (4–6) with population of an intermediate (refs. 7 and 8 and reviewed by Daggett and Fersht in ref. 9). On the other hand, Krantz et al. (10) conclude that there is little support in the literature for early accumulation of an intermediate before an initial search-dependent nucleation barrier. They argue that intermediates that are stable relative to the unfolded state cannot form and accumulate before the initial nucleation step. They propose that intermediates can form only later, and can then accumulate only when blocked by later barriers, which they believe are often due to nonobligatory misfolding reorganization events.
Classical evidence for the presence of an on-pathway intermediate requires three criteria to be fulfilled: (i) the intermediate forms rapidly enough to be on the pathway; (ii) it reacts (or interconverts) rapidly enough to be on pathway; and (iii) it can be isolated and characterized (11). Although there is much compelling evidence for intermediates (8, 12–19), it is difficult to refute the arguments of Krantz et al. (10) because the elusive nature of protein-folding intermediates makes them very difficult to characterize and to be shown to be on-pathway rather than off. The challenge, therefore, is to design equivalent experiments that can detect unambiguously whether proteins can form productive early intermediates that are more stable than the unfolded state. In one of the most convincing studies, the laboratories of Radford and Roder (12) find that the Im7 protein folds in two distinct first-order phases by a combination of stopped-flow and continuous-flow fluorimetry. The observed rate constants for the slower phase (10- to 100-ms time scale) follow a classical V-shaped chevron plot against denaturant concentration. The faster phase (t1/2 ≈ 500 μs) is for a rapidly formed intermediate that potentially could be either on- or off-pathway. They eliminated the off-pathway route by analyzing the chevron plot for the intermediate by using the criterion that it unfolds too slowly in water to be off-pathway (19, 20). All of the kinetic criteria, including an amplitude analysis of the traces, support an on-pathway intermediate of lower energy than the unfolded state for Im7, and so the classical criteria i and ii are fulfilled. Importantly, the intermediate can be engineered to be the most stable species under folding conditions, with kinetics and fluorescent properties similar to those for the evanescent intermediate for wild-type folding, thus becoming close to fulfilling criterion iii (21).
Here, we demonstrate all three of the classical criteria for a low-energy intermediate in the folding of an FF domain from human HYPA/FBP11, a protein with 71 residues that forms three α-helices and one 310-helix (22). An intermediate was also detected by a procedure in multiple independent molecular dynamics (MD) simulations of thermal unfolding, thereby providing atomic-resolution models for the unfolding pathway and intermediate state ensemble.
Materials and Methods
Protein Expression and Purification. Wild-type FF domain and mutants were expressed from a modified pRSET vector (Invitrogen) and purified by using a modification of the published procedure (22).
Kinetic and Equilibrium Experiments. Buffer urea solutions containing 50 mM sodium acetate, pH 5.7, with either 100 mM NaCl or 400 mM sodium sulfate were used. Equilibrium and kinetic experiments monitored tryptophan fluorescence on excitation at 280 nm and CD at 222 nm at 283 K or 298 K. Stopped-flow experiments were performed on an SX.18MV or a PiStar-180 spectrometer (Applied Photophysics, Leatherhead, U.K.). There are two proline residues in the structured part of the FF structure. The rate constants for the slow cis-trans interconversion in refolding experiments, and their amplitudes, were determined in double jump unfolding/refolding experiments (23). The amplitudes of the cis-trans isomerization, 15% of the total, were accounted for in the amplitude analysis of stopped-flow traces (data not shown). Continuous-flow experiments were conducted at room temperature (≈298 K) on an in-house-built apparatus of similar design and methodology to that published (24). Temperature-jump fluorescence measurements were performed on a Hi-Tech PTJ-64 capacitor-discharge T-jump apparatus (Hi-Tech, Salisbury, U. K.) from 21.5 to 25°C.
MD Simulation. Nine 30-ns simulations of the FF domain were performed by using encad (25, 26). The protein was solvated in a box of F3C waters (27), and its unfolding was simulated by raising the temperature to 498 K. Multiple independent trajectories were determined by varying the number of minimization steps on the initial NMR structure (PDB code 1UZC). Asp and Glu side chains were protonated to simulate low pH in five simulations of the wild-type protein. They remained unprotonated to simulate neutral pH in two other simulations of the wild-type protein as well as two simulations of the L25A/D46N double mutant. A control simulation was also performed at 298 K, neutral pH.
Results
Kinetic and Equilibrium Measurements on Wild-Type FF. Urea-induced denaturation of FF was monitored both by far-UV CD at 222 nm and Trp fluorescence emission (Fig. 1), at 330 ± 25 nm (bandpass) or >360 nm (cut-off filter). Equilibrium unfolding fitted well to a two-state transition when measured independently by CD and fluorescence, with good agreement in the calculated thermodynamic parameters. {By CD, [urea]50% = 2.81 ± 0.04 M and mD-N = 1.08 ± 0.02 kcal·mol-1·M-1 (mean and SDs of four independent experiments); and by fluorescence, [urea]50% = 2.81 ± 0.05 M and mD—N = 0.99 ± 0.05 kcal·mol-1·M-1, as determined from global fitting of the urea dependence at 100 individual wavelengths (301–400 nm, error estimated by boot strap analysis)}. However, intermediates may be missed in equilibrium denaturation experiments because most of the denaturation curve is followed at high denaturant concentrations where the intermediate is not populated.
Fig. 1.

Equilibrium data for wild-type FF and the L25A/D46N mutant in 50 mM sodium acetate, pH 5.7/100 mM NaCl. (a) Fluorescence spectra: wild-type FF (Native), L25A/D46N (Intermediate), and the denatured protein in water (calculated from a global fit to urea denaturation curves of L25A/D46N at 100 different wavelengths). (b and c) Endpoints from stopped-flow refolding and unfolding traces at different [urea] at 10°C for wild-type FF (open circles) and L25A/D46N (filled circles) using a 330 ± 25 nm bandpass (b) or a 360 nm cut-off (c) filter. Equilibrium urea denaturation curves were qualitatively similar. The dashed lines are the slopes of the respective denatured baseline as given by the fit (solid lines) to a two-state model. Open triangles are the starting points for refolding calculated by using the refolding amplitudes (corrected for the cis-Pro phase and the dead-time of the stopped flow). Assuming linear dependence for the fluorescence of the unfolded species with [urea], these points should fall on the dashed line in a two-state system. With the 330-nm bandpass emission filter, a burst phase manifested as loss of amplitude is observed whereas the 360-nm filter yields a hyperfluorescent burst phase. (d) Far-UV CD spectra of wild-type FF and L25A/D46N at 3°C. (Inset) CD-monitored urea titrations at 222 nm of wild-type FF (open circles) and L25A/D46N (filled) at 25°C.
The refolding and unfolding kinetics of wild-type FF were measured by stopped-flow, continuous-flow, and T-jump techniques (Fig. 2). Excluding the cis-trans proline isomerization, stopped-flow millisecond time courses were well described by single exponentials. But, submillisecond continuous-flow experiments revealed a faster phase, consistent with a burst phase detected in amplitude analyses of stopped-flow experiments using both far-UV CD at 222 nm (data not shown) and Trp fluorescence emission (Fig. 1).
Fig. 2.

(a) Kinetic traces showing refolding of wild-type FF at 2.0 M urea, a concentration at which both continuous (Left) and stopped-flow (Right) measurements are feasible (50 mM sodium acetate, 100 mM NaCl; 330 ± 25-nm emission filter). The solid line is a bi-exponential function fitted to both phases simultaneously. (b and c) Kinetic refolding and unfolding traces of L25A/D46N measured by using continuous flow fluorimetry (50 mM sodium acetate, 0.4 M Na2SO4; 330 ± 25-nm emission filter). (d) Chevron plots of the slow (circles) and fast phases of wild-type FF (open triangles) and the L25A/D46N mutant (filled triangles) recorded at 25°C by T-jump (filled circles), stopped flow (open circles), and continuous flow (triangles) fluorimetry. The error bars for stopped-flow data are SDs calculated from five to six experimental traces. The errors for the continuous flow data were estimated as 10%. The kobss were fitted to a two-state model (32), yielding the following parameters: for λ1: kf = 19,000 ± 2,000 s-1, ku = 1,500 ± 2,300 s-1, mf = 0.29 ± 0.16 kcal·mol-1·M-1, mu = 0.35 ± 0.22 kcal·mol-1·M-1; for λ2: kf = 3,100 ± 200 s-1, ku = 29 ± 0.8 s-1, mf = 0.86 ± 0.02 kcal·mol-1·M-1, mu = 0.15 ± 0.002 kcal·mol-1·M-1. Experiments were performed in 50 mM sodium acetate, pH 5.7/100 mM NaCl. (e) Amplitudes from kinetic traces measured with the 330 ± 25-nm filter. The fluorescence of the stopped-flow unfolding (open circles) and refolding (filled circles) amplitudes (right axis) can only be qualitatively compared with the amplitudes from continuous flow fluorimetry (triangles, left axis). The solid lines are free fits to the standard solvent denaturation equation, assuming that the refolding and unfolding amplitudes are zero at high and low [urea], respectively.
The urea dependence of the relaxation rate constants for the fast (λ1), and slow (λ2) phases is shown in Fig. 2. The two phases were monitored by fluorescence by using both a 330 ± 25-nm bandpass emission filter and a 360-nm cut-off filter. The fluorescence change of the fast phase followed by the 360-nm cut-off filter was in the opposite direction from that of the slow phase followed by the same filter, showing that the fast phase is distinct from the phase attributed to formation of N (compare Fig. 1). This distinction is crucial to the interpretation of the kinetic data. By using the 330 ± 25 nm bandpass filter, the formation of I and N both showed increasing fluorescence in a refolding experiment (Fig. 2). With the 360-nm cut-off filter, however, formation of I still had a positive change in fluorescence, but the formation of N resulted in a decrease in fluorescence (compare Figs. 1 b and c and 4). Therefore, the formation of I can unequivocally be distinguished from the formation of N in the kinetic experiments.
Fig. 4.

(a) Traces of refolding of L25A/D46N and I44A/V67A in 1.6 M urea in continuous flow spectrofluorometric experiments using a 360-nm cutoff filter. The line is a fit to a double exponential where the rate constant for the fast phase was constrained by data obtained by using a 330 ± 25 nm bandpass filter (Inset). The fluorescence of U (0.85) and N (0.65) were obtained from this fit. The fluorescence of I (1.01) was calculated by using the measured fluorescence for L25A/D46N and the equilibrium constant obtained from a chevron plot of the mutant (not shown). (b) Simulated refolding traces at 1.6 M urea using best-fit parameters for on-pathway (solid line), and off-pathway (dashed line) models (Table 1), compared with the experimentally observed trace. (c and d) Progress curves for U, I, and N at 1.6 M urea generated by using best-fit parameters for on and off-pathway models (Table 1).
An Experimental Model for the Intermediate. The mutations L25A and D46N each destabilized FF by 2.5–3 kcal·mol-1. As ΔGD—N at 298 K for wild-type FF domain is 2.8–3 kcal·mol-1, the native form of the double mutant L25A/D46N should not be populated to more than ≈1–2% as energetics are typically additive for noninteracting residues (28, 29).
CD measurements indicated that the double mutant had ≈20–40% of the helical content of wild-type FF (Fig. 1d), depending on the experimental conditions. Its fluorescence spectrum (Fig. 1a) had a higher quantum yield than native and denatured states at >360 nm. Importantly, the fluorescence change on denaturation of L25A/D46N using the 360-nm cut-off filter is qualitatively distinct from that of the wild type (Fig. 1c); by changing in the opposite direction, it showed that it is not the native state that is lost on denaturation of L25A/D46N, and so the mutant is not a mixture of native and denatured states in equilibrium.
Additional evidence that L25A/D46N is a model for the intermediate came from its folding kinetics. First, the chevron plot for L25A/D46N was identical to that of the fast phase of wild type under similar conditions (Fig. 2); and there was no detectable slow phase by either temperature-jump or rapid-mixing techniques. Second, kinetic traces of L25A/D46N could be measured not only under refolding conditions but also under unfolding conditions (Fig. 2). If the native state is populated, as for wild-type FF, the slow N to I transition obscures the fast I to U.
Kinetic Evidence for an On-Pathway Intermediate. If the fast phase is on a much faster time scale than the slow phase, then it is very difficult to distinguish between on- and off-pathway sequences because there is effectively a preequilibrium (11) (Scheme 1). To differentiate between the on/off-pathway scenarios, λ1 and λ2 must be close enough to allow coupling of the rate constants under the experimental conditions (see Fig. 3). If U and I are in fast preequilibrium [i.e., kUI and kIU >> kIN and kNI (or kUN and kNU)] λ1 is determined by kUI + kIU. The observed refolding rate constant λ2, at low denaturant concentrations, would then be slowed by a factor equal to (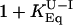 ). Under these conditions, the solutions for on- and off-pathway schemes are kinetically equivalent, and the order of the events cannot be resolved. But, as λ1 and λ2 become more similar, kUN (in the off-path scheme) will begin to influence λ1. So, when λ1 is not dependent solely on kUI and kIU, and λ2 on kIN and kNI, the four microscopic rate constants, with their associated m values, should be fitted simultaneously to the observed rate constants of the two phases λ1 and λ2. Linear free energy relationships with urea are assumed for each of the rate constants, i.e.,
). Under these conditions, the solutions for on- and off-pathway schemes are kinetically equivalent, and the order of the events cannot be resolved. But, as λ1 and λ2 become more similar, kUN (in the off-path scheme) will begin to influence λ1. So, when λ1 is not dependent solely on kUI and kIU, and λ2 on kIN and kNI, the four microscopic rate constants, with their associated m values, should be fitted simultaneously to the observed rate constants of the two phases λ1 and λ2. Linear free energy relationships with urea are assumed for each of the rate constants, i.e., 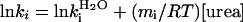 , where R is the gas constant and T the absolute temperature.
, where R is the gas constant and T the absolute temperature.
Scheme 1.

Schematic of on-pathway and off-pathway models.
Fig. 3.

Chevron plots of the slow and fast phases of I44A/V67A recorded at 25°C in 50 mM sodium acetate, pH 5.7/0.4 M Na2SO4. The slow phase λ2 was measured by stopped-flow and temperature-jump and the fast phase λ1 by continuous flow fluorimetry. The solid lines are fits to on-pathway and off-pathway models, respectively. Best-fit parameters are listed in Table 1. The dashed lines are the dependences of the respective microscopic rate constants on urea as given by the parameters in Table 1.
Mutation of two exposed hydrophobic residues into alanine residues yielded a protein, I44A/V67A, with a folding rate constant for the slower phase ≈2- to 4-fold higher than that of wild type, depending on the conditions (data not shown). The kobs values for the fast phase λ1 of the I44A/V67A mutant in the presence of 400 mM sodium sulfate, as measured by continuous flow fluorimetry, are plotted in Fig. 3 together with values for the slow phase λ2, measured by stopped-flow and T-jump fluorimetry under the same conditions. The observed λ1 and λ2 were fitted globally to the two roots of the quadratic equation describing a two-step reaction (11). The best-fit parameters from fitting on- and off pathway models, respectively, are shown in Table 1. The fit to an on-pathway scheme is slightly better than to an off-pathway, as judged by analysis of the residuals and the fitting errors.
Table 1. I44A/V67A: best-fit parameters for on- and off-pathway models.
| s-1
|
kcal·mol-1·M-1
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kUI | kIU | kIN | kUN | kNI | kNU | mUI | mIU | mIN | mUN | mNI | mNU | |
| On-path | 41,000 ± 6,000 | 1,400 ± 500 | 5,900 ± 1,100 | 23 ± 12 | 0.52 ± 0.09 | 0.25 ± 0.06 | 0.10 ± 0.10 | 0.12 ± 0.04 | ||||
| Off-path | 3,000 ± 24,000 | 5,000 ± 1,500 | 48,000 ± 12,000 | 7.2 ± 6.6 | 0 ± 3 | 0.08 ± 0.5 | 0.7 ± 0.6 | 0.20 ± 0.07 | ||||
From simultaneous fits of the fast and slow phases to the general solution of a three-state model (11).
The fluorescence of L25A/D46N enabled analysis of the amplitudes of continuous-flow refolding traces as a decisive test to judge whether I is on- or off-pathway (Fig. 4). The simulated trace, as predicted by the on-pathway model, fits almost perfectly with the experimental one. On the other hand, the off-pathway scheme yields a trace where the amplitude of λ1 is negative (the U to N transition dominates the phase) whereas the experimental amplitude is positive (U to I dominates, see Fig. 3). The off-pathway model would not account for the observed trace even if the fluorescence of I were seriously underestimated (even by >100%). This result rules out the off-pathway scheme as a model to describe the folding of the FF domain.
MD. Transition and intermediate states during thermal denaturation simulations are generally identified by conformational clustering methodologies in which similar structures are clustered near one another in a Euclidean space (30, 31). A fundamental difficulty in labeling such states from non-equilibrium unfolding simulations is determining their relative free energies. We have defined the transition state of unfolding to be the point of no return when the simulation leaves the extended native cluster. A more complex procedure is needed to define states as low free energy intermediates with any confidence.
The population of a given state is related to its free energy, such that a state with low free energy should have a higher population than a state with high free energy. In non-equilibrium unfolding simulations, however, the population of a given state can be dependent on the simulation time in addition to the problems with obtaining reliable populations with only limited sampling. In the absence of multiple exchanges between states, it is difficult to define the relative populations of those states. To identify relatively low free energy states, we consider the fact that high free energy states are unlikely to be populated significantly. The probability that a high free energy state will be observed in multiple simulations decreases exponentially with the number of simulations. If a state has a probability, p, that it will be observed in one simulation, it has a probability p2 of being observed in two, p3 of being observed in three, and so on. For high free energy states, we expect that p << 1 and the probability of being significantly populated in multiple simulations vanishes rapidly.
The results of an all-vs.-all structural comparison of the five, low pH, wild-type simulations are given in Fig. 5. The presence of on-diagonal clusters indicates regions of local similarity during unfolding. The presence of off-diagonal clusters indicates that similar conformations are being sampled in multiple simulations. This concurrence suggests that a relatively low free energy intermediate conformation is present. As indicated in the figure legend, we label these states as I1, I2, and I3, based on their structural similarity to one another. These should be seen as rather imprecise classifications, as a given cluster may be almost equally similar to two other clusters that are not similar to one another. The distinction between I1 and I2 is particularly fuzzy.
Fig. 5.

Pairwise Cα-RMSD plots for multiple simulations. Each structure is compared with all other structures in all simulations at 50 ps resolution over the 30-ns simulations. The x and y coordinates indicate the time points being compared, with color-coded RMSDs. (a) Pairwise comparisons of the five simulations of the protein at low pH. The magnitude of the Cα-RMSD is given in the color scale to the right of the plot. On-diagonal clusters, indicative of local clusters in a simulation, are boxed off. Off-diagonal clusters are circled. The presence of off-diagonal clusters suggests that the on-diagonal clusters belong to a common unfolding intermediate. On-diagonal clusters are grouped into three structurally distinct intermediates as indicated by the labels I1,I2, and I3.(b) Pairwise comparison between two simulations at low pH, two at neutral pH, and two of the L25A/D46N double mutant at neutral pH. An intermediate that is structurally similar to that seen at low pH (I1/2) is seen in all neutral pH simulations, and a new intermediate (Imut) is seen in the mutant simulations.
Of the three intermediate states identified, only I1 seems to be obligatory. In two simulations (simulations 4 and 5), the protein passed from the native state through I1, and directly to a fully unfolded state (Fig. 5a). In both simulations 1 and 3, the protein unfolded through I1 then I2, and, in simulation 2, it unfolded through I1 followed by I3 before becoming more fully unfolded. In simulations 2, 4, and 5, the protein collapsed from an unfolded state back to one of the intermediate conformations, but only transitions back to I1 and I3 were observed. Fig. 6a summarizes the transitions observed in the low pH simulations.
Fig. 6.

The structures and reaction schemes observed from the low (a) and neutral (b) pH simulations. The green arrows and text indicate a transition to Imut that is observed only in the simulations of the mutant protein. Arrows between states/structures indicate transitions seen in the simulations. Other transitions may be possible but were not observed directly. The structures shown are the mean structures from each state. The thickness of the ribbon is based on the standard deviation for the position of each α-carbon in the intermediate ensemble indicated. They are colored to show the location of the native helices. The N-terminal helix is red, the middle helix green, and the C-terminal helix is blue. The native ensemble is taken from a 30-ns simulation at neutral pH. Intermediate ensembles are pooled from the clusters indicated in Fig. 5. The denatured ensembles are taken from regions of their respective simulations that do not fall into intermediate clusters. The structure from the denatured ensemble that is most similar to the mean structure and that is most dissimilar is overlaid over the unfolded mean structure to give an idea of range.
The three intermediate conformational states defined here are virtually identical in terms of overall properties and those typically measured by experiment. I3 is slightly less structured than I1 or I2 based on packing and helix content. These low pH intermediates contain a roughly native-like amount of α-helix. The situation was similar at neutral pH for the wild-type protein (Fig. 5b), but it only populated I1 or I2 or what was really a combination of the two, I1/2 (because of the increased overlap of I1 and I2 at neutral pH, for simplicity we just denote it I1/2 in Figs. 5 and 6b). The mutant unfolded through I1, and then oscillated between I1/2 and a distinct conformation not seen in wild-type, labeled Imut. The intermediate conformations observed at neutral pH had lower helical content (≈50%) than those at low pH (≈90%). Nevertheless, all of the intermediate conformations were similar, with I2, I3, and Imut as variations on I1 with altered helix packing and helix content (Fig. 6).
Discussion
Experimental Evidence for an On-Pathway Intermediate. The wild-type FF domain folded in two phases, with rate constants of 19,000 s-1 and 3,000 s-1, each of which fitted a classical two-state chevron plot (32). To determine whether the intermediate was on- or off-pathway, two mutants were constructed. The double mutant I44A/V67A under suitable conditions had the two phases coupled. The unfolding rate constant for the faster phase in water (extrapolated from the chevron plot) was less than the folding rate constant for the slower phase; this result is inconsistent with an off-pathway intermediate (18–20). It could be argued that this analysis assumes a linear dependence of the logarithm of each individual rate constant with denaturant concentration and so the result is not beyond doubt. However, we constructed the double mutant L25A/D46N, which folded only to the intermediate state, so we could use its fluorescence properties for a quantitative amplitude analysis. It had a higher fluorescence yield than either the native and denatured states at higher wavelengths (>360 nm) (Fig. 1). An intermediate of high fluorescence was clearly seen to form and decay in the kinetic experiments with I44A/V67A (Fig. 4). The microscopic rate constants for the trace fitted well to an on-pathway scheme but there were anomalies assuming an off-pathway intermediate. Thus, the simplest scheme describing the data is a three-state on-pathway model (Scheme 1). The very nature of kinetic analysis requires that more complex schemes must also fit. But, similar analysis as in Fig. 4 suggests that an on-pathway scheme fits the data better than a triangular (parallel) scheme although this result is highly dependent on the fluorescence yield of I (not shown). Even if there is a direct route from U to N, only 10% of the molecules would follow this path, as judged by fitting a triangular scheme (19) to the kinetic data.
Evidence for an On-Pathway Intermediate by MD. There was an obligatory intermediate (I1) during the unfolding of the FF domain in all nine MD simulations, independent of pH and sequence. Other intermediates were also visited besides I1, and there were interconversions between the various conformations. Intermediates were identified by all-vs.-all structural comparisons. Local clusters were identified within each simulation as possible intermediate conformations. However, we also required correspondence between different simulations, such that multiple simulations are visiting the same region of conformational space, consistent with a relative minimum on the free energy landscape, or a low free energy intermediate. Interestingly, the intermediate conformations identified in the simulations may help to explain the distinctive fluorescent properties observed experimentally. The two Trp residues are partially exposed in the native state, and Trp-11 is in close proximity to several main chain carbonyl groups as well as Tyr-9. Trp-11 is in close contact with Tyr-9 ≈64% of the time in the native simulation, which could lead to partial quenching of the Trp signal. In contrast, these residues are in contact ≈10% of the time in the combined intermediate state, which might explain the hyperfluorescence of the intermediate.
Although we observed several distinct intermediates, particularly for the wild-type protein at low pH, they had very similar physical properties, similar populations, and interconverted readily, at least under these conditions, suggesting that they may be conformational substates within the intermediate state. In addition to populating the intermediates observed for wild type, the mutant populated a distinct intermediate in conformational exchange with the other intermediates. Finally, the intermediates at neutral pH had disrupted secondary structure, with ≈50% of the native helical structure.
Acknowledgments
We thank Dr. Neil Ferguson for valuable comments on the work. P.J. is supported by a European Molecular Biology Organization long-term fellowship. S.G. is supported by a fellowship from the Instituto Pasteur-Fondazione Cenci Bolognetti (Rome). Financial support for the computational studies was provided by the National Institutes of Health (Grant GM 50789 to V.D. and Molecular Biophysics Training Grant T32 GM 08268 to R.D.).
Abbreviations: MD, molecular dynamics; I, intermediate; N, native; U, unfolded.
References
- 1.Fersht, A. R. (1995) Proc. Natl. Acad. Sci. USA 92, 10869-10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itzhaki, L. S., Otzen, D. E. & Fersht, A. R. (1995) J. Mol. Biol. 254, 260-288. [DOI] [PubMed] [Google Scholar]
- 3.Otzen, D. E., Itzhaki, L. S., ElMasry, N. F., Jackson, S. E. & Fersht, A. R. (1994) Proc. Natl. Acad. Sci. USA 91, 10422-10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ptitsyn, O. B. (1995) Trends Biochem. Sci. 20, 376-379. [DOI] [PubMed] [Google Scholar]
- 5.Karplus, M. & Weaver, D. L. (1994) Protein Sci. 3, 650-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baldwin, R. L. & Rose, G. D. (1999) Trends Biochem. Sci. 24, 77-83. [DOI] [PubMed] [Google Scholar]
- 7.Gianni, S., Guydosh, N. R., Khan, F., Caldas, T. D., Mayor, U., White, G. W. N., DeMarco, M. L., Daggett, V. & Fersht, A. R. (2003) Proc. Natl. Acad. Sci. USA 100, 13286-13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayor, U., Guydosh, N. R., Johnson, C. M., Grossmann, J. G., Sato, S., Jas, G. S., Freund, S. M. V., Alonso, D. O. V., Daggett, V. & Fersht, A. R. (2003) Nature 421, 863-867. [DOI] [PubMed] [Google Scholar]
- 9.Daggett, V. & Fersht, A. (2003) Nat. Rev. Mol. Cell. Biol. 4, 497-502. [DOI] [PubMed] [Google Scholar]
- 10.Krantz, B. A., Mayne, L., Rumbley, J., Englander, S. W. & Sosnick, T. R. (2002) J. Mol. Biol. 324, 359-371. [DOI] [PubMed] [Google Scholar]
- 11.Fersht, A. (1999) Structure and Mechanism in Protein Science (Freeman, New York).
- 12.Capaldi, A. P., Shastry, M. C. R., Kleanthous, C., Roder, H. & Radford, S. E. (2001) Nat. Struct. Biol. 8, 68-72. [DOI] [PubMed] [Google Scholar]
- 13.Ferguson, N., Capaldi, A. P., James, R., Kleanthous, C. & Radford, S. E. (1999) J. Mol. Biol. 286, 1597-1608. [DOI] [PubMed] [Google Scholar]
- 14.Radford, S. E., Dobson, C. M. & Evans, P. A. (1992) Nature 358, 302-307. [DOI] [PubMed] [Google Scholar]
- 15.Sánchez, I. E. & Kiefhaber, T. (2003) J. Mol. Biol. 325, 367-376. [DOI] [PubMed] [Google Scholar]
- 16.Shastry, M. C. R. & Roder, H. (1998) Nat. Struct. Biol. 5, 385-392. [DOI] [PubMed] [Google Scholar]
- 17.Teilum, K., Maki, K., Kragelund, B. B., Poulsen, F. M. & Roder, H. (2002) Proc. Natl. Acad. Sci. USA 99, 9807-9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Travaglini-Allocatelli, C., Gianni, S., Morea, V., Tramontano, A., Soulimane, T. & Brunori, M. (2003) J. Biol. Chem. 278, 41136-41140. [DOI] [PubMed] [Google Scholar]
- 19.Wildegger, G. & Kiefhaber, T. (1997) J. Mol. Biol. 270, 294-304. [DOI] [PubMed] [Google Scholar]
- 20.Bai, Y. (1999) Proc. Natl. Acad. Sci. USA 96, 477-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capaldi, A. P., Kleanthous, C. & Radford, S. E. (2002) Nat. Struct. Biol. 9, 209-216. [DOI] [PubMed] [Google Scholar]
- 22.Allen, M., Friedler, A., Schon, O. & Bycroft, M. (2002) J. Mol. Biol. 323, 411-416. [DOI] [PubMed] [Google Scholar]
- 23.Brandts, J. F., Halvorson, H. R. & Brennan, M. (1975) Biochemistry 14, 4953-4963. [DOI] [PubMed] [Google Scholar]
- 24.Shastry, M. C. R., Luck, S. D. & Roder, H. (1998) Biophys. J. 74, 2714-2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levitt, M. (1990) encad Computer Program, Energy Calculations and Dynamics (Molecular Applications Group, Palo Alto, CA).
- 26.Levitt, M., Hirshberg, M., Sharon, R. & Daggett, V. (1995) Comput. Phys. Commun. 91, 215-221. [Google Scholar]
- 27.Levitt, M., Hirshberg, M., Sharon, R., Laidig, K. E. & Daggett, V. (1997) J. Phys. Chem. B 101, 5051-5061. [Google Scholar]
- 28.Carter, P. J., Winter, G., Wilkinson, A. J. & Fersht, A. R. (1984) Cell 38, 835-840. [DOI] [PubMed] [Google Scholar]
- 29.Serrano, L., Day, A. G. & Fersht, A. R. (1993) J. Mol. Biol. 233, 305-312. [DOI] [PubMed] [Google Scholar]
- 30.Li, A. & Daggett, V. (1998) J. Mol. Biol. 275, 677-694. [DOI] [PubMed] [Google Scholar]
- 31.Li, A. & Daggett, V. (1994) Proc. Natl. Acad. Sci. USA 91, 10430-10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson, S. E. & Fersht, A. R. (1991) Biochemistry 30, 10428-10435. [DOI] [PubMed] [Google Scholar]