

Abstract
The complete set of viable deletion strains in Saccharomyces cerevisiae was screened for sensitivity of mutants to five oxidants to identify cell functions involved in resistance to oxidative stress. This screen identified a unique set of mainly constitutive functions providing the first line of defense against a particular oxidant; these functions are very dependent on the nature of the oxidant. Most of these functions are distinct from those involved in repair and recovery from damage, which are generally induced in response to stress, because there was little correlation between mutant sensitivity and the reported transcriptional response to oxidants of the relevant gene. The screen identified 456 mutants sensitive to at least one of five different types of oxidant, and these were ranked in order of sensitivity. Many genes identified were not previously known to have a role in resistance to reactive oxygen species. These encode functions including protein sorting, ergosterol metabolism, autophagy, and vacuolar acidification. Only two mutants were sensitive to all oxidants examined, only 12 were sensitive to at least four, and different oxidants had very different spectra of deletants that were sensitive. These findings highlight the specificity of cellular responses to different oxidants: No single oxidant is representative of general oxidative stress. Mitochondrial respiratory functions were overrepresented in mutants sensitive to H2O2, and vacuolar protein-sorting mutants were enriched in mutants sensitive to diamide. Core functions required for a broad range of oxidative-stress resistance include transcription, protein trafficking, and vacuolar function.
Cells growing aerobically are exposed to reactive oxygen species (ROS) generated during metabolism. These include hydrogen peroxide (H2O2), the hydroxyl radical (OH•), and the superoxide anion (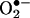 ), which can damage proteins, lipids, carbohydrates, and DNA. Oxidative stress occurs when cellular defense mechanisms are unable to cope with existing ROS, and it has been associated with a number of pathologies including cancer, cardiovascular disease, Down's syndrome, Friedreich's ataxia, aging, and age-related diseases (1, 2). ROS have been implicated in a caspase-independent mechanism activating apoptosis (3). In respiring cells, the primary source of ROS is leakage of electrons from the mitochondrial respiratory chain (4). Saccharomyces cerevisiae cells that lack functional mitochondria or an intact electron transport chain or that have been treated with mitochondrial inhibitors are viable, but sensitive to ROS (5–8).
), which can damage proteins, lipids, carbohydrates, and DNA. Oxidative stress occurs when cellular defense mechanisms are unable to cope with existing ROS, and it has been associated with a number of pathologies including cancer, cardiovascular disease, Down's syndrome, Friedreich's ataxia, aging, and age-related diseases (1, 2). ROS have been implicated in a caspase-independent mechanism activating apoptosis (3). In respiring cells, the primary source of ROS is leakage of electrons from the mitochondrial respiratory chain (4). Saccharomyces cerevisiae cells that lack functional mitochondria or an intact electron transport chain or that have been treated with mitochondrial inhibitors are viable, but sensitive to ROS (5–8).
ROS can lead to cell death; however, cells possess a variety of defenses including cell-cycle delay (9–11), the induction of enzymes such as catalases, peroxidases, and superoxide dismutases, and the synthesis of antioxidants such as glutathione, vitamins C and E, and ubiquinol (12). Human and yeast cells can mount an adaptive response in which exposure to a low dose of an oxidant induces resistance to a higher dose (5, 6, 13). In S. cerevisiae, there is overlap in the stress systems induced by treating cells with various oxidants. However, differences have been noted in sensitivity, in adaptive and cell-cycle responses to different oxidants or toxic products of ROS damage (5, 9, 10, 14, 15), and in activation of the transcription factor Yap1p by H2O2 or diamide (16).
Most studies of oxidative stress at the molecular level have used a range of external oxidants or free-radical-generating compounds. Genomewide surveys of changes in transcripts and proteins have identified many genes that are activated or repressed in response to a few oxidants (17, 18). These studies have provided insight on regulatory responses, but they have not revealed the relevance of these genes to survival, repair of damage, or cellular recovery. A number of antioxidant and repair processes have been characterized by isolating mutants affected by antioxidants or resistant to oxidants, or genes that confer resistance when overexpressed (19, 20). Despite these studies, many mechanisms whereby cells are damaged by particular oxidants or protect themselves from damage remain to be identified.
The availability of a genomewide set of deletion strains (21) has enabled comprehensive screening of cellular functions involved in stress responses. Some genes that play a role may, when deleted, confer no specific phenotype because of gene redundancy or compensatory parallel pathways. However, this approach relies on the sheer number of genes under study and the fact that mutations affecting some components of an important function or pathway will show a phenotype of sensitivity or resistance. A preliminary screen has been performed for three types of oxidative stress on ≈600 strains of the EUROFAN collection (14). Because this collection consists mainly of strains that were individually deleted for unknown function, the results of that survey were more relevant to identifying gene function than to identifying cell functions important in stress resistance. These results also indicated that many more genes involved in maintaining cellular resistance remained to be identified.
This study aimed at obtaining a comprehensive view of the cellular functions needed to maintain cellular viability to ROS by screening the S. cerevisiae deletion mutant collection (21) by using five different ROS: H2O2, linoleic acid 13-hydroperoxide (LoaOOH, a product of lipid peroxidation), menadione (superoxide-generating agent), cumene hydroperoxide (CHP, an aromatic hydroperoxide), and the thiol oxidant diamide.
Methods
Yeast Strains and Growth Conditions. BY4743 homozygous diploid deletions for all nonessential genes from the Saccharomyces Gene Deletion Project (21) were obtained from the European Saccharomyces Cerevisiae Archive for Functional Analysis (EURO-SCARF; www.uni-frankfurt.de/fb15/mikro/euroscarf). Diploids were used to minimize the effect of secondary mutations in haploid cells. Cells were grown in yeast extract/peptone/dextrose (YEPD) [2% (wt/vol) d-glucose, 2% (wt/vol) bacteriological peptone, and 1% yeast extract], synthetic complete (SC) medium [2% (wt/vol) d-glucose, 0.17% yeast nitrogen base, 0.5% ammonium sulfate, and 0.074% complete supplement mixture (Difco)], and yeast extract/peptone/glycerol (YEPG) [3% (vol/vol) glycerol, 2% (wt/vol) bacteriological peptone, and 1% yeast extract]. Agar plates were solidified with 2% (wt/vol) agar. All cells were incubated at 30°C.
Preparation of Oxidant Plates. All plates were prepared 1 d before use. A single batch of SC agar was prepared and cooled to 50°C, oxidant (made fresh) was added, and the agar was immediately mixed and poured. CHP was prepared as a concentrated stock in N,N-dimethyl formamide. LoaOOH was synthesized by using the method of Evans et al. (22). Plates were stored overnight at 4°C in the dark.
Oxidative-Stress Sensitivity Screening. Screening of sensitivity to oxidants was performed by modifying the method of Higgins et al. (14). Strains were thawed and replicated to liquid YEPD in 96-well plates sealed with Breathe-Easy sealing membranes (Sigma–Aldrich) and incubated at 30°C for 2 d without stacking. Strains were diluted 1/10 in 0.17% yeast nitrogen base/0.5% ammonium sulfate by using a Biomek 2000 robotic work station, and OD600 was read to ensure an equal concentration of cells in each well. By using a 96-pin replicator, cells were spotted on SC agar plates containing a range of concentrations of each oxidant, an SC control plate, and a YEPG plate to test respiratory function. Plates were incubated at 30°C for 2 d, and the growth of each mutant was compared with that of the WT on the same plate and with that of the mutant on the control SC plate. Mutants that showed no or severely impaired growth compared with the WT on oxidant plates were scored as sensitive and graded for sensitivity according to the highest concentrations on which the mutant could grow.
Computer Analysis. Statistical analysis of overrepresentation of functional groups was performed by using funspec (23). All available databases were addressed by using a probability cutoff of 0.01 and the Bonferroni correction for multiple testing. Hierarchical clustering analysis of sensitivity and transcriptional data was performed by using genespring version 5 (Silicon Genetics, Redwood City, CA). Mutant sensitivity data were imported into genespring and represented in the same way as gene induction data. Sensitive deletion strains were represented by red, with color intensity decreasing with decreasing sensitivity. Hierarchical clustering was performed in genespring by using the Pearson correlation coefficient.
Results
Identification of 675 Deletant Strains with Altered Sensitivity to at Least One Form of Oxidative Stress. Initially, concentrations of the different oxidants to which the WT showed sensitivity were determined by spot test analysis using a range of concentrations up to 3.5 mM H2O2, 0.08 mM LoaOOH, 5 mM menadione, 0.12 mM CHP, and 1.5 mM diamide. Known oxidative-stress-sensitive mutants were shown to be sensitive over this range (Fig. 1).
Fig. 1.

Determination of oxidant concentrations for mutant screening. The WT and known oxidant-sensitive mutants were inoculated in yeast extract/peptone/dextrose (YEPD) medium in a 96-well plate and grown to stationary phase. The cultures were replicated on plates containing different concentrations of H2O2, diamide, CHP, LoaOOH, and menadione. Representative examples of H2O2, menadione (Men.), and CHP plates are shown. The first two columns in each panel represent the WT strain.
Deletants from the genomewide set (n = 4,546) were tested for sensitivity to five oxidants and resistance to two (H2O2 and diamide). At least three concentrations of each oxidant were used to test for sensitivity, and one was used to test for resistance. This screen identified 675 deletants as sensitive and 45 as resistant to at least one oxidant. To rank the deletants in terms of their sensitivity, they were rescreened in a single experiment over a more extensive set of oxidant concentrations. In all, for each oxidant, seven concentrations were used: 0.75–1.6 mM H2O2, 0.5–1.2 mM menadione, 0.045–0.08 mM LoaOOH, 0.8–1.2 mM diamide, and 0.035–0.11 mM CHP. These data can be found in Table 2, which is published as supporting information on the PNAS web site. Sensitive strains were ranked from one to six, with one denoting the most sensitive.
Identification of Genes and Functions Required for Oxidative-Stress Resistance. Of the 675 sensitive deletants, 219 were sensitive only to the highest concentration of at least one oxidant. Although these included GLR1 and HAP1 deletants, the majority of mutants known to be affected by oxidative stress were sensitive to more than one concentration. These included those affected in glutathione peroxidase (GPX3), thioredoxin (TRX2), cytochrome c peroxidase (CCP1), pentose phosphate pathway enzymes (GND1 and RPE1) and transcription factors for response to oxidative stress (YAP1 and SKN7). To ensure that mutants only weakly affected did not bias the analysis, the 456 that showed sensitivity to at least two concentrations of any oxidant were chosen for subsequent analysis.
The sensitivity data showed several striking features, the first of which was the number of genes involved. Relatively few genes were previously known for their involvement in protection from ROS, but this study identified many hundreds of other genes of equal importance. Secondly, no two oxidants gave the same or a similar profile of sensitivity, with more deletion strains sensitive to only one oxidant than to multiple oxidants (Fig. 2A). Therefore, there were very few mutants in which sensitivity could be ascribed to a general weakening of the cell by virtue of mutation.
Fig. 2.

Hierarchical clustering of deletion mutant sensitivity data. (A) Sensitivity data (obtained as described in Methods) were imported into genespring 5 so that highest sensitivity was represented by the most intense red color. Hierarchical clustering was performed by using the Pearson correlation, and clusters were analyzed for an overrepresentation of biological functions. (B) Hierarchical clustering of the microarray data from Gasch et al. (18) and our own data on LoaOOH (N.A., V.J.H., and I.W.D., unpublished data) was combined with the deletion mutant data. Transcriptional data were given a weight of 75, relative to 1 for the sensitivity data. HS, heat shock; C, CHP; L, LoaOOH; M, menadione; H, H2O2; D, diamide.
The majority of the 456 deletions were of genes with no previously known association with oxidative-stress tolerance, including 157 genes of unknown function. To identify cell functions important in maintaining resistance, genes were grouped into functional categories (Table 1) according to funspec (23).
Table 1. Functional categories overrepresented in the sensitive mutant data.
| Database | Annotation | Genes conferring resistance | P value |
|---|---|---|---|
| H2O2 | |||
| MIPS protein complex | Mitochondrial ribosomes | 17 genes | <1 e-14 |
| Mitochondrial ribosomal large subunit | 13 genes | <1 e-14 | |
| Cytochrome bc1 | COR1, QCR7, QCR8, CYT1 | 1.8 e-5 | |
| MIPS phenotype | Respiratory deficiency | 18 genes | 8.4 e-10 |
| GO for cellular component | Mitochondrial ribosomes | 21 genes | <1 e-14 |
| Mitochondrial large ribosomal subunit | 14 genes | <1 e-14 | |
| Mitochondrial small ribosomal subunit | 6 genes | 5.9 e-6 | |
| Respiratory chain complex III | COR1, QCR7, QCR8, CYT1 | 1.8 e-5 | |
| GO for biological process | Mitochondrial genome maintenance | RIM1, YHM1, MGM101, MRPL20, ABF2, HMI1 | 2.9 e-7 |
| Aerobic respiration | 10 genes | 3.0 e-8 | |
| Diamide | |||
| MIPS functional classification | Vacuolar transport | VPS8, STP22, PEP7, FAB1, DID4, SNF7, VPS9, TLG2, VMA4, VPS30, VMA13 | 2.3 e-7 |
| Protein targeting, sorting, and translocation | VPS8, SEC66, PEP7, VPS3, FAB1, DID4, VPS1, SNF7, PEP3, SEC72, VPS33, MFT1, VPS9, YDJ1, TLG2, VPS17, TIM18 | 4.7 e-7 | |
| GO for biological process | Vacuole organization and biogenesis | BSD2, STP22, VPS3, FAB1, VPS25, VPS1, VPS67, PEP3, VPS65, VPS33, VPS9, VPS75, VMA4, VPS30, VMA13 | 3.3 e-7 |
| Vacuolar protein targeting | BSD2, STP22, VPS3, VPS25, VPS1, VPS67, VPS65, VPS9, VPS75, VPS30 | 4.1 e-6 | |
| GO for molecular function | RNA polymerase II transcription elongation factor | PAF1, SPT4, ELP2, IKI3, ELP3, ELP4 | 4.0 e-6 |
| GO for cellular component | Transcription elongation factor complex | PAF1, SPT4, ELP2, IKI3, ELP3, ELP4 | 8.4 e-6 |
| CHP | |||
| GO for molecular function | Transcriptional regulator | ROX3, PAF1, SNF5, ADA2, PGD1, SPT4, SRB5, SNF6, SKN7, SWI3, YAP1, URE2, LEM3, GAL11, HFI1 | 2.7 e-7 |
| RNA polymerase II transcription factor | ROX3, SNF5, PGD1, SRB5, SNF6, SWI3, GAL11 | 3.2 e-6 | |
| GO for biological process | Vacuole organization and biogenesis | VMA2, TRX2, VMA10, VPS25, PEP3, VPS33, VPS75, VPH1, VMA4, TFP3 | 2.5 e-7 |
| Vacuolar acidification | VMA2, VMA10, VPH1, VMA4, TFP3 | 1.3 e-6 | |
| LoaOOH | |||
| MIPS functional classification | Regulation of C-compound and carbohydrate utilization | TPS1, SNF5, PTC1, REG1, SNF6, YHR155W, PFK26, IMP2, FBP26, SWI3, GAL11, PHO85 | 8.3 e-7 |
Gene lists were analyzed by using funspec (23). Pvalues represent the probability that the intersection of a given list of genes with any given functional category occurs by chance. e is the base of natural logarithms. MIPS, Munich Information Center for Protein Sequences; GO, Gene Ontology.
The Electron Transport Chain Is Vital for H2O2 Tolerance. The most striking feature of H2O2 sensitivity was the relative abundance of genes involved in mitochondrial function (Fig. 3). This function had the greatest significance (P < 1.0 e-14) of any gene set examined (Table 1). For all other oxidants, 21–34% of mutants were respiratorily deficient, compared with 88% of the H2O2-sensitive deletion strains. The main cluster of genes that when deleted gave sensitivity to only H2O2 was primarily involved in mitochondrial function (Fig. 2A).
Fig. 3.

Functional grouping of deletion mutant sensitivity data. Sensitive strains were organized in functional groups based on the categories of genes identified in the Munich Information Center for Protein Sequences (MIPS) database and Saccharomyces Genome Database, combined with visual inspection. The relative contribution of each functional group to sensitivity is shown for each oxidative-stress condition examined. The number of sensitive strains is shown next to each condition. More detailed functional grouping is available in Table 2. Mit, mitochondria; Vac, VPS and vacuolar function; Met, metabolism; Pro, mRNA/protein synthesis; CW, cell wall; O/U, other/unknown.
Respiratory chain functions were those required for H2O2 tolerance. Strains with defects in mitochondrial protein synthesis or mitochondrial DNA maintenance were significantly overrepresented according to Munich Information Center for Protein Sequences (MIPS) classifications and Gene Ontology (GO) annotations (Table 1), whereas strains with disruptions of the electron transport chain were strongly represented. Mutants affected in ubiquinone biosynthesis, subunits of complexes III and IV, and in assembly of complex III were sensitive. Complex III was of central importance (Table 1) because many strains deleted for its subunits (COR1, QCR7, QCR8, and CYT1) were sensitive. Additionally, deletion of CBP3, a complex III assembly gene, conferred sensitivity to H2O2. All of these results heavily implicate the need for a complete respiratory chain, in particular complexes III and IV, in maintaining resistance to H2O2.
Only five deletants showed resistance to H2O2. These included two subunits (THP2 and MFT1) of the four-member THO complex, which functions in transcriptional elongation and genome stability. Deletion of these genes results in transcriptionally activated hyperrecombination phenotypes (24), which may implicate DNA recombination repair in correction of DNA damage known to be induced by H2O2 (25).
Lipid and Carbohydrate Metabolism Is Crucial for LoaOOH Tolerance. S. cerevisiae does not synthesize polyunsaturated lipids, but it can take them up from the environment and incorporate them into membranes (26). Hence, lipid peroxidation is a major source of membrane damage in these cells (22). Mutants affected in ergosterol biosynthesis (ERG3), synthesis of mannosylated sphingolipids (SUR1 and CSG2), sterol uptake (ARV1 and PDX3), peroxisome function (PEX4, PEX19, and POT1), and vacuolar lipid degradation (AUT1 and CVT17) were mainly sensitive to LoaOOH, but not other oxidants. Peroxisome function is of central importance because strains lacking it were sensitive to only LoaOOH, and the peroxisomal biogenesis gene PEX17 is essential for cell-cycle delay during LoaOOH-induced stress (10). Peroxisome may be the site of LoaOOH detoxification through its breakdown, or it may be needed for breakdown of other damaged lipids. Another potential site for the degradation of LoaOOH and other damaged lipids is the vacuole, because CVT17 is required for the degradation of subvacuolar lipid vesicles and has a role in the vacuolar breakdown of membranes (27).
The most significant functional category of the LoaOOH-sensitive mutants was involvement in regulation of C-compound and carbohydrate utilization (Table 1 and Fig. 3). The pentose phosphate pathway (GND1, RPE1, and TKL1) and control of the balance between glycolysis and gluconeogenesis were essential for resistance. Strains deleted for either phosphofructokinase 2 or fructose bisphosphatase 2 activities encoded by FBP26 and PFK26 (28) showed a high degree of sensitivity only to LoaOOH. Specificity to this stress was found for other aspects of carbohydrate metabolism, including trehalose metabolism (TPS1) and utilization of maltose, galactose, and raffinose (IMP2′). LoaOOH sensitivity was enhanced when genes involved in the TCA cycle, isocitrate dehydrogenase (IDH2), coenzyme Q biosynthesis (COQ1), cytochrome oxidase assembly (COX15), and FO–F1 ATP synthase (ATP7), were deleted. It is proposed that energy is required to maintain active defense against lipid peroxidation in contrast to the need to avoid free-radical leakage from the electron transport chain during H2O2-induced stress (see below).
The Pentose Phosphate Pathway Is Required for Menadione Tolerance. The superoxide-generating agent menadione gave the fewest sensitive deletion strains. A striking feature of menadione-sensitive mutants was the extreme sensitivity of those deleted for pentose phosphate pathway genes. Strains deleted for GND1, RPE1, and TKL1 were sensitive to the physiological oxidants H2O2 and LoaOOH, but these were among the five most sensitive to menadione of all strains tested. This finding indicates that NADPH and possibly antioxidant functions that use NADPH have a more significant role in the detoxification of the superoxide ion than do other ROS.
Diamide Tolerance Needs Vacuolar Protein-Sorting (VPS) Functions. Diamide preferentially oxidizes small (i.e., in terms of molecular weight) thiols and rapidly decreases the intracellular glutathione pool (29). This may account for the distinctive pattern of mutant functions that led to diamide sensitivity relative to other oxidants (Fig. 3 and Table 1). Many of the mutants displaying the phenotype of extreme sensitivity to diamide were impaired in VPS (Fig. 3). The most sensitive mutants were those affected in endoplasmic reticulum (ER) protein sorting (SEC66 and SEC72), VPS class E (VPS25), vacuolar protein targeting (VPS3), and vacuolar import (VID28). Ergosterol metabolism, represented by ERG3, ERG4, and ARV1, is important during diamide stress. Deficiency in VPS is a common feature of these two groups (30) and highlights the importance of this process in a thiol-oxidizing environment.
Mutants elp2, elp3, elp4, and iki3, affected in the ELP transcriptional elongation complex, were conspicuous for their hypersensitivity and specificity to diamide (Table 1). The ELP complex may be involved in the elongation of specific transcripts that are especially needed to combat thiol oxidation.
This curious spectrum of diamide-resistance genes may reflect the lack of free-radical generation, differentiating it from the other ROS. In Penicillium chrysogenum, diamide oxidizes glutathione and induces its synthesis, with no corresponding increase in intracellular peroxide or catalase activity (31). It is likely, therefore, that diamide treatment affects cellular redox potential specifically, because glutathione is an abundant redox buffer and diamide can rapidly cross biological membranes (29). Surprisingly, the mutations causing resistance to diamide included many affecting mitochondrial functions that, when deleted, led to H2O2 sensitivity. Diamide-resistant mutants included HUT1, which encodes a putative disulfide isomerase located in the ER (32). Diamide toxicity may therefore be mediated in part by disruption of appropriate disulfide isomerization in the ER. Disulfide bond formation of proteins in the ER therefore seems to be the main function that is sensitive to changes in cellular redox potential.
Transcriptional Regulation and Vacuolar Acidification Are Needed for CHP Tolerance. Transcription functions were significantly overrepresented in deletants sensitive to CHP, but not other oxidants (Table 1). As for other oxidants, the mutant lacking Yap1p was sensitive to CHP. However, strains with defective regulation of Yap1p were very sensitive to CHP. Thioredoxins negatively regulate Yap1p (33), and the TRX2 deletion was sensitive. Strains deleted for GPX3 and YBP1, whose products are peroxide-signal transducers for Yap1p (34, 35), were also sensitive.
Vacuolar acidification mutants were also overrepresented in those oxidants more sensitive to CHP than others, and this function constituted a major proportion of the overall vacuole grouping (Table 1 and Fig. 3). Genes involved in cell wall function, including CWH36, SSD1, and ECM33 for synthesis and PAF1 and FYV3 for maintenance, were important in combating CHP-induced stress (Fig. 3). Defects in the cell wall may increase permeability to this bulky molecule, resulting in increased toxicity.
Cellular Functions Needed for General Oxidative-Stress Resistance. Whereas the extensive differences between deletion strains sensitive to ROS were striking, the mutants identified had common functions. Core functions required for general tolerance of oxidative damage were identified by those needed for resistance to at least four of the oxidants. These functions could be grouped into three broad categories: protein synthesis (transcription and translation), VPS, and cell wall biosynthesis and maintenance.
The most important cellular functions for all types of oxidative stress included those involved in gene expression; this finding highlights the requirement for de novo transcription in response to oxidative damage. Many strains lacking proteins associated with, or part of, the RNA polymerase II complex were sensitive to most of the oxidants, including those deleted for GAL11, SRB5, PGD1, ROX3, PAF1, and SIN3. Chromatin remodeling by means of the ADA and SAGA histone acetyltransferase complexes was important (SPT8, HFI1, and ADA2). The SWI/SNF nucleosome remodeling complex was highly represented (SNF2, SNF5, SNF6, and SWI3). As expected, genes encoding the specific oxidative-stress transcription factors YAP1 and SKN7 were found to be essential for viability during all forms of oxidative stress. The YAP1 deletion mutant was more sensitive than that deleted for SKN7. Yap1p is known to have a larger role than Skn7p during oxidative stress and to be more specific to ROS than Skn7p (36). Mutations resulting in defective translation initiation were sensitive to most ROS (DOM34, HCR1, EAP1, TIF3, and PAT1).
Protein sorting and vacuole function (VID28, PEP3, VID30, and LUV1), vacuolar acidification (VMA2 and VMA4), and ergosterol metabolism (ERG3, ERG4, and ERG5) were all VPS functions found to be required for broad resistance to oxidative stress. VPS follows protein synthesis and may be important during oxidative stress for processing and trafficking of response proteins and for removal of damaged proteins. Cell wall maintenance functions also were essential. Genes that when deleted conferred sensitivity included those directly affecting cell wall maintenance (CWH8 and CWH36) and others involved in cellular functions known to affect the cell wall (ADA2, FAB1, and PAT1). This may account for the importance of the VPS pathways, because they would be required for synthesis of cell surface components.
Most Genes Required to Maintain Resistance Are Not Induced in Response to Oxidative Stress. All genes whose deletion conferred sensitivity were analyzed for their transcriptional response to oxidative and heat stress by using the data of Gasch et al. (18), and our results for LoaOOH (N.A., V.J.H., and I.W.D., unpublished data). The sensitivity results and transcriptional data were clustered together, giving the transcriptional data the most weight to show the relationship between mutant sensitivity and expression of the relevant gene (Fig. 2B). Genes important for resistance to oxidative stress were randomly distributed when compared with the clustered transcriptional data. Such a discrepancy between transcription and sensitivity has been shown in the case of DNA-damaging agents (37). One of the very few exceptions was YAP1, which was upregulated during all stresses and which when deleted conferred sensitivity to all forms of oxidative stress.
Discussion
To date, ≈30 S. cerevisiae genes have been studied for their antioxidant function (1, 12). This study has increased the number of genes involved in ROS resistance by at least 10-fold, to 456, including 157 genes of unknown function. These numbers are remarkable because lethal and multiple gene deletions were not examined; this result demonstrates the power of genomewide mutational analysis. Moreover, few results could be ascribed to mutations causing a general weakening of the cell, because few mutants were sensitive to all oxidants. This finding, coupled with the differences in functions identified, highlights the specificity of deletion mutant analysis. Functions whose role in ROS resistance have not previously been emphasized include VPS, vacuolar acidification, and ergosterol metabolism. Previously, a range of different oxidants has been used to study oxidative stress. The data presented here show that oxidative stress encompasses a broad range of cellular insults that have profoundly different and very specific physiological consequences for the cell, because many functions were uniquely required for resistance to only one ROS.
This specificity contrasts with the data obtained from gene expression studies using transcriptional or proteomic analysis (17, 18, 38). These approaches are therefore very complementary. Analysis of protein changes in response to H2O2, and transcriptional changes in response to diamide, H2O2, menadione, and a shift in carbon source from glucose to oleate all showed a strong induction of classic Yap1p targets. These included the glutathione and thioredoxin detoxification systems and other well characterized antioxidant functions such as catalase and superoxide dismutase (17, 18, 38). Gasch et al. (18) showed the existence of a large regulon of genes responding to a broad range of stress conditions including oxidative stress. The genes involved in this general stress response are controlled by the Msn2p/Msn4p transcription factors. Therefore, there is a set of largely constitutive cell processes needed for maintenance of cell viability in the face of different oxidants; such functions depend on the nature of the oxidant. After these processes are overwhelmed by ROS, the cell has more general functions that are induced for detoxification, repair, or recovery from damage.
More detailed insight was gained on functions with a known role in oxidative stress. There is a remarkably specific requirement for an intact respiratory chain in defense against H2O2-induced stress. Whereas rho0 petite mutants lacking all mitochondrial DNA are known to be sensitive to H2O2 (6), the extent of mitochondrial involvement in defense against H2O2 had not been previously understood. A requirement for energy was proposed to explain why petites are more sensitive than respiratorily competent strains (8). This scenario seems less likely because H2O2-sensitive deletants did not identify a deficiency in other energy-generating reactions. The requirement for a complete respiratory chain is more likely to be caused by the mechanism of toxicity of H2O2. Respiratory chain complex III is responsible for >80% of the superoxide produced (39), and the presence of H2O2 during enhanced superoxide production may be critical. The simultaneous presence of superoxide and H2O2 in the cell would catalyze production of the reactive hydroxy radical by Fenton chemistry. Any transition metal ions would be reduced by the superoxide anion, leading to hydroxyl radical formation from H2O2 (4). Additionally, protein oxidation is linked to ROS generated by semiquinones of the Q-cycle (40). Despite the superoxide radical's conversion to H2O2 during its detoxification (4), there was little overlap between the defense mechanisms for H2O2 and menadione, especially in mitochondrial functions. This highlights the fact that superoxide and H2O2 effects on the cell are fundamentally different, and it is consistent with the above hypothesis concerning the toxicity of superoxide in the presence of added H2O2 and with the differences seen in cross-adaptation between superoxide and H2O2 (7).
Because protein metabolism was a core function for survival of all oxidants, protein damage would seem to be an essential component of their toxicity. Very few mutants affected in DNA repair were sensitive, excluding those involved in the repair of mitochondrial DNA. Protein, not DNA, damage may therefore be the major factor in the loss of cell viability as a result of oxidative stress.
These findings have wider implications, because ROS produced in the mitochondria have been linked to aging in a range of organisms including Caenorhabditis elegans and in S. cerevisiae (41, 42). Further understanding of the role of the respiratory chain in the tolerance of ROS could provide insights on the role of ROS in the aging process. Mitochondrially produced ROS have been implicated in a caspase-independent mechanism of apoptotic induction in mammals (43); however, much of the work has been performed in yeast (1). Yeast has been used as a model for the study of mitochondrial biogenesis in humans and to identify human disease genes involving mitochondria through the identification of yeast mitochondrial homologues that when deleted reduce fitness (44, 45). Clearly the study of oxidative stress in yeast, especially the role of mitochondria, offers exciting insight on mechanisms involved in aging, apoptosis, and disease in higher organisms.
Supplementary Material
Acknowledgments
We thank Gabriel Perrone, Anthony Beckhouse, Cristy Gelling, Jia Qi Wang, and Thomas Felder for assistance and helpful discussions. This research was supported by grants from the Australian Research Council.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ROS, reactive oxygen species; VPS, vacuolar protein sorting; CHP, cumene hydroperoxide; LoaOOH, linoleic acid 13-hydroperoxide.
References
- 1.Costa, V. & Moradas-Ferreira, P. (2001) Mol. Aspects Med. 22, 217-246. [DOI] [PubMed] [Google Scholar]
- 2.Busciglio, J. & Yankner, B. A. (1995) Nature 378, 776-779. [DOI] [PubMed] [Google Scholar]
- 3.Fleury, C., Pampin, M., Tarze, A. & Mignotte, B. (2002) Biosci. Rep. 22, 59-79. [DOI] [PubMed] [Google Scholar]
- 4.Gille, G. & Sigler, K. (1995) Folia Microbiol. (Prague) 40, 131-152. [DOI] [PubMed] [Google Scholar]
- 5.Jamieson, D. J. (1992) J. Bacteriol. 174, 6678-6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collinson, L. P. & Dawes, I. W. (1992) J. Gen. Microbiol. 138, 329-335. [DOI] [PubMed] [Google Scholar]
- 7.Flattery-O'Brien, J. A., Collinson, L. P. & Dawes, I. W. (1993) J. Gen. Microbiol. 139, 501-507. [DOI] [PubMed] [Google Scholar]
- 8.Grant, C. M., MacIver, F. H. & Dawes, I. W. (1997) FEBS Lett. 410, 219-222. [DOI] [PubMed] [Google Scholar]
- 9.Flattery-O'Brien, J. A. & Dawes, I. W. (1998) J. Biol. Chem. 273, 8564-8571. [DOI] [PubMed] [Google Scholar]
- 10.Alic, N., Higgins, V. J. & Dawes, I. W. (2001) Mol. Biol. Cell 12, 1801-1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wanke, V., Accorsi, K., Porro, D., Esposito, F., Russo, T. & Vanoni, M. (1999) Mol. Microbiol. 32, 753-764. [DOI] [PubMed] [Google Scholar]
- 12.Jamieson, D. J. (1998) Yeast 14, 1511-1527. [DOI] [PubMed] [Google Scholar]
- 13.Marini, M., Frabetti, F., Musiani, D. & Franceschi, C. (1996) Int. J. Radiat. Biol. 70, 337-350. [DOI] [PubMed] [Google Scholar]
- 14.Higgins, V. J., Alic, N., Thorpe, G. W., Breitenbach, M., Larsson, V. & Dawes, I. W. (2002) Yeast 19, 203-214. [DOI] [PubMed] [Google Scholar]
- 15.Turton, H. E., Dawes, I. W. & Grant, C. M. (1997) J. Bacteriol. 179, 1096-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wemmie, J. A., Steggerda, S. M. & Moye-Rowley, W. S. (1997) J. Biol. Chem. 272, 7908-7914. [DOI] [PubMed] [Google Scholar]
- 17.Godon, C., Lagniel, G., Lee, J., Buhler, J. M., Kieffer, S., Perrot, M., Boucherie, H., Toledano, M. B. & Labarre, J. (1998) J. Biol. Chem. 273, 22480-22489. [DOI] [PubMed] [Google Scholar]
- 18.Gasch, A. P., Spellman, P. T., Kao, C. M., Carmel-Harel, O., Eisen, M. B., Storz, G., Botstein, D. & Brown, P. O. (2000) Mol. Biol. Cell 11, 4241-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krems, B., Charizanis, C. & Entian, K. D. (1995) Curr. Genet. 27, 427-434. [DOI] [PubMed] [Google Scholar]
- 20.Cohen, G., Fessel, F., Traczyk, A., Rytka, J. & Ruis, H. (1985) Mol. Gen. Genet. 200, 74-79. [DOI] [PubMed] [Google Scholar]
- 21.Winzeler, E. A., Shoemaker, D. D., Astromoff, A., Liang, H., Anderson, K., Andre, B., Bangham, R., Benito, R., Boeke, J. D., Bussey, H., et al. (1999) Science 285, 901-906. [DOI] [PubMed] [Google Scholar]
- 22.Evans, M. V., Turton, H. E., Grant, C. M. & Dawes, I. W. (1998) J. Bacteriol. 180, 483-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson, M. D., Grigull, J., Mohammad, N. & Hughes, T. R. (2002) BMC Bioinformatics 3, 35-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chavez, S., Beilharz, T., Rondon, A. G., Erdjument-Bromage, H., Tempst, P., Svejstrup, J. Q., Lithgow, T. & Aguilera, A. (2000) EMBO J. 19, 5824-5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imlay, J. A., Chin, S. M. & Linn, S. (1988) Science 240, 640-642. [DOI] [PubMed] [Google Scholar]
- 26.Avery, S. V., Howlett, N. G. & Radice, S. (1996) Appl. Environ. Microbiol. 62, 3960-3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epple, U. D., Eskelinen, E. L. & Thumm, M. (2003) J. Biol. Chem. 278, 7810-7821. [DOI] [PubMed] [Google Scholar]
- 28.Paravicini, G. & Kretschmer, M. (1992) Biochemistry 31, 7126-7133. [DOI] [PubMed] [Google Scholar]
- 29.Kosower, N. S. & Kosower, E. M. (1995) Methods Enzymol. 251, 123-133. [DOI] [PubMed] [Google Scholar]
- 30.Seeley, E. S., Kato, M., Margolis, N., Wickner, W. & Eitzen, G. (2002) Mol. Biol. Cell 13, 782-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emri, T., Pocsi, I. & Szentirmai, A. (1997) Free Radical Biol. Med. 23, 809-814. [DOI] [PubMed] [Google Scholar]
- 32.Nakanishi, H., Nakayama, K., Yokota, A., Tachikawa, H., Takahashi, N. & Jigami, Y. (2001) Yeast 18, 543-554. [DOI] [PubMed] [Google Scholar]
- 33.Izawa, S., Maeda, K., Sugiyama, K., Mano, J., Inoue, Y. & Kimura, A. (1999) J. Biol. Chem. 274, 28459-28465. [DOI] [PubMed] [Google Scholar]
- 34.Delaunay, A., Pflieger, D., Barrault, M. B., Vinh, J. & Toledano, M. B. (2002) Cell 111, 471-481. [DOI] [PubMed] [Google Scholar]
- 35.Veal, E. A., Ross, S. J., Malakasi, P., Peacock, E. & Morgan, B. A. (2003) J. Biol. Chem. 278, 30896-30904. [DOI] [PubMed] [Google Scholar]
- 36.Lee, J., Godon, C., Lagniel, G., Spector, D., Garin, J., Labarre, J. & Toledano, M. B. (1999) J. Biol. Chem. 274, 16040-16046. [DOI] [PubMed] [Google Scholar]
- 37.Birrell, G. W., Brown, J. A., Wu, H. I., Giaever, G., Chu, A. M., Davis, R. W. & Brown, J. M. (2002) Proc. Natl. Acad. Sci. USA 99, 8778-8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koerkamp, M. G., Rep, M., Bussemaker, H. J., Hardy, G. P., Mul, A., Piekarska, K., Szigyarto, C. A., De Mattos, J. M. & Tabak, H. F. (2002) Mol. Biol. Cell 13, 2783-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chance, B., Sies, H. & Boveris, A. (1979) Physiol. Rev. 59, 527-605. [DOI] [PubMed] [Google Scholar]
- 40.Aguilaniu, H., Gustafsson, L., Rigoulet, M. & Nystrom, T. (2001) J. Biol. Chem. 276, 35396-35404. [DOI] [PubMed] [Google Scholar]
- 41.Hekimi, S. & Guarente, L. (2003) Science 299, 1351-1354. [DOI] [PubMed] [Google Scholar]
- 42.Laun, P., Pichova, A., Madeo, F., Fuchs, J., Ellinger, A., Kohlwein, S., Dawes, I., Frohlich, K. U. & Breitenbach, M. (2001) Mol. Microbiol. 39, 1166-1173. [PubMed] [Google Scholar]
- 43.Fleury, C., Mignotte, B. & Vayssiere, J. L. (2002) Biochimie 84, 131-141. [DOI] [PubMed] [Google Scholar]
- 44.Foury, F. & Kucej, M. (2002) Curr. Opin. Chem. Biol. 6, 106-111. [DOI] [PubMed] [Google Scholar]
- 45.Steinmetz, L. M., Scharfe, C., Deutschbauer, A. M., Mokranjac, D., Herman, Z. S., Jones, T., Chu, A. M., Giaever, G., Prokisch, H., Oefner, P. J., et al. (2002) Nat. Genet. 31, 400-404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.